
Hyperbaric Oxygen Treatment: Effects on Mitochondrial Function and Oxidative Stress



Abstract
1. Introduction
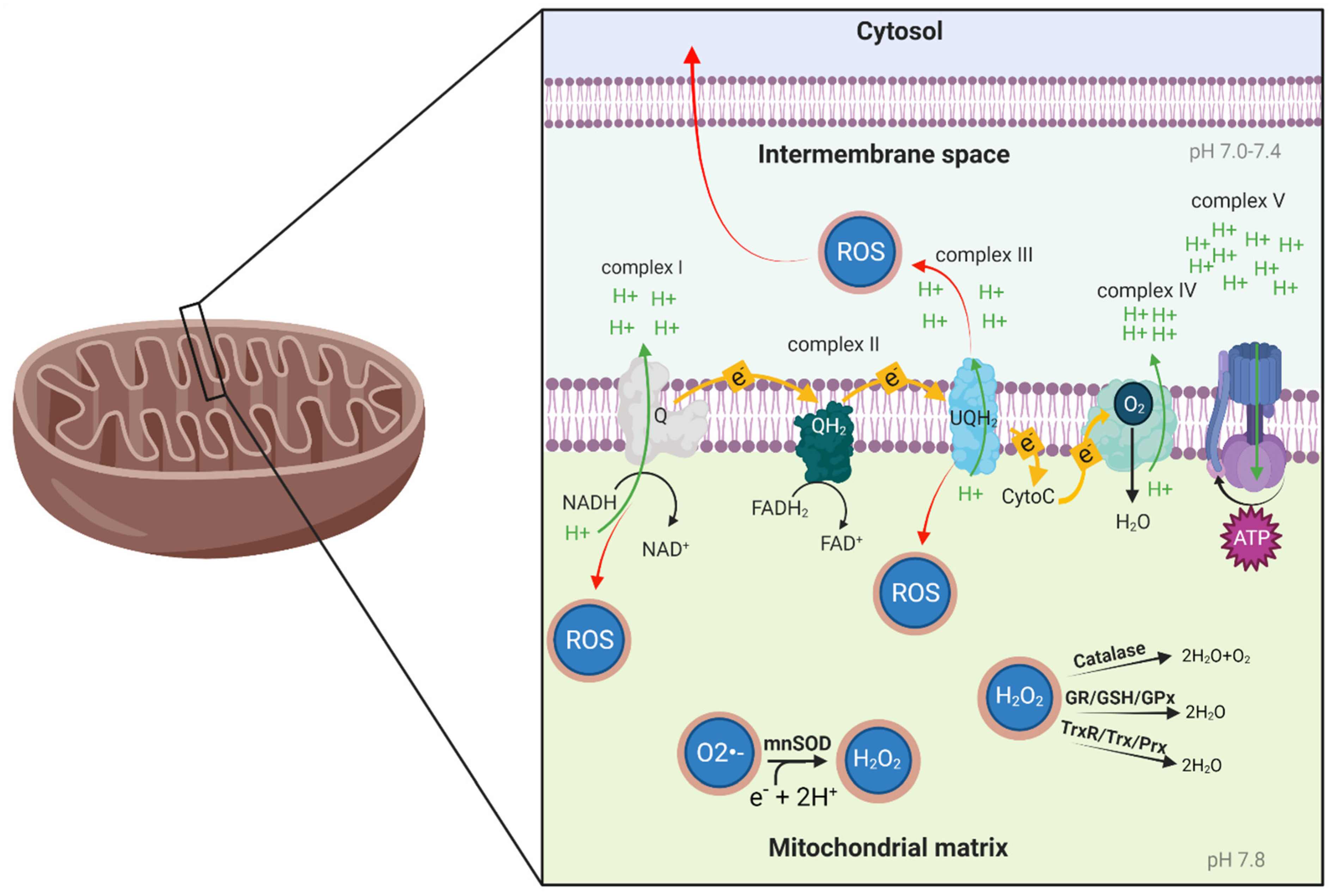
2. Mitochondrial Function and Oxidative Stress
2.1. Mitochondrial Function
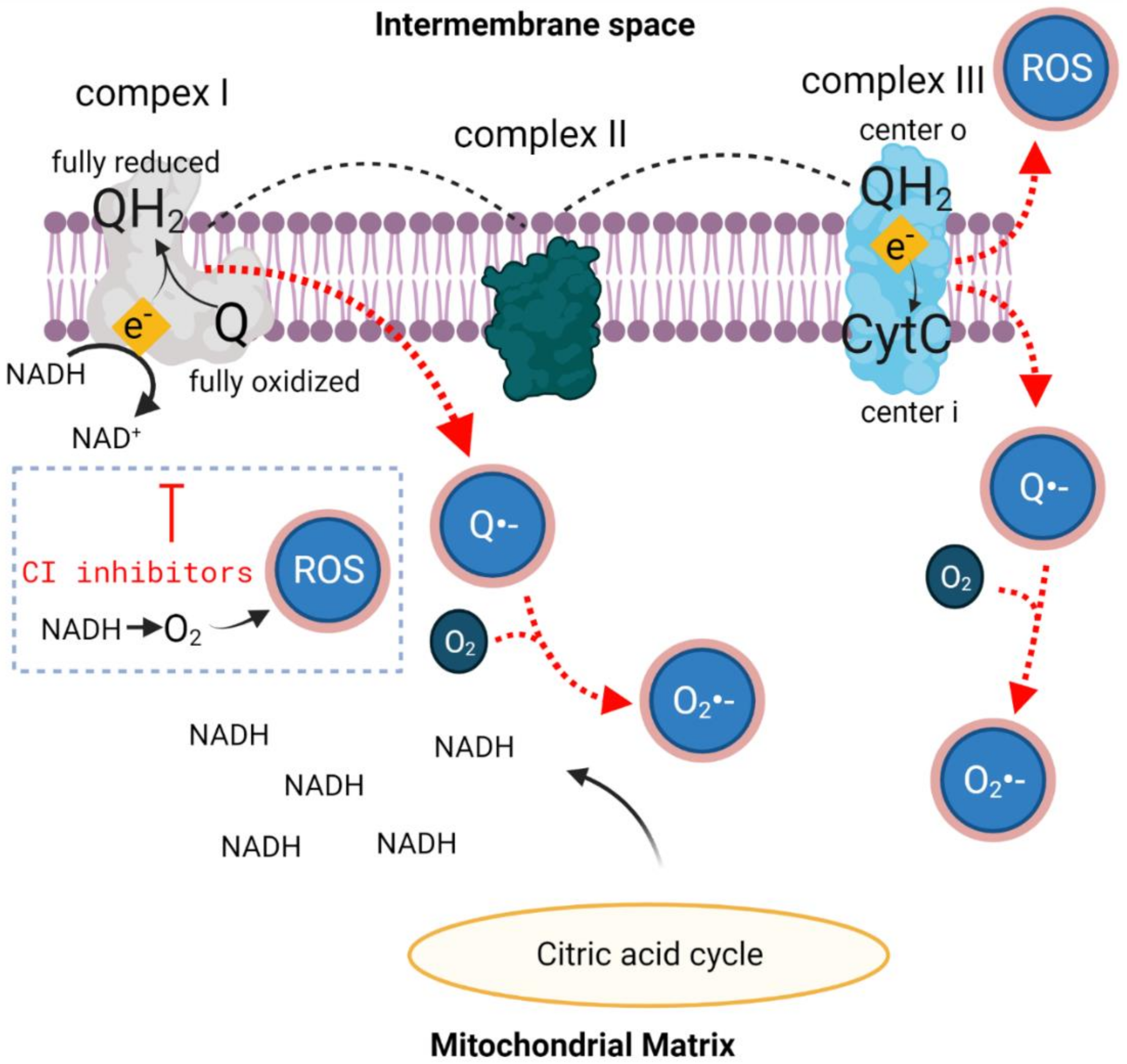
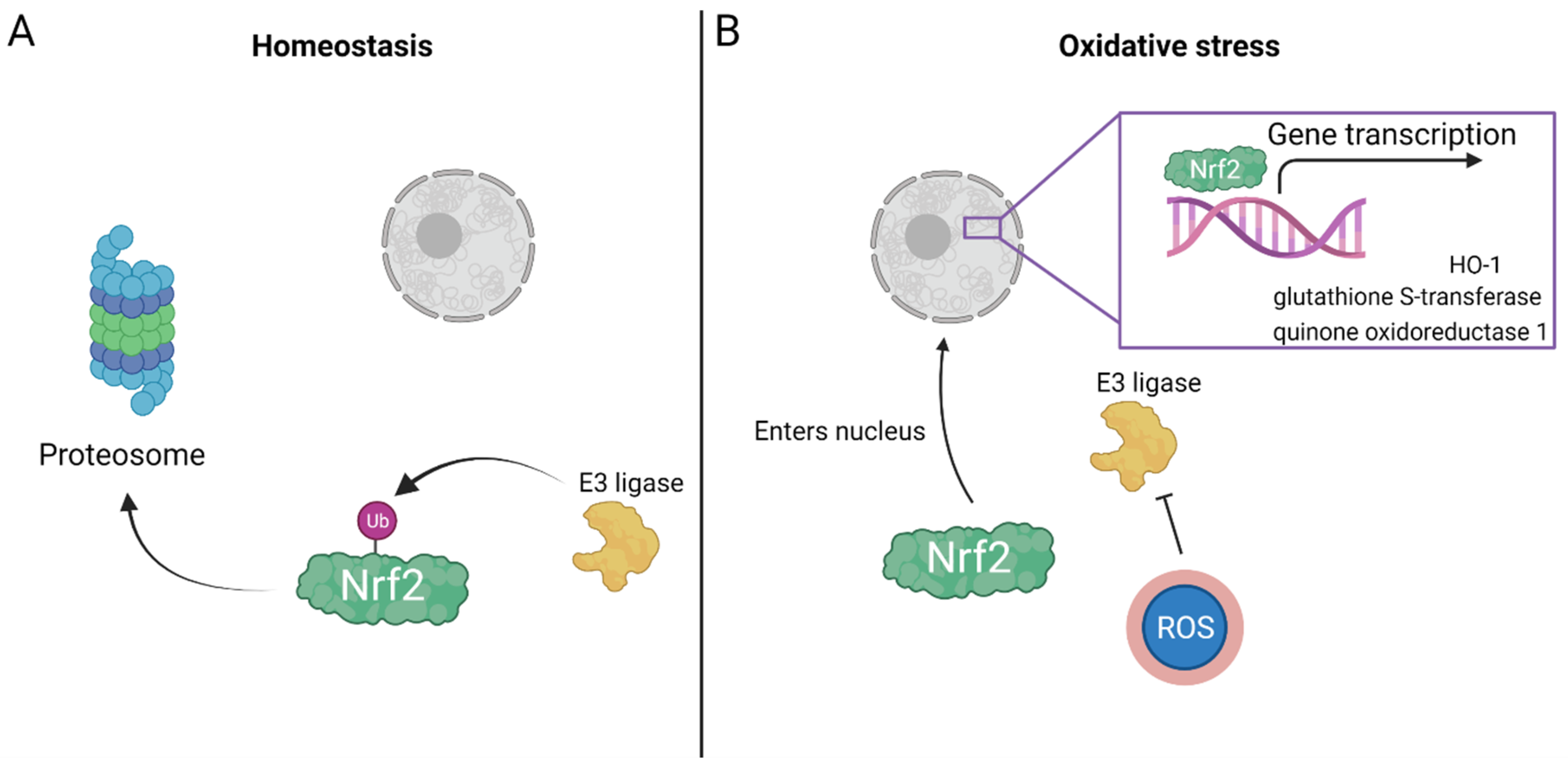
2.2. Oxidative Stress
3. HBOT, Mitochondrial Function and Oxidative Stress
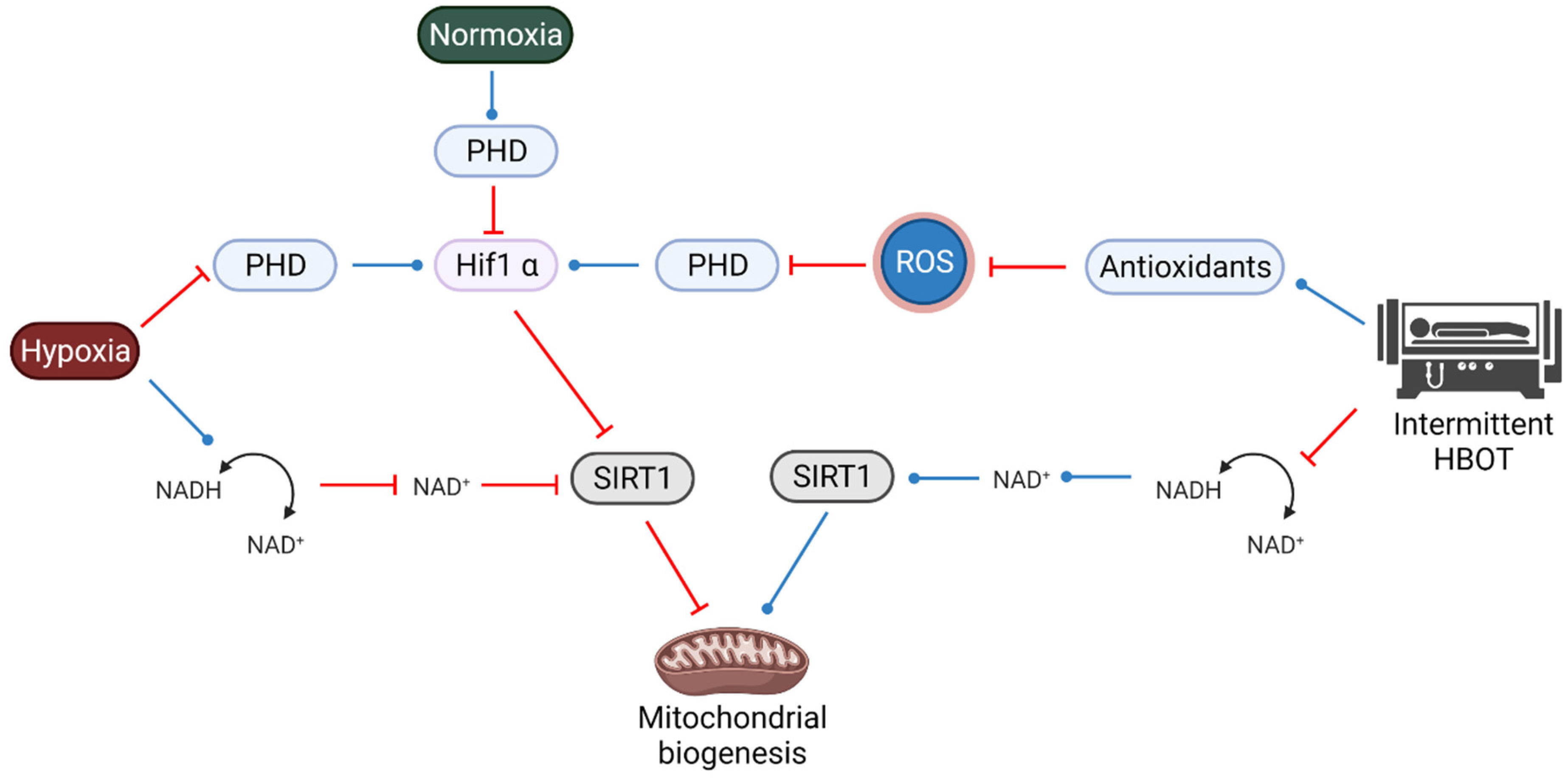
4. The Interplay between SIRT1, HIF1a and ROS during HBOT
5. Disease and Oxidative Stress
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Calvert, J.W.; Cahill, J.; Zhang, J.H. Hyperbaric oxygen and cerebral physiology. Neurol. Res. 2007, 29, 132–141. [Google Scholar] [CrossRef]
- Glik, J.; Cholewka, A.; Stanek, A.; Englisz, B.; Sieroń, K.; Mikuś-Zagórska, K.; Knefel, G.; Nowak, M.; Kawecki, M. Thermal imaging and planimetry evaluation of the results of chronic wounds treatment with hyperbaric oxygen therapy. Adv. Clin. Exp. Med. 2019, 28, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Kasprzyk-Kucewicz, T.; Cholewka, A.; Englisz-Jurgielewicz, B.; Mucha, R.; Relich, M.; Kawecki, M.; Sieroń, K.; Onak, P.; Stanek, A. Thermal effects of topical hyperbaric oxygen therapy in hard-to-heal wounds—A pilot study. Int. J. Environ. Res. Public Health 2021, 18, 6737. [Google Scholar] [CrossRef]
- Gebala-Prajsnar, K.; Stanek, A.; Pasek, J.; Prajsnar, G.; Berszakiewicz, A.; Sieron, A.; Cholewka, A. Selected physical medicine interventions in the treatment of diabetic foot syndrome. Acta Angiol. 2015, 21, 140–145. [Google Scholar] [CrossRef]
- Lin, P.Y.; Sung, P.H.; Chung, S.Y.; Hsu, S.L.; Chung, W.J.; Sheu, J.J.; Hsueh, S.K.; Chen, K.H.; Wu, R.W.; Yip, H.K. Hyperbaric oxygen therapy enhanced circulating levels of endothelial progenitor cells and angiogenesis biomarkers, blood flow, in ischemic areas in patients with peripheral arterial occlusive disease. J. Clin. Med. 2018, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Carturan, D.; Boussuges, A.; Vanuxem, P.; Bar-Hen, A.; Burnet, H.; Gardette, B. Ascent rate, age, maximal oxygen uptake, adiposity, and circulating venous bubbles after diving. J. Appl. Physiol. 2002, 93, 1349–1356. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Edwards, M.; Singh, M.; Selesny, S.; Cooper, J.S. Hyperbaric Treatment of Thermal Burns; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Cooper, J.S.; Hanley, M.E. Hyperbaric Treatment of Radiation Proctitis; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hanley, M.E.; Manna, B. Hyperbaric Treatment of Diabetic Foot Ulcer; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Gottfried, I.; Schottlender, N.; Ashery, U. Hyperbaric Oxygen Treatment—From Mechanisms to Cognitive Improvement. Biomolecules 2021, 11, 1520. [Google Scholar] [CrossRef]
- Shapira, R.; Efrati, S.; Ashery, U. Hyperbaric oxygen therapy as a new treatment approach for Alzheimer’s disease. Neural Regen. Res. 2018, 13, 817. [Google Scholar] [CrossRef]
- Shapira, R.; Gdalyahu, A.; Gottfried, I.; Sasson, E.; Hadanny, A.; Efrati, S.; Blinder, P.; Ashery, U. Hyperbaric oxygen therapy alleviates vascular dysfunction and amyloid burden in an Alzheimer’s disease mouse model and in elderly patients. Aging (Albany N. Y.) 2021. [Google Scholar] [CrossRef] [PubMed]
- Kamat, S.M.; Mendelsohn, A.R.; Larrick, J.W. Rejuvenation through Oxygen, More or Less. Rejuvenation Res. 2021, 24, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Somaa, F. A Review of the Application of Hyperbaric Oxygen Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 81, 1361–1367. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Mates, J.M.; Perez-Gomez, C.; Nunez de Castro, I. Antioxidant Enzymes and Human Diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, K.H.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Rodriguez-enriquez, S.; He, L.; Lemasters, J.J. Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2463–2472. [Google Scholar] [CrossRef]
