Computational Insights into the Potential of Withaferin-A, Withanone and Caffeic Acid Phenethyl Ester for Treatment of Aberrant-EGFR Driven Lung Cancers



Abstract
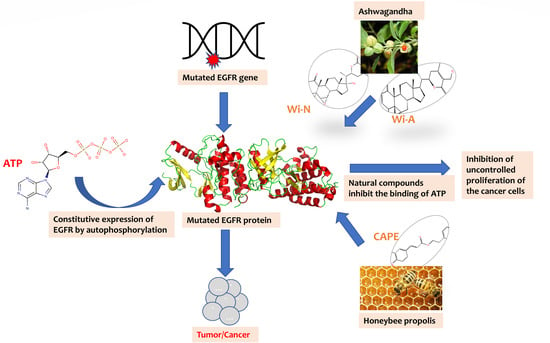
1. Introduction
2. Materials and Methods
2.1. Structure Preparation of Proteins and Ligands
2.2. Molecular Docking and MD Simulations to Check the Potential of Natural Compounds to Serve as ATP Competitive Inhibitors of EGFR Mutants
2.3. Analysis of MD Simulated Systems
3. Results
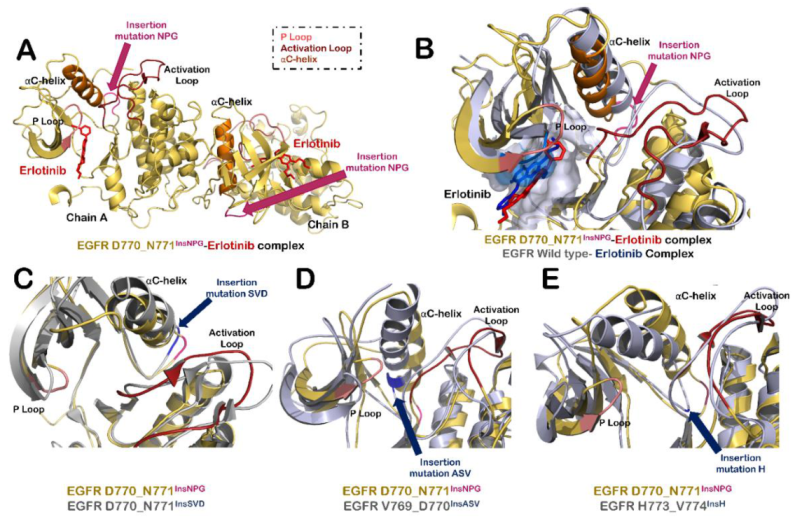
3.1. Computational Modeling of Exon 20 Insertion Mutants of EGFR
3.2. Structural Differences between the Active and Inactive Conformation of EGFR Protein
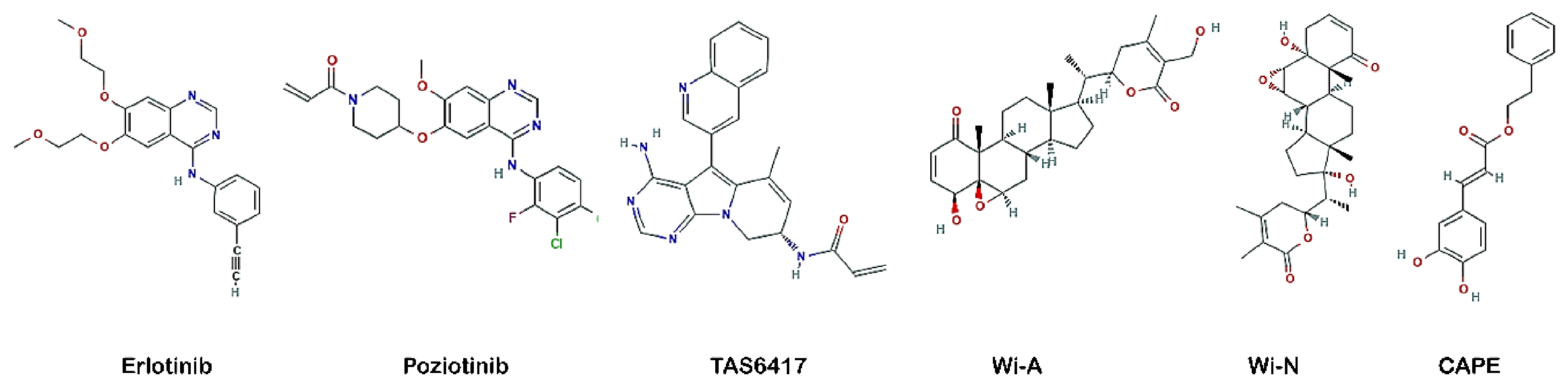
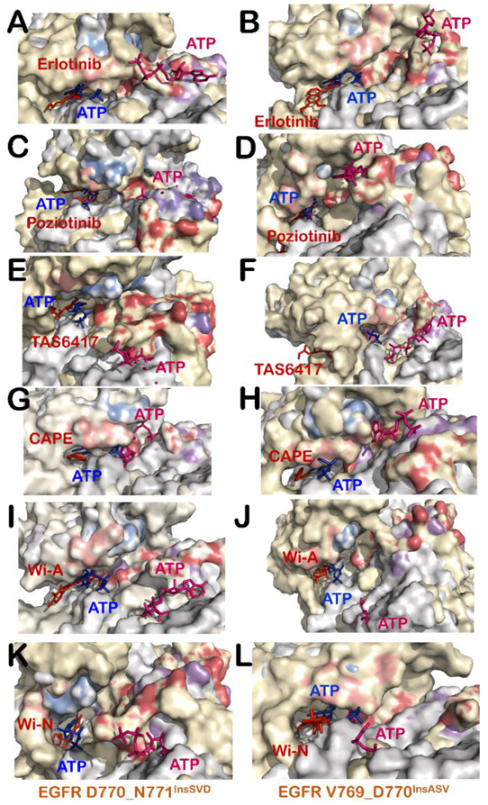
3.3. Wi-A, Wi-N and CAPE Showed Potential as Competitive Inhibitors of ATP for EGFR Mutants
3.4. All Compounds Showed Potential as ATP Competitive Inhibitors for EGFR Exon 20 Insertion Mutants (D770_N771InsSVD and V769_D770InsASV)
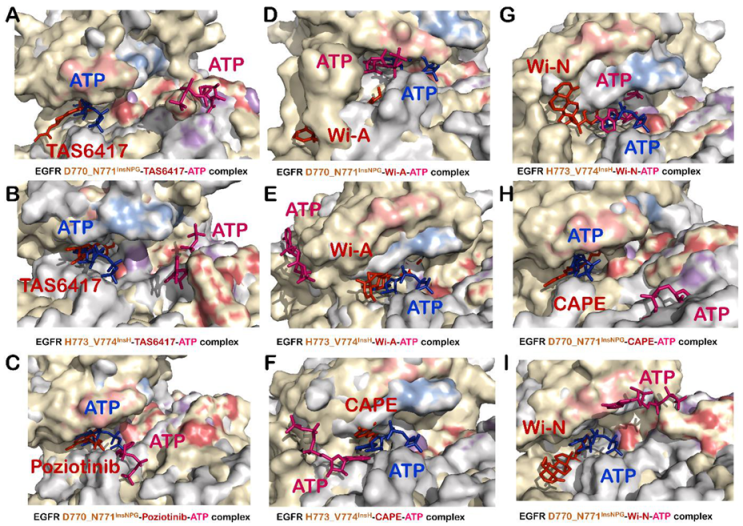
3.5. TAS6417 Could Serve as ATP Competitive Inhibitor of All the Four Exon 20 Insertion Mutants
3.6. Poziotinib and Wi-A Could Not Serve as ATP Competitive Inhibitors for D770_N771InsNPG Mutant, Whereas CAPE and Wi-N Could Not Inhibit H773_V774InsH Mutant of EGFR
3.7. Combination of Wi-A and Wi-N Could Target Activity of All Four Exon 20 Insertion Mutants, L858R and Exon19del Mutants as Well as Wildtype EGFR
3.8. CAPE Could Serve as Inhibitors for the Activity of Wildtype EGFR and Exon 20 Insertion Mutants Only
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. S1), S24–S31. [Google Scholar] [CrossRef]
- Li, E.; Hristova, K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 2006, 45, 6241–6251. [Google Scholar] [CrossRef]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef]
- Katz, M.; Amit, I.; Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 2007, 1773, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Vyse, S.; Huang, P.H. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Huang, Y.; Hong, S.; Zhang, Z.; Wang, M.; Gan, J.; Wang, W.; Guo, H.; Wang, K.; Zhang, L. EGFR exon 20 insertion mutations and response to osimertinib in non-small-cell lung cancer. BMC Cancer 2019, 19, 595. [Google Scholar] [CrossRef]
- Engelman, J.A.; Janne, P.A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 2895–2899. [Google Scholar] [CrossRef]
- Liao, B.C.; Lin, C.C.; Yang, J.C. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr. Opin. Oncol. 2015, 27, 94–101. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Floc’h, N.; Martin, M.J.; Riess, J.W.; Orme, J.P.; Staniszewska, A.D.; Menard, L.; Cuomo, M.E.; O’Neill, D.J.; Ward, R.A.; Finlay, M.R.V.; et al. Antitumor Activity of Osimertinib, an Irreversible Mutant-Selective EGFR Tyrosine Kinase Inhibitor, in NSCLC Harboring EGFR Exon 20 Insertions. Mol. Cancer Ther. 2018, 17, 885–896. [Google Scholar] [CrossRef]
