
L-Type Ca2+ Channel Regulation by Calmodulin and CaBP1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
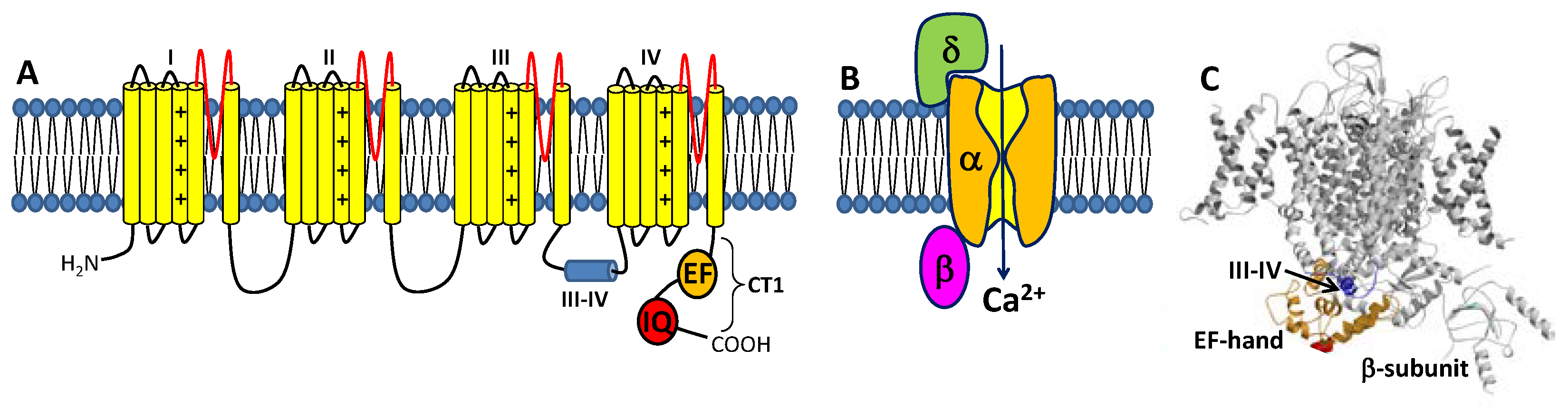
1.1. Voltage-Gated L-Type Ca2+ Channel Structure and Function
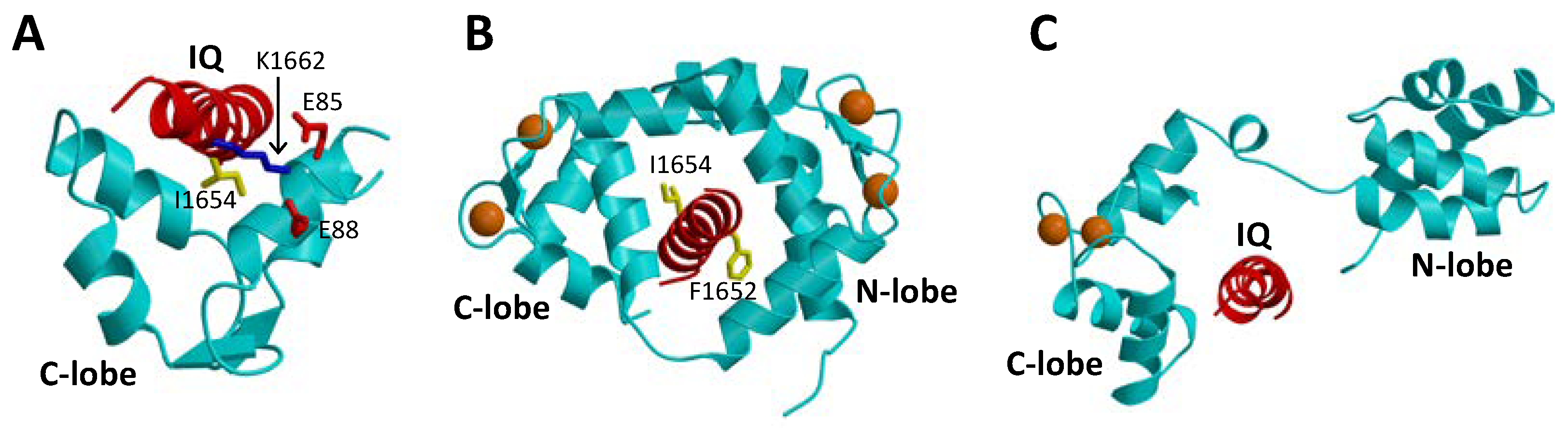
1.2. CaM Is a Ca2+ Sensor for CaVs
1.3. CaBP1 Promotes Activation of CaVs
2. CaV Channel Function Regulated by CaM and CaBP1
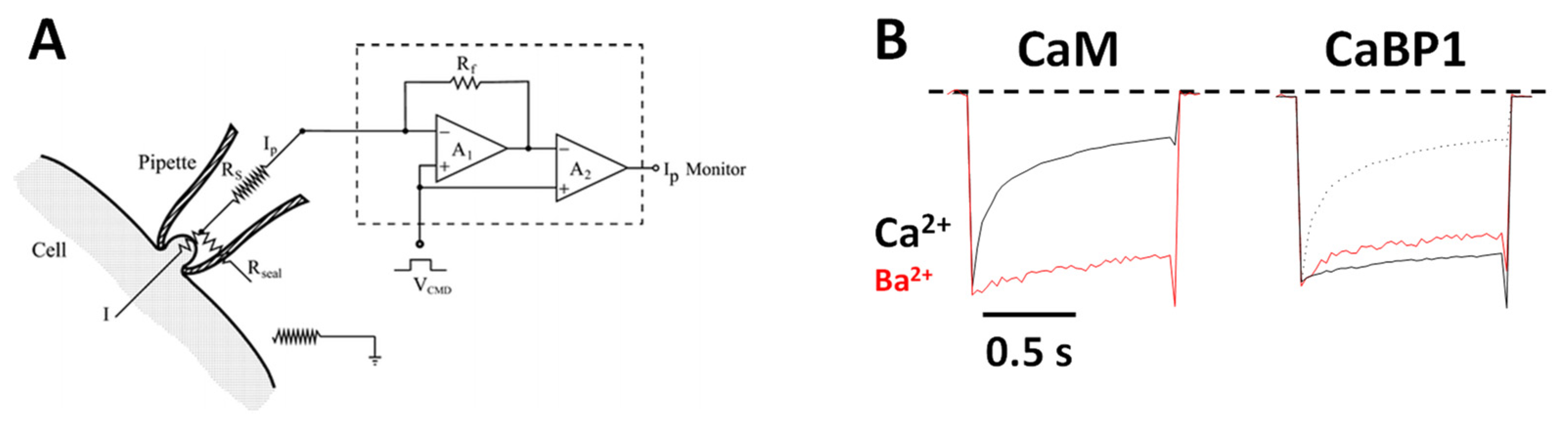
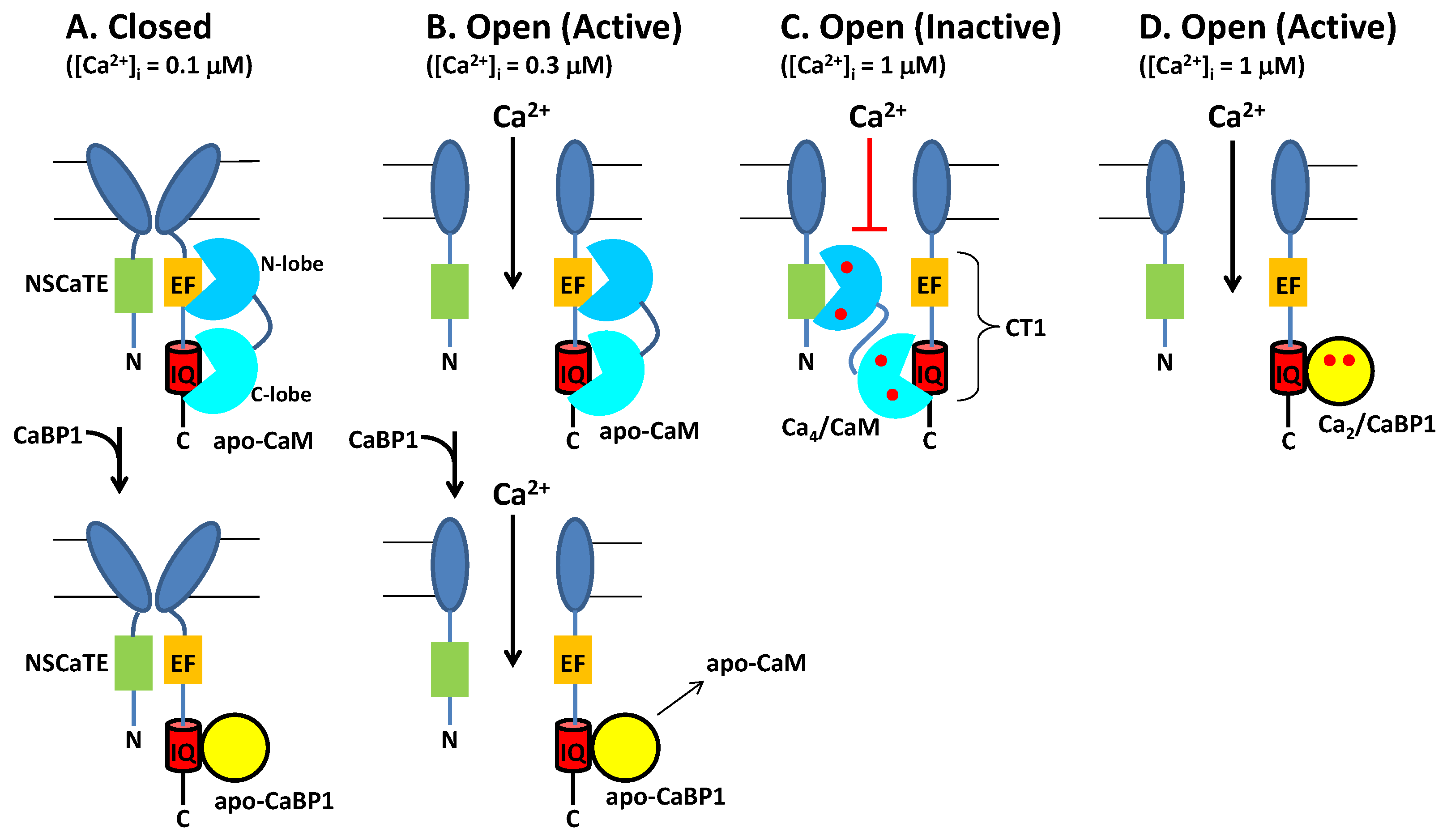
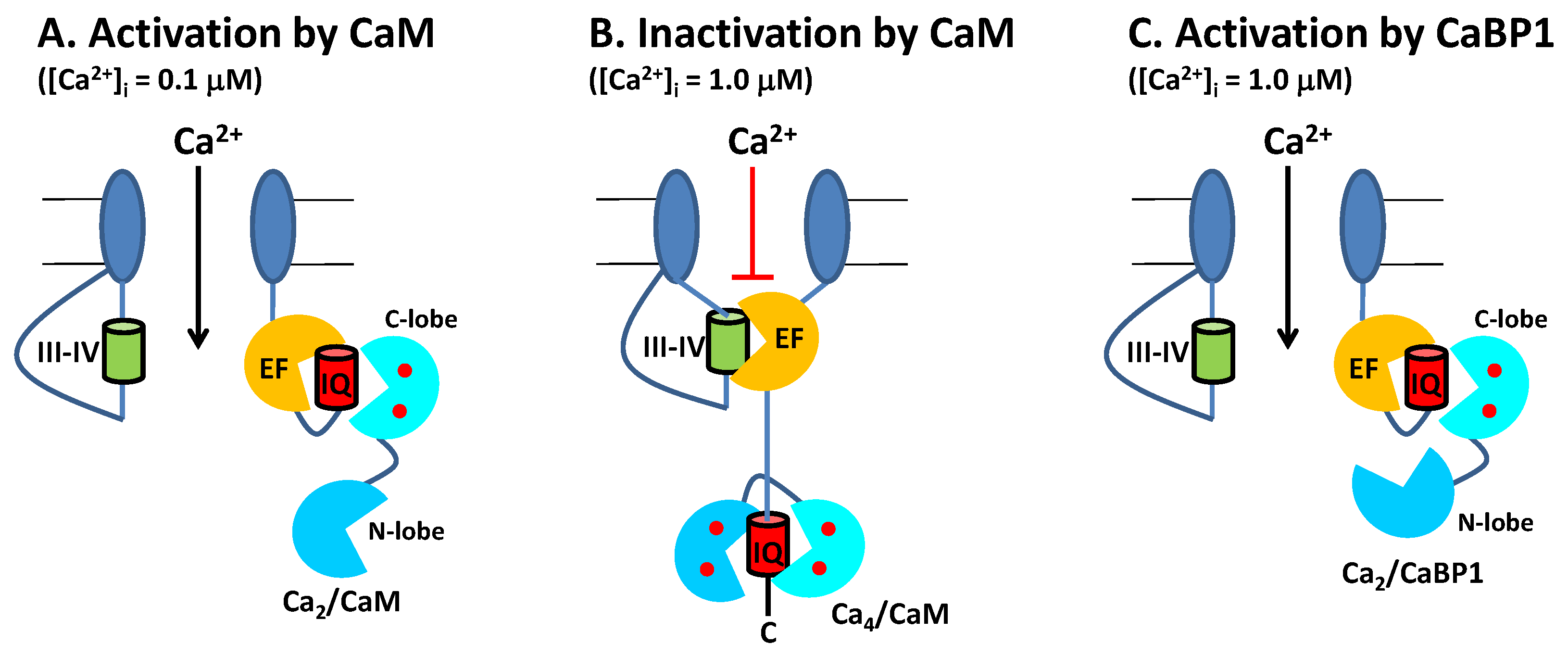
2.1. CaM Is Both an Accelerator and a Brake for CaV Channel Activity
2.2. CaBP1 Binding to CaV Prevents CDI and Activates Channel Opening
3. Functional Role of Ca2+-Bound Forms of CaM and CaBP1 under Basal Conditions
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Clapham, D.E. Calcium signaling. Cell 2007, 6, 1047–1058. [Google Scholar] [CrossRef]
- Hardie, J.; Lee, A. Decalmodulation of Cav1 channels by CaBPs. Channels 2015, 10, 33–37. [Google Scholar] [CrossRef]