- Mcbride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Topical Review Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 591, 57–68. [Google Scholar] [CrossRef]
- Levy, M.; Faas, G.C.; Saggau, P.; Craigen, W.J.; Sweatt, J.D. Mitochondrial Regulation of Synaptic Plasticity in the Hippocampus. J. Biol. Chem. 2003, 278, 17727–17734. [Google Scholar] [CrossRef]
- Huang, L.; Obenaus, A. Hyperbaric oxygen therapy for traumatic brain injury. Med. Gas Res. 2011, 1, 21. [Google Scholar] [CrossRef]
- Duan, S.; Shao, G.; Yu, L.; Ren, C.; Duan, S.; Shao, G.; Yu, L.; Ren, C. Angiogenesis contributes to the neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats Angiogenesis contributes to the neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischem. Int. J. Neurosci. 2015, 125, 625–634. [Google Scholar] [CrossRef]
- Schulze, J.; Kaiser, O.; Paasche, G.; Lamm, H.; Pich, A.; Hoffmann, A.; Lenarz, T.; Warnecke, A. Effect of hyperbaric oxygen on BDNF-release and neuroprotection: Investigations with human mesenchymal stem cells and genetically modified NIH3T3 fibroblasts as putative cell therapeutics. PLoS ONE 2017, 12, e0178182. [Google Scholar] [CrossRef]
- Calbet, J.A.L.; Martin-Rodriguez, S.; Martin-Rincon, M.; Morales-Alamo, D. An integrative approach to the regulation of mitochondrial respiration during exercise: Focus on high-intensity exercise. Redox Biol. 2020, 101478. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Turrens, J.F.; Chang, L.Y.; Bush, K.M.; Crapo, J.D.; Freeman, B.A. Detection of catalase in rat heart mitochondria. J. Biol. Chem. 1991, 266, 22028–22034. [Google Scholar] [CrossRef]
- Salvi, M.; Battaglia, V.; Brunati, A.M.; La Rocca, N.; Tibaldi, E.; Pietrangeli, P.; Marcocci, L.; Mondoví, B.; Rossi, C.A.; Toninello, A. Catalase takes part in rat liver mitochondria oxidative stress defense. J. Biol. Chem. 2007, 282, 24407–24415. [Google Scholar] [CrossRef]
- Sinet, P.M.; Heikkila, R.E.; Cohen, G. Hydrogen Peroxide Production by Rat Brain In Vivo. J. Neurochem. 1980, 34, 1421–1428. [Google Scholar] [CrossRef]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta—Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications review-Article. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative Stress in Neurodegenerative Diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; D’Adda Di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/Reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef]
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver. Involvement of complex I, reactive oxygen species and cardiolipin. Biochim. Biophys. Acta—Bioenerg. 2007, 1767, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta—Bioenerg. 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Parameshwaran, K.; Irwin, M.H.; Steliou, K.; Pinkert, C.A. Protection by an antioxidant of rotenone-induced neuromotor decline, reactive oxygen species generation and cellular stress in mouse brain. Pharmacol. Biochem. Behav. 2012, 101, 487–492. [Google Scholar] [CrossRef]
- MacKenzie, E.L.; Ray, P.D.; Tsuji, Y. Role and regulation of ferritin H in rotenone-mediated mitochondrial oxidative stress. Free Radic. Biol. Med. 2008, 44, 1762–1771. [Google Scholar] [CrossRef]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta—Bioenerg. 2016, 1857, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor Rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, L.; Shulmeister, V.M.; Chi, Y.I.; Kim, K.K.; Hung, L.W.; Crofts, A.R.; Berry, E.A.; Kim, S.H. Electron transfer by domain movement in cytochrome bc1. Nature 1998, 392, 677–684. [Google Scholar] [CrossRef]