- Cha, M.Y.; Lee, K.O.; Kim, M.; Song, J.Y.; Lee, K.H.; Park, J.; Chae, Y.J.; Kim, Y.H.; Suh, K.H.; Lee, G.S.; et al. Antitumor activity of HM781-36B, a highly effective pan-HER inhibitor in erlotinib-resistant NSCLC and other EGFR-dependent cancer models. Int. J. Cancer 2012, 130, 2445–2454. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Elamin, Y.Y.; Tan, Z.; Carter, B.W.; Zhang, S.; Liu, S.; Li, S.; Chen, T.; Poteete, A.; Estrada-Bernal, A.; et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat. Med. 2018, 24, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Hasako, S.; Terasaka, M.; Abe, N.; Uno, T.; Ohsawa, H.; Hashimoto, A.; Fujita, R.; Tanaka, K.; Okayama, T.; Wadhwa, R.; et al. TAS6417, A Novel EGFR Inhibitor Targeting Exon 20 Insertion Mutations. Mol. Cancer Ther. 2018, 17, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bhalla, M.; de Jager, P.; Gilca, M. An overview on ashwagandha: A Rasayana (rejuvenator) of Ayurveda. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Ven Murthy, M.R.; Ranjekar, P.K.; Ramassamy, C.; Deshpande, M. Scientific basis for the use of Indian ayurvedic medicinal plants in the treatment of neurodegenerative disorders: Ashwagandha. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.C.; Wadhwa, R. Science of Ashwagandha: Preventive and Therapeutic Potentials; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Grover, A.; Priyandoko, D.; Gao, R.; Shandilya, A.; Widodo, N.; Bisaria, V.S.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Withanone binds to mortalin and abrogates mortalin-p53 complex: Computational and experimental evidence. Int. J. Biochem. Cell Biol. 2012, 44, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lim, I.H.; Sung, E.G.; Kim, J.Y.; Song, I.H.; Park, Y.K.; Lee, T.J. Withaferin A inhibits matrix metalloproteinase-9 activity by suppressing the Akt signaling pathway. Oncol. Rep. 2013, 30, 933–938. [Google Scholar] [CrossRef]
- Gao, R.; Shah, N.; Lee, J.S.; Katiyar, S.P.; Li, L.; Oh, E.; Sundar, D.; Yun, C.O.; Wadhwa, R.; Kaul, S.C. Withanone-rich combination of Ashwagandha withanolides restricts metastasis and angiogenesis through hnRNP-K. Mol. Cancer Ther. 2014, 13, 2930–2940. [Google Scholar] [CrossRef]
- Yu, Y.; Katiyar, S.P.; Sundar, D.; Kaul, Z.; Miyako, E.; Zhang, Z.; Kaul, S.C.; Reddel, R.R.; Wadhwa, R. Withaferin-A kills cancer cells with and without telomerase: Chemical, computational and experimental evidences. Cell Death Dis. 2017, 8, e2755. [Google Scholar] [CrossRef]