- Botigeli Baldim, L.; Nejo, R.J.; Souza Jordani, E.M.; Gomes Jordani, C.M.; Neves Cardoso Picinato, M.A.; Fleury Fina, C.; Castro-e-silva, O. Effect of hyperbaric oxygen therapy on liver function during intermittent ischemia 1 Efeito da oxigenoterapia hiperbárica sobre a função hepática na isquemia intermitente. Acta Cirúrgica Bras. 2013, 28, 61–65. [Google Scholar] [CrossRef][Green Version]
- Palzur, E.; Zaaroor, M.; Vlodavsky, E.; Milman, F.; Soustiel, J.F. Neuroprotective effect of hyperbaric oxygen therapy in brain injury is mediated by preservation of mitochondrial membrane properties. Brain Res. 2008, 1221, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Daugherty, W.P.; Sun, D.; Levasseur, J.E.; Altememi, N.; Hamm, R.J.; Rockswold, G.L.; Bullock, M.R. Protection of mitochondrial function and improvement in cognitive recovery in rats treated with hyperbaric oxygen following lateral fluid-percussion injury. J. Neurosurg. 2007, 106, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Liu, K.; Li, L.; Li, X.; Zhao, P. Effects of hyperbaric oxygen therapy on neuropathic pain via mitophagy in microglia. Mol. Pain 2017, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kurt, B.; Kurt, Y.; Karslioǧlu, Y.; Topal, T.; Erdamar, H.; Korkmaz, A.; Türközkan, N.; Yaman, H.; Odabaşi, Z.; Günhan, Ö. Effects of hyperbaric oxygen on energy production and xanthine oxidase levels in striated muscle tissue of healthy rats. J. Clin. Neurosci. 2008, 15, 445–450. [Google Scholar] [CrossRef]
- Dave, K.R.; Prado, R.; Busto, R.; Raval, A.P.; Bradley, W.G.; Torbati, D.; Perez-Pinzón, M.A. Hyperbaric oxygen therapy protects against mitochondrial dysfunction and delays onset of motor neuron disease in Wobbler mice. Neuroscience 2003, 120, 113–120. [Google Scholar] [CrossRef]
- Tian, X.Q.; Zhang, L.D.; Wang, J.M.; Dai, J.G.; Shen, S.S.; Yang, L.; Huang, P.L. The protective effect of hyperbaric oxygen and Ginkgo biloba extract on Aβ25-35-induced oxidative stress and neuronal apoptosis in rats. Behav. Brain Res. 2013, 242, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, C.; Huang, J.; Wei, H.; Fan, Q. Neuroprotective effect of combined therapy with hyperbaric oxygen and madopar on 6-hydroxydopamine-induced Parkinson’s disease in rats. Neurosci. Lett. 2015, 600, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ge, B.; Yuan, Y.; Wang, G. Hyperbaric Oxygen Ameliorated Acute Pancreatitis in Rats via the Mitochondrial Pathway. Dig. Dis. Sci. 2020, 65, 3558–3569. [Google Scholar] [CrossRef] [PubMed]
- Shams, Z.; Khalatbary, A.R.; Ahmadvand, H.; Zare, Z.; Kian, K. Neuroprotective effects of hyperbaric oxygen (HBO) therapy on neuronal death induced by sciatic nerve transection in rat. BMC Neurol. 2017, 17, 220. [Google Scholar] [CrossRef]
- Tezgin, D.; Giardina, C.; Perdrizet, G.A.; Hightower, L.E. The effect of hyperbaric oxygen on mitochondrial and glycolytic energy metabolism: The caloristasis concept. Cell Stress Chaperones 2020, 25, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Godman, C.A.; Chheda, K.P.; Hightower, L.E.; Perdrizet, G.; Shin, D.G.; Giardina, C. Hyperbaric oxygen induces a cytoprotective and angiogenic response in human microvascular endothelial cells. Cell Stress Chaperones 2010, 15, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.E.; Collins, S.; Andersen, M.E. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharmacol. 2010, 244, 77–83. [Google Scholar] [CrossRef]
- Gao, J.; Liu, S.; Xu, F.; Liu, Y.; Lv, C.; Deng, Y.; Shi, J.; Gong, Q. Trilobatin Protects Against Oxidative Injury in Neuronal PC12 Cells Through Regulating Mitochondrial ROS Homeostasis Mediated by AMPK/Nrf2/Sirt3 Signaling Pathway. Front. Mol. Neurosci. 2018, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Xian, Z.; Choi, Y.H.; Zheng, M.; Jiang, J.; Zhao, Y.; Wang, C.; Li, J.; Li, Y.; Li, L.; Piao, H.; et al. Imperatorin alleviates ROS-mediated airway remodeling by targeting the Nrf2/HO-1 signaling pathway. Biosci. Biotechnol. Biochem. 2020, 84, 898–910. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, J.K.; Kim, H. ABCB7 simultaneously regulates apoptotic and non-apoptotic cell death by modulating mitochondrial ROS and HIF1α-driven NFκB signaling. Oncogene 2020, 39, 1969–1982. [Google Scholar] [CrossRef]
- Djordjevic, J.; Roy Chowdhury, S.; Snow, W.M.; Perez, C.; Cadonic, C.; Fernyhough, P.; Albensi, B.C. Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice. Cells 2020, 9, 1541. [Google Scholar] [CrossRef]
- Bosco, G.; Paganini, M.; Giacon, T.A.; Oppio, A.; Vezzoli, A.; Dellanoce, C.; Moro, T.; Paoli, A.; Zanotti, F.; Zavan, B.; et al. Oxidative stress and inflammation, microRNA, and hemoglobin variations after administration of oxygen at different pressures and concentrations: A randomized trial. Int. J. Environ. Res. Public Health 2021, 18, 9755. [Google Scholar] [CrossRef] [PubMed]
- Simsek, K.; Ozler, M.; Yildirim, A.O.; Sadir, S.; Demirbas, S.; Oztosun, M.; Korkmaz, A.; Ay, H.; Oter, S.; Yildiz, S. Evaluation of the oxidative effect of long-term repetitive hyperbaric oxygen exposures on different brain regions of rats. Sci. World J. 2012, 2012, 849183. [Google Scholar] [CrossRef]
- Oter, S.; Korkmaz, A.; Topal, T.; Ozcan, O.; Sadir, S.; Ozler, M.; Ogur, R.; Bilgic, H. Correlation between hyperbaric oxygen exposure pressures and oxidative parameters in rat lung, brain, and erythrocytes. Clin. Biochem. 2005, 38, 706–711. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Oxygen toxicity in aerobes. In Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 1999; pp. 17–21. ISBN 9780198500445. [Google Scholar]
- Yildiz, Ş.; Aktaş, Ş.; Cimşit, M.; Ay, H.; Toǧrol, E. Seizure incidence in 80,000 patient treatments with hyperbaric oxygen. Aviat. Space. Environ. Med. 2004, 75, 992–994. [Google Scholar]
- Hadanny, A.; Meir, O.; Bechor, Y.; Fishlev, G.; Bergan, J.; Efrati, S. The safety of hyperbaric oxygen treatment–retrospective analysis in 2334 patients. Undersea Hyperb. Med. 2016, 43, 113–122. [Google Scholar]
- Hadanny, A.; Efrati, S. The Hyperoxic—Hypoxic Paradox. Biomolecules 2020, 10, 958. [Google Scholar] [CrossRef]
- Ay, H.; Topal, T.; Ozler, M.; Uysal, B.; Korkmaz, A.; Oter, S.; Ogur, R.; Dundar, K. Persistence of hyperbaric oxygen-induced oxidative effects after exposure in rat brain cortex tissue. Life Sci. 2007, 80, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Ay, H.; Topal, T.; Uysal, B.; Ozler, M.; Oter, S.; Korkmaz, A.; Dundar, K. Time-dependent course of hyperbaric oxygen-induced oxidative effects in rat lung and erythrocytes. Clin. Exp. Pharmacol. Physiol. 2007, 34, 787–791. [Google Scholar] [CrossRef]
- Zhou, Q.; Huang, G.; Yu, X.; Xu, W. A Novel Approach to Estimate ROS Origination by Hyperbaric Oxygen Exposure, Targeted Probes and Specific Inhibitors. Cell. Physiol. Biochem. 2018, 47, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Dennog, C.; Hartmann, A.; Frey, G.; Speit, G. Detection of DNA damage after hyperbaric oxygen (HBO) therapy. Mutagenesis 1996, 11, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Topuz, K.; Colak, A.; Cemil, B.; Kutlay, M.; Demircan, M.N.; Simsek, H.; Ipcioglu, O.; Kucukodaci, Z.; Uzun, G. Combined hyperbaric oxygen and hypothermia treatment on oxidative stress parameters after spinal cord injury: An experimental study. Arch. Med. Res. 2010, 41, 506–512. [Google Scholar] [CrossRef]