- Bhargava, P.; Malik, V.; Liu, Y.; Ryu, J.; Kaul, S.C.; Sundar, D.; Wadhwa, R. Molecular Insights Into Withaferin-A-Induced Senescence: Bioinformatics and Experimental Evidence to the Role of NFkappaB and CARF. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 74, 183–191. [Google Scholar] [CrossRef]
- Sundar, D.; Yu, Y.; Katiyar, S.P.; Putri, J.F.; Dhanjal, J.K.; Wang, J.; Sari, A.N.; Kolettas, E.; Kaul, S.C.; Wadhwa, R. Wild type p53 function in p53(Y220C) mutant harboring cells by treatment with Ashwagandha derived anticancer withanolides: Bioinformatics and experimental evidence. J. Exp. Clin. cancer Res. CR 2019, 38, 103. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Kalra, R.S.; Malik, V.; Katiyar, S.P.; Sundar, D.; Kaul, S.C.; Wadhwa, R. 2, 3-Dihydro-3beta-methoxy Withaferin-A Lacks Anti-Metastasis Potency: Bioinformatics and Experimental Evidences. Sci. Rep. 2019, 9, 17344. [Google Scholar] [CrossRef] [PubMed]
- Sari, A.N.; Bhargava, P.; Dhanjal, J.K.; Putri, J.F.; Radhakrishnan, N.; Shefrin, S.; Ishida, Y.; Terao, K.; Sundar, D.; Kaul, S.C.; et al. Combination of Withaferin-A and CAPE Provides Superior Anticancer Potency: Bioinformatics and Experimental Evidence to Their Molecular Targets and Mechanism of Action. Cancers 2020, 12, 1160. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sundar, D. Mechanistic Insights into Withanolides Action Against Cancer. In Proceedings of the Proceedings of International Conference on Drug Discovery (ICDD) 2020, Hyderabad, India, 29 February–2 March 2020. [Google Scholar]
- Malik, V.; Garg, S.; Afzal, S.; Dhanjal, J.K.; Yun, C.O.; Kaul, S.C.; Sundar, D.; Wadhwa, R. Bioinformatics and Molecular Insights to Anti-Metastasis Activity of Triethylene Glycol Derivatives. Int. J. Mol. Sci. 2020, 21, 5463. [Google Scholar] [CrossRef]
- Dutta, R.; Khalil, R.; Green, R.; Mohapatra, S.S.; Mohapatra, S. Withania Somnifera (Ashwagandha) and Withaferin A: Potential in Integrative Oncology. Int. J. Mol. Sci. 2019, 20, 5310. [Google Scholar] [CrossRef] [PubMed]
- Kunimasa, K.; Nagano, T.; Shimono, Y.; Dokuni, R.; Kiriu, T.; Tokunaga, S.; Tamura, D.; Yamamoto, M.; Tachihara, M.; Kobayashi, K.; et al. Glucose metabolism-targeted therapy and withaferin A are effective for epidermal growth factor receptor tyrosine kinase inhibitor-induced drug-tolerant persisters. Cancer Sci. 2017, 108, 1368–1377. [Google Scholar] [CrossRef]
- Watabe, M.; Hishikawa, K.; Takayanagi, A.; Shimizu, N.; Nakaki, T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J. Biol. Chem. 2004, 279, 6017–6026. [Google Scholar] [CrossRef]