- Oscarsson, N.; Ny, L.; Mölne, J.; Lind, F.; Ricksten, S.E.; Seeman-Lodding, H.; Giglio, D. Hyperbaric oxygen treatment reverses radiation induced pro-fibrotic and oxidative stress responses in a rat model. Free Radic. Biol. Med. 2017, 103, 248–255. [Google Scholar] [CrossRef]
- Matsunami, T.; Sato, Y.; Sato, T.; Yukawa, M. Antioxidant status and lipid peroxidation in diabetic rats under hyperbaric oxygen exposure. Physiol. Res. 2010, 59, 97–104. [Google Scholar] [CrossRef]
- Rothfuß, A.; Dennog, C.; Speit, G. Adaptive protection against the induction of oxidative DNA damage after hyperbaric oxygen treatment. Carcinogenesis 1998, 19, 1913–1917. [Google Scholar] [CrossRef]
- Körpınar, S.; Uzun, H. The Effects of Hyperbaric Oxygen at Different Pressures on Oxidative Stress and Antioxidant Status in Rats. Medicina 2019, 55, 205. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Liang, X.; Chen, D.; Chen, Y.; Doycheva, D.; Tang, J.; Tang, J.; Zhang, J.H. Delayed hyperbaric oxygen therapy promotes neurogenesis through reactive oxygen species/hypoxia-inducible factor-1α/β-catenin pathway in middle cerebral artery occlusion rats. Stroke 2014, 45, 1807–1814. [Google Scholar] [CrossRef]
- Tonelli, C.; In, I.; Chio, C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Balestra, C.; Lambrechts, K.; Mrakic-Sposta, S.; Vezzoli, A.; Levenez, M.; Germonpré, P.; Virgili, F.; Bosco, G.; Lafère, P. Hypoxic and hyperoxic breathing as a complement to low-intensity physical exercise programs: A proof-of-principle study. Int. J. Mol. Sci. 2021, 22, 9600. [Google Scholar] [CrossRef] [PubMed]
- Cimino, F.; Speciale, A.; Anwar, S.; Canali, R.; Ricciardi, E.; Virgili, F.; Trombetta, D.; Saija, A. Anthocyanins protect human endothelial cells from mild hyperoxia damage through modulation of Nrf2 pathway. Genes Nutr. 2013, 8, 391–399. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Z.; Li, Q.; Li, W.; Xu, J.; Cao, H. Hyperbaric oxygen preconditioning protects lung against hyperoxic acute lung injury in rats via heme oxygenase-1 induction. Biochem. Biophys. Res. Commun. 2015, 456, 549–554. [Google Scholar] [CrossRef]
- Chen, J. Heme oxygenase in neuroprotection: From mechanisms to therapeutic implications. Rev. Neurosci. 2014, 25, 269–280. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1—Nrf2 pathway in stress response and cancer evolution. Genes Cell 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Yadav, A.; Singh, S.V.; Mishra, M.; Singh, K.S.; Rath, S.K. Redox Biology Isoniazid prevents Nrf2 translocation by inhibiting ERK1 phosphorylation and induces oxidative stress and apoptosis. Redox Biol. 2015, 6, 80–92. [Google Scholar] [CrossRef]
- Dhamodharan, U.; Karan, A.; Sireesh, D.; Vaishnavi, A.; Somasundar, A.; Rajesh, K.; Rmkumar, K.M. Free Radical Biology and Medicine Tissue-speci fi c role of Nrf2 in the treatment of diabetic foot ulcers during hyperbaric oxygen therapy. Free Radic. Biol. Med. 2019, 138, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Fratantonio, D.; Cimino, F.; Speciale, A.; Virgili, F. Need (more than) two to Tango: Multiple tools to adapt to changes in oxygen availability. BioFactors 2018, 44, 207–218. [Google Scholar] [CrossRef]
- Fratantonio, D.; Virgili, F.; Zucchi, A.; Lambrechts, K.; Latronico, T.; Lafère, P.; Germonpré, P.; Balestra, C. Increasing oxygen partial pressures induce a distinct transcriptional response in human PBMC: A pilot study on the “normobaric oxygen paradox”. Int. J. Mol. Sci. 2021, 22, 458. [Google Scholar] [CrossRef]
- van Vliet, T.; Casciaro, F.; Demaria, M. To breathe or not to breathe: Understanding how oxygen sensing contributes to age-related phenotypes. Ageing Res. Rev. 2021, 67, 101267. [Google Scholar] [CrossRef]
- Bernaudin, M.; Bellail, A.; Marti, H.H.; Yvon, A.; Vivien, D.; Duchatelle, I.; Mackenzie, E.T.; Petit, E. Neurons and astrocytes express EPO mRNA: Oxygen-sensing mechanisms that involve the redox-state of the brain. Glia 2000, 30, 271–278. [Google Scholar] [CrossRef]
- Hassan, A.; Arnold, B.M.; Caine, S.; Toosi, B.M.; Verge, V.M.K.; Muir, G.D. Acute intermittent hypoxia and rehabilitative training following cervical spinal injury alters neuronal hypoxia- and plasticity-associated protein expression. PLoS ONE 2018, 13, e0197486. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef]
- Ryou, M.G.; Chen, X.; Cai, M.; Wang, H.; Jung, M.E.; Metzger, D.B.; Mallet, R.T.; Shi, X. Intermittent Hypoxia Training Prevents Deficient Learning-Memory Behavior in Mice Modeling Alzheimer’s Disease: A Pilot Study. Front. Aging Neurosci. 2021, 13, 674688. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Manaenko, A.; Ye, Z.H.; Sun, X.J.; Hu, Q. Hypoxia therapy—A new hope for the treatment of mitochondrial dysfunctions. Med. Gas Res. 2016, 6, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen consumption and usage during physical exercise: The balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Suzuki, K.; Posa, A.; Petrovszky, Z.; Koltai, E.; Boldogh, I. The systemic role of SIRT1 in exercise mediated adaptation. Redox Biol. 2020, 35, 101467. [Google Scholar] [CrossRef]
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018. [Google Scholar] [CrossRef]
- Ohlsson, H.; Karlsson, K.; Edlund, T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 1993, 12, 4251–4259. [Google Scholar] [CrossRef] [PubMed]
- Robson, R.; Kundur, A.R.; Singh, I. Oxidative stress biomarkers in type 2 diabetes mellitus for assessment of cardiovascular disease risk. Diabetes Metab. Syndr. Clin. Res. Rev. 2018, 12, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Gürdöl, F.; Cimşit, M.; Öner-Iyidoǧan, Y.; Körpinar, S.; Yalçinkaya, S.; Koçak, H. Early and late effects of hyperbaric oxygen treatment on oxidative stress parameters in diabetic patients. Physiol. Res. 2008, 57, 41–47. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative Stress and Cancer: Have We Moved Forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, M.C. Serial Review: Causes and Consequences of Oxidative Stress in Alzheimer’s Disease. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging 2006, 27, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Yatin, S.M.; Aksenov, M.; Butterfield, D.A. The antioxidant vitamin E modulates amyloid beta-peptide-induced creatine kinase activity inhibition and increased protein oxidation: Implications for the free radical hypothesis of Alzheimer’s disease. Neurochem. Res. 1999, 24, 427–435. [Google Scholar] [CrossRef]
- Clementi, M.E.; Pezzotti, M.; Orsini, F.; Sampaolese, B.; Mezzogori, D.; Grassi, C.; Giardina, B.; Misiti, F. Alzheimer’s amyloid β-peptide (1-42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: An intriguing role for methionine 35. Biochem. Biophys. Res. Commun. 2006, 342, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Pogocki, D.; Schöneich, C. Redox properties of Met(35) in neurotoxic beta-amyloid peptide. A molecular modeling study. Chem. Res. Toxicol. 2002, 15, 408–418. [Google Scholar] [CrossRef]
- Polyzos, A.A.; McMurray, C.T. The chicken or the egg: Mitochondrial dysfunction as a cause or consequence of toxicity in Huntington’s disease. Mech. Ageing Dev. 2017, 161, 181–197. [Google Scholar] [CrossRef]
- Tian, X.Q.; Wang, J.M.; Dai, J.G.; Yang, L.; Zhang, L.D.; Shen, S.S.; Huang, P.L. Hyperbaric oxygen and ginkgo biloba extract inhibit Aβ25-35-induced toxicity and oxidative stress in vivo: A potential role in Alzheimer’s disease. Int. J. Neurosci. 2012, 122, 563–569. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, F.; Zhao, L.; Cheng, C.; Zhong, R.; Dong, C.; Le, W. Hyperbaric oxygen ameliorates cognitive impairment in patients with Alzheimer’s disease and amnestic mild cognitive impairment. Alzheimer’s Dement. 2020, 6, e12030. [Google Scholar] [CrossRef]