- Ozen, S.; Akyol, O.; Iraz, M.; Sogut, S.; Ozugurlu, F.; Ozyurt, H.; Odaci, E.; Yildirim, Z. Role of caffeic acid phenethyl ester, an active component of propolis, against cisplatin-induced nephrotoxicity in rats. J. Appl. Toxicol. JAT 2004, 24, 27–35. [Google Scholar] [CrossRef]
- Khalil, M.L. Biological activity of bee propolis in health and disease. Asian Pac. J. Cancer Prev. 2006, 7, 22–31. [Google Scholar]
- Chen, M.J.; Chang, W.H.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Shih, S.C.; Chen, Y.J. Caffeic acid phenethyl ester induces apoptosis of human pancreatic cancer cells involving caspase and mitochondrial dysfunction. Pancreatology 2008, 8, 566–576. [Google Scholar] [CrossRef]
- Izuta, H.; Shimazawa, M.; Tsuruma, K.; Araki, Y.; Mishima, S.; Hara, H. Bee products prevent VEGF-induced angiogenesis in human umbilical vein endothelial cells. BMC Complement. Altern. Med. 2009, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Kumari, A.; Putri, J.F.; Ishida, Y.; Terao, K.; Kaul, S.C.; Sundar, D.; Wadhwa, R. Caffeic acid phenethyl ester (CAPE) possesses pro-hypoxia and anti-stress activities: Bioinformatics and experimental evidences. Cell Stress Chaperones 2018, 23, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, S.; Zhang, L.; Qiao, X.; Zhang, Y.; Zhao, X.; Xiao, G.; Li, Z. CAPE-pNO2 Inhibited the Growth and Metastasis of Triple-Negative Breast Cancer via the EGFR/STAT3/Akt/E-Cadherin Signaling Pathway. Front. Oncol. 2019, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 2011, 308, 43–53. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; Stricker, K.; Grauslund, M.; Sorensen, J.B. Intrinsic resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Willmore-Payne, C.; Holden, J.A.; Wittwer, C.T.; Layfield, L.J. The use of EGFR exon 19 and 21 unlabeled DNA probes to screen for activating mutations in non-small cell lung cancer. J. Biomol. Tech. 2008, 19, 217–224. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-2: Protein Preparation Wizard, Epik, Impact, Prime, LigPrep, Glide; Desmond Molecular Dynamics System, D. E. Shaw Research; Maestro-Desmond Interoperability Tools; Schrödinger, LLC: New York, NY, USA, 2018.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. “VMD—Visual Molecular Dynamics”. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.C.; Wolf, R.M. MM/GBSA binding energy prediction on the PDBbind data set: Successes, failures, and directions for further improvement. J. Chem. Inf. Model. 2013, 53, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Eck, M.J.; Hahn, W.C. EGFR in limbo. Cell 2012, 149, 735–737. [Google Scholar] [CrossRef]
- Schlessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, K.H.; Kim, S.W.; Min, Y.J.; Cho, E.; Lee, Y.; Lee, S.H.; Kim, H.Y.; Lee, G.K.; Nam, B.H.; et al. A Phase II Study of Poziotinib in Patients with Epidermal Growth Factor Receptor (EGFR)-Mutant Lung Adenocarcinoma Who Have Acquired Resistance to EGFR-Tyrosine Kinase Inhibitors. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2017, 49, 10–19. [Google Scholar] [CrossRef][Green Version]
- Kitazaki, T.; Oka, M.; Nakamura, Y.; Tsurutani, J.; Doi, S.; Yasunaga, M.; Takemura, M.; Yabuuchi, H.; Soda, H.; Kohno, S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 2005, 49, 337–343. [Google Scholar] [CrossRef]
- Le, T.; Gerber, D.E. Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used? Cancers 2019, 11, 366. [Google Scholar] [CrossRef] [PubMed]
- Schettino, C.; Bareschino, M.A.; Ricci, V.; Ciardiello, F. Erlotinib: An EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment. Expert Rev. Respir. Med. 2008, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Jazieh, A.R.; Al Sudairy, R.; Abu-Shraie, N.; Al Suwairi, W.; Ferwana, M.; Murad, M.H. Erlotinib in wild type epidermal growth factor receptor non-small cell lung cancer: A systematic review. Ann. Thorac. Med. 2013, 8, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Piperdi, B.; Perez-Soler, R. Role of erlotinib in the treatment of non-small cell lung cancer: Clinical outcomes in wild-type epidermal growth factor receptor patients. Drugs 2012, 72 (Suppl. S1), 11–19. [Google Scholar] [CrossRef]
- Udagawa, H.; Hasako, S.; Ohashi, A.; Fujioka, R.; Hakozaki, Y.; Shibuya, M.; Abe, N.; Komori, T.; Haruma, T.; Terasaka, M.; et al. TAS6417/CLN-081 Is a Pan-Mutation-Selective EGFR Tyrosine Kinase Inhibitor with a Broad Spectrum of Preclinical Activity against Clinically Relevant EGFR Mutations. Mol. Cancer Res. 2019, 17, 2233–2243. [Google Scholar] [CrossRef]
- Kumar, V.; Dhanjal, J.K.; Bhargava, P.; Kaul, A.; Wang, J.; Zhang, H.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Withanone and Withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- White, P.T.; Subramanian, C.; Motiwala, H.F.; Cohen, M.S. Natural Withanolides in the Treatment of Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 329–373. [Google Scholar] [CrossRef]
- Garg, S.; Huifu, H.; Kumari, A.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Induction of Senescence in Cancer Cells by a Novel Combination of Cucurbitacin B and Withanone: Molecular Mechanism and Therapeutic Potential. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2020, 75, 1031–1041. [Google Scholar] [CrossRef]
- Ozturk, G.; Ginis, Z.; Akyol, S.; Erden, G.; Gurel, A.; Akyol, O. The anticancer mechanism of caffeic acid phenethyl ester (CAPE): Review of melanomas, lung and prostate cancers. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 2064–2068. [Google Scholar]