- Harch, P.G.; Fogarty, E.F. Hyperbaric oxygen therapy for Alzheimer’s dementia with positron emission tomography imaging: A case report. Med. Gas Res. 2018, 8, 181–184. [Google Scholar] [CrossRef]
- Xu, J.J.; Yang, S.T.; Sha, Y.; Ge, Y.Y.; Wang, J.M. Hyperbaric oxygen treatment for Parkinson’s disease with severe depression and anxiety. Medicine 2018, 97, 2017–2019. [Google Scholar] [CrossRef]
- Phillips, G.R.; Huang, J.K.; Wang, Y.; Tanaka, H.; Shapiro, L.; Zhang, W.; Shan, W.S.; Arndt, K.; Frank, M.; Gordon, R.E.; et al. The presynaptic particle web: Ultrastructure, composition, dissolution, and reconstitution. Neuron 2001, 32, 63–77. [Google Scholar] [CrossRef]
- Chen, C.; Chen, W.; Nong, Z.; Nie, Y.; Chen, X.; Pan, X.; Guo, Y.; Yao, M.; Deng, W. Hyperbaric oxygen alleviated cognitive impairments in mice induced by repeated cerebral ischemia-reperfusion injury via inhibition of autophagy. Life Sci. 2020, 241, 117170. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kwon, H.; Han, P.L. Hyperoxygenation treatment reduces beta-amyloid deposition via mecp2-dependent upregulation of MMP-2 and MMP-9 in the hippocampus of Tg-APP/PS1 mice. Exp. Neurobiol. 2021, 30, 294–307. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- Dornand, J.; Gerber, M. Inhibition of murine T-cell responses by anti-oxidants: The targets of lipo-oxygenase pathway inhibitors. Immunology 1989, 68, 384–391. [Google Scholar]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of Reactive Oxygen Species in Toll-Like Receptor 4-Dependent Activation of NF-κB. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox Regulation of NF-kB Activation: Distinct Redox Regulation between the Cytoplasm and the Nucleus. Antioxid. Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef]
- Dwyer, D.J.; Kohanski, M.A.; Collins, J.J. Role of reactive oxygen species in antibiotic action and resistance. Curr. Opin. Microbiol. 2009, 12, 482–489. [Google Scholar] [CrossRef]
- Paiva, C.N.; Bozza, M.T. Are Reactive Oxygen Species always detrimental to Pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef]
- Cimsit, M.; Uzun, G.; Yildiz, S. Hyperbaric oxygen therapy as an anti-infective agent. Expert Rev. Anti. Infect. Ther. 2009, 7, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Meffert, M.K. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006, 13, 852–860. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Treatment Length | Pressure | Effect on Mitochondria | Disease/Condition |
|---|---|---|---|---|
| Kurt et al., 2008 [51] | 4 weeks | 3 ATA | Increased energy production | Healthy |
| Dave et al., 2003 [52] | 30 days | 2 ATA | Improved complex IV activity | Wobbler mice (model for amyotrophic lateral sclerosis (ALS)) |
| Tian et al., 2013 [53] | 20 days of 1 h treatments | 2 ATA | Reduced mitochondria-mediated apoptosis signaling (increased Bcl-2 and decreased Bax) | Amyloid-β25-35-injected rats |
| Pan et al., 2015 [54] | 14 days of 1 h treatments | 2.5 ATA | Reduced mitochondria-mediated apoptosis signaling (increased Bcl-2 and decreased Bax) | Rat model of Parkinson’s disease |
| Botigeli Baldim et al., 2013 [47] | A single treatment of 1 h | 2 ATA | Reduced baseline mitochondrial consumption rate | Ischemia-induced rats |
| Zhou et al., 2007 [49] | A single treatment of 1 h | 1.5 ATA | Increased ATP levels | Traumatic brain injury |
| Palzur et al., 2008 [48] | 4 treatments (twice, 2 consecutive) of 45 min each | 2.8 ATA | Reduction in mitochondrial membrane potential and an increase in caspase 8 activity level | Focal brain injury |