- Widodo, N.; Priyandoko, D.; Shah, N.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by Ashwagandha leaf extract and its component Withanone involves ROS signaling. PLoS ONE 2010, 5, e13536. [Google Scholar] [CrossRef]
- Grover, A.; Singh, R.; Shandilya, A.; Priyandoko, D.; Agrawal, V.; Bisaria, V.S.; Wadhwa, R.; Kaul, S.C.; Sundar, D. Ashwagandha derived withanone targets TPX2-Aurora A complex: Computational and experimental evidence to its anticancer activity. PLoS ONE 2012, 7, e30890. [Google Scholar] [CrossRef] [PubMed]
- Wadegaonkar, V.P.; Wadegaonkar, P.A. Withanone as an inhibitor of survivin: A potential drug candidate for cancer therapy. J. Biotechnol. 2013, 168, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, K.; Saxena, N.; Shah, N.; Singh, R.; Manjunath, K.; Uthayakumar, M.; Kanaujia, S.P.; Kaul, S.C.; Sekar, K.; Wadhwa, R. Differential activities of the two closely related withanolides, Withaferin A and Withanone: Bioinformatics and experimental evidences. PLoS ONE 2012, 7, e44419. [Google Scholar] [CrossRef] [PubMed]
- Ekhteiari Salmas, R.; Durdagi, S.; Gulhan, M.F.; Duruyurek, M.; Abdullah, H.I.; Selamoglu, Z. The effects of pollen, propolis, and caffeic acid phenethyl ester on tyrosine hydroxylase activity and total RNA levels in hypertensive rats caused by nitric oxide synthase inhibition: Experimental, docking and molecular dynamic studies. J. Biomol. Struct. Dyn. 2018, 36, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Moujir, L.M.; Llanos, G.G.; Araujo, L.; Amesty, A.; Bazzocchi, I.L.; Jimenez, I.A. Withanolide-Type Steroids from Withania aristata as Potential Anti-Leukemic Agents. Molecules 2020, 25, 5744. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Kumar, S.; Saloni, H.S.; Kim, M.H.; Sharma, P.; Misra, S.; Khan, F. Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Des. Dev. Ther. 2017, 11, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Mulakala, C.; Viswanadhan, V.N. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J. Mol. Graph. Model. 2013, 46, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EGFR Exon 20 Insertion Mutant | D770_N771InsNPG | D770_N771InsSVD | V769_D770InsASV | H773_V774InsH | |||||
|---|---|---|---|---|---|---|---|---|---|
| Inhibitor | Properties | Chain A | Chain B | Chain A | Chain B | Chain A | Chain B | Chain A | Chain B |
| Erlotinib | Binding Energy of inhibitor | −42.68 | −41.12 | −66.41 | −64.28 | −46.87 | −61.40 | −46.35 | −45.27 |
| Binding Energy of ATP | −32.37 | −24.83 | −16.17 | −27.34 | −22.62 | −27.44 | −20.40 | −25.51 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 3.0 and 3.0 | 2.8 and 3.4 | 2.8 and 3.5 | 2.8 and 3.7 | 2.8 and 4.2 | 2.8 and 3.6 | 2.8 and 3.2 | 4.3 and 4.8 | |