| Zhao et al., 2020 [55] | Twice daily 90-min treatments for either 1, 2 or 3 days | 2.5 ATA | Day 1—increased mitochondrial apoptosis activation mediated by Bcl2/Bax ratio. Reduced caspase 3 and 9 activity in damaged area. Increased ATP levels. Day 2—similar results. Day 3—no activation of protein apoptosis or differences in energy production | Pancreatitis-induced rats |
| Han et al., 2017 [50] | Five 1 h treatments | 2.4 ATA | Reduction in ∆ψ after HBOT in control group, but not in the mitophagy-inhibited group | Mitophagy-inhibited rats |
| Shams et al., 2017 [56] | Five 1 h treatments | 2 ATA | Reduced apoptosis in HBOT rats (compared to nontreated sciatic nerve-damaged rats) | Rats with sciatic nerve damage |
| Study | Treatment Length | Pressure | Effect on Oxidative Stress | Disease/Condition |
|---|---|---|---|---|
| Zhou et al., 2018 [73] | One treatment of 60 min | 2.7 ATA | ROS were significantly elevated in the mitochondria and in the cell | HUVEC culture |
| Dennog et al., 1996 [74] | A total of 75 min—three 20 min treatments with a 5 min break between | 2.5 ATA | Oxidative DNA damage in the leukocytes | Healthy human volunteers |
| Topuz et al., 2010 [75] | One treatment of 90 min | 2.4 ATA | Prevented elevation in lipid peroxidation observed in nontreated group and increased antioxidant activity | Spinal cord injury in mice |
| Oscarsson et al., 2017 [76] | 20 treatments of 90 min | 2 ATA | Elevated levels of DNA oxidation and of SOD2, HO-1 and Nrf2 expression | Irradiated rats |
| Matsunami et al., 2010 [77] | 7 days of 2 h treatment | 2.8 ATA | Elevated lipid peroxidation and decreased SOD activity in treated diabetic rats | Induced diabetic rats |
| Simsek et al., 2012 [65] | 1, 2, 3, 4, 6 or 8 weeks of 90 min treatments | 2.8 ATA | ROS and radical scavenger enzyme levels in rat brain were not significantly altered | Healthy rats |
| Rothfuß et al., 1998 [78] | A total of 75 min—three 20 min treatments with a 5 min break between | 2.5 ATA | Increased antioxidant activity lasting for at least 1 week | Healthy human volunteers |
| Körpınar and Uzun, 2019 [79] | 3 treatments of 1 h (within 24 h) | 2 ATA | Significant elevation of lipid peroxidation (MDA) and reduced antioxidant SOD levels in the plasma | Healthy rats |
| 3 treatments of 1 h (within 24 h) | 2.4 ATA | |||
| 15 treatments of 1 h (within 10 days) | 2 ATA | No significant change in lipid peroxidation (MDA) and antioxidant SOD levels in the plasma | ||
| 15 treatments of 1 h (within 10 days) | 2.4 ATA | |||
| Hu et al., 2014 [80] | 1–4 days of 1 h treatments twice a day | 2 ATA | Decreased levels of lipid peroxidation. GPx, SOD and Gr(glutathione reductase) levels increased after 1 treatment, and these levels increased further on days 2, 3 and 4 compared to the first day | Cerebral artery occlusion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schottlender, N.; Gottfried, I.; Ashery, U. Hyperbaric Oxygen Treatment: Effects on Mitochondrial Function and Oxidative Stress. Biomolecules 2021, 11, 1827. https://doi.org/10.3390/biom11121827
Schottlender N, Gottfried I, Ashery U. Hyperbaric Oxygen Treatment: Effects on Mitochondrial Function and Oxidative Stress. Biomolecules. 2021; 11(12):1827. https://doi.org/10.3390/biom11121827
Chicago/Turabian StyleSchottlender, Nofar, Irit Gottfried, and Uri Ashery. 2021. "Hyperbaric Oxygen Treatment: Effects on Mitochondrial Function and Oxidative Stress" Biomolecules 11, no. 12: 1827. https://doi.org/10.3390/biom11121827
APA StyleSchottlender, N., Gottfried, I., & Ashery, U. (2021). Hyperbaric Oxygen Treatment: Effects on Mitochondrial Function and Oxidative Stress. Biomolecules, 11(12), 1827. https://doi.org/10.3390/biom11121827