| Downward shift of P loop | No | No | No | yes | yes | yes | No | No | |
| Poziotinib | Binding Energy of inhibitor | −59.55 | −70.14 | −48.17 | −61.34 | −69.83 | −52.96 | −60.82 | −63.21 |
| Binding Energy of ATP | −20.56 | −25.61 | −22.40 | −26.60 | −33.42 | −11.91 | −53.86 | −33.76 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 3.1 and 3.0 | 3.3 and 3.0 | 2.9 and 5 | 2.9 and 3.7 | 3.5 and 5.6 | 2.9 and 3.1 | 2.8 and 4.6 | 2.8 and 3.3 | |
| Downward shift of P loop | yes | yes | yes | No | No | No | No | No | |
| TAS6417 | Binding Energy of inhibitor | −39.31 | −43.73 | −71.92 | −43.88 | −54.74 | −62.26 | −47.06 | −57.40 |
| Binding Energy of ATP | −34.17 | −20.42 | −32.55 | −13.39 | −36.68 | −35.79 | −16.83 | −22.77 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 2.8 and 3.6 | 2.8 and 4.2 | 6.4 and 8.3 | 3.0 and 3.0 | 2.8 and 3.9 | 2.9 and 3.2 | 4.5 and 6.0 | 2.9 and 3.3 | |
| Downward shift of P loop | No | yes | No | yes | yes | yes | No | No | |
| CAPE | Binding Energy of inhibitor | −56.48 | −46.79 | −70.04 | −52.52 | −63.94 | −57.29 | −44.17 | −35.49 |
| Binding Energy of ATP | −28.92 | −28.82 | −20.88 | −36.59 | −29.20 | −17.95 | −39.10 | −40.25 | |
| DFG motif and αC-helix orientation * | In and Out | In | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 10.6 and 12.7 | 2.9 and 3.0 | 3.4 and 4.8 | 2.8 and 3.1 | 2.8 and 4.2 | 2.8 and 5.0 | 3.9 and 4.4 | 2.8 and 3.5 | |
| Downward shift of P loop | No | yes | No | No | yes | yes | No | No | |
| Wi-A | Binding Energy of inhibitor | −86.90 | −46.23 | −30.44 | −25.60 | −53.51 | −45.11 | −50.64 | −42.62 |
| Binding Energy of ATP | −45.06 | −30.59 | −56.43 | −62.40 | −20.97 | −38.84 | −23.17 | −35.66 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 7.3 and 9.2 | 3.2 and 4.2 | 4.1 and 5.8 | 4.1 and 5.8 | 2.8 and 4.6 | 2.8 and 3.9 | 4.9 and 6.0 | 2.8 and 4.3 | |
| Downward shift of P loop | No | No | No | No | yes | yes | No | No | |
| Wi-N | Binding Energy of inhibitor | −48.28 | −39.06 | −62.79 | −58.97 | −38.08 | −9.87 | −35.58 | −37.44 |
| Binding Energy of ATP | −20.17 | −26.49 | −26.82 | −33.29 | −60.96 | −50.39 | −13.47 | −33.15 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 5.6 and 7.5 | 2.8 and 3.5 | 2.8 and 3.1 | 7.8 and 9.5 | 2.8 and 4.7 | 3.2 and 5.0 | 6.0 and 8.1 | 2.9 and 3.0 | |
| Downward shift of P loop | No | Yes | No | Yes | Yes | Yes | No | No | |
| EGFR Mutants | exon19del | EGFR_L858R | Wildtype EGFR | ||||
|---|---|---|---|---|---|---|---|
| Inhibitor | Properties | Chain A | Chain B | Chain A | Chain B | Chain A | Chain B |
| Erlotinib | Binding Energy of inhibitor | −46.61 | −36.29 | −63.30 | −56.41 | −62.06 | −40.37 |
| Binding Energy of ATP | −35.92 | −32.26 | −38.51 | −37.26 | −38.94 | −56.57 | |
| DFG motif and αC-helix orientation * | Out | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 3.6 and 3.1 | 3.4 and 3.2 | 3.9 and 2.8 | 9.6 and 6.4 | 6.4 and 4.7 | 6.5 and 7.5 | |
| Downward movement of P loop | No | Yes | Yes | No | No | Yes | |
| CAPE | Binding Energy of inhibitor | −38.15 | − 58.83 | −45.43 | −49.22 | −65.68 | −63.23 |
| Binding Energy of ATP | −37.61 | −34.63 | −26.92 | −23.62 | −58.39 | −30.15 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 3.6 and 3.5 | 3.4 and 3.7 | 3.9 and 3.1 | 9.6 and 6.5 | 6.4 and 3.9 | 6.5 and 10.1 | |
| Downward movement of P loop | Yes | yes | Yes | Yes | No | Yes | |
| Wi-A | Binding Energy of inhibitor | −49.35 | −46.23 | −22.52 | −44.50 | −49.50 | −48.78 |
| Binding Energy of ATP | −45.06 | −30.59 | −56.43 | −62.40 | −35.78 | −35.78 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 3.6 and 3.4 | 3.4 and 3.2 | 3.9 and 3.1 | 9.6 and 6.4 | 6.4 and 3.5 | 6.5 and 6.4 | |
| Downward movement of P loop | No | No | No | No | No | Yes | |
| Wi-N | Binding Energy of inhibitor | −59.83 | −22.21 | −45.76 | −51.86 | −44.88 | −51.70 |
| Binding Energy of ATP | −14.15 | −23.45 | −20.82 | −20.29 | −35.42 | −37.25 | |
| DFG motif and αC-helix orientation * | In | In | In | In | In | In | |
| Distance between Lys745 and Glu762 | 3.6 and 3.6 | 3.4 and 3.2 | 3.9 and 3.7 | 9.6 and 6.4 | 6.4 and 3.3 | 6.5 and 7.8 | |
| Downward movement of P loop | Yes | Yes | Yes | Yes | No | No | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, V.; Kumar, V.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Computational Insights into the Potential of Withaferin-A, Withanone and Caffeic Acid Phenethyl Ester for Treatment of Aberrant-EGFR Driven Lung Cancers. Biomolecules 2021, 11, 160. https://doi.org/10.3390/biom11020160
Malik V, Kumar V, Kaul SC, Wadhwa R, Sundar D. Computational Insights into the Potential of Withaferin-A, Withanone and Caffeic Acid Phenethyl Ester for Treatment of Aberrant-EGFR Driven Lung Cancers. Biomolecules. 2021; 11(2):160. https://doi.org/10.3390/biom11020160
Chicago/Turabian StyleMalik, Vidhi, Vipul Kumar, Sunil C. Kaul, Renu Wadhwa, and Durai Sundar. 2021. "Computational Insights into the Potential of Withaferin-A, Withanone and Caffeic Acid Phenethyl Ester for Treatment of Aberrant-EGFR Driven Lung Cancers" Biomolecules 11, no. 2: 160. https://doi.org/10.3390/biom11020160
APA StyleMalik, V., Kumar, V., Kaul, S. C., Wadhwa, R., & Sundar, D. (2021). Computational Insights into the Potential of Withaferin-A, Withanone and Caffeic Acid Phenethyl Ester for Treatment of Aberrant-EGFR Driven Lung Cancers. Biomolecules, 11(2), 160. https://doi.org/10.3390/biom11020160