- Minor, D.L.; Findeisen, F., Jr. Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin) 2010, 4, 459–474. [Google Scholar] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef]
- Randall, A.; Tsien, R.W. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J. Neurosci. 1995, 15, 2995–3012. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.; Roderick, H. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Wadel, K.; Neher, E.; Sakaba, T. The Coupling between Synaptic Vesicles and Ca2+ Channels Determines Fast Neurotransmitter Release. Neuron 2007, 53, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.G.; Groth, R.D.; Ma, H.; Barrett, C.F.; Owen, S.F.; Safa, P.; Tsien, R.W. CaV1 and CaV2 Channels Engage Distinct Modes of Ca2+ Signaling to Control CREB-Dependent Gene Expression. Cell 2012, 149, 1112–1124. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.B.; Randall, A.; Tsien, R.W. Roles of N-Type and Q-Type Ca2+ Channels in Supporting Hippocampal Synaptic Transmission. Science 1994, 264, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.B.; Wester, M.R.; Miller, L.E.; Nagarkar, M.D.; Johnson, M.B.; Saha, M.S. Cloning and characterization of voltage-gated calcium channel alpha1 subunits in Xenopus laevis during development. Dev. Dyn. 2009, 238, 2891–2902. [Google Scholar] [CrossRef]
- Simms, B.A.; Souza, I.A.; Rehak, R.; Zamponi, G.W. The Cav1.2 N terminus contains a CaM kinase site that modulates channel trafficking and function. Pflüg. Arch. Eur. J. Physiol. 2014, 467, 677–686. [Google Scholar] [CrossRef]
- Stanika, R.; Villanueva, I.; Kazanina, G.; Andrews, S.B.; Pivovarova, N.B. Comparative Impact of Voltage-Gated Calcium Channels and NMDA Receptors on Mitochondria-Mediated Neuronal Injury. J. Neurosci. 2012, 32, 6642–6650. [Google Scholar] [CrossRef] [PubMed]
- Christel, C.; Lee, A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. et Biophys. Acta (BBA) Gen. Subj. 2011, 1820, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Dick, I.E.; Tadross, M.R.; Liang, H.; Tay, L.H.; Yang, W.; Yue, D.T. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 2008, 451, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Zühlke, R.D.; Pitt, G.S.; Deisseroth, K.; Tsien, R.W.; Reuter, H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 1999, 399, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.M.; Snutch, T.P. Voltage-gated calcium channels and disease. BioFactors 2011, 37, 197–205. [Google Scholar] [CrossRef]
- Catterall, W.A. Ion Channel Voltage Sensors: Structure, Function, and Pathophysiology. Neuron 2010, 67, 915–928. [Google Scholar] [CrossRef]
- Patton, D.E.; West, J.W.; Catterall, W.A.; Goldin, A.L. A peptide segment critical for sodium channel inactivation functions as an inactivation gate in a potassium channel. Neuron 1993, 11, 967–974. [Google Scholar] [CrossRef]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Dai, S.; Hall, D.D.; Hell, J.W. Supramolecular Assemblies and Localized Regulation of Voltage-Gated Ion Channels. Physiol. Rev. 2009, 89, 411–452. [Google Scholar] [CrossRef]
- Hall, D.D.; Dai, S.; Tseng, P.-Y.; Malik, Z.; Nguyen, M.; Matt, L.; Schnizler, K.; Shepherd, A.; Mohapatra, D.P.; Tsuruta, F.; et al. Competition between α-actinin and Ca2+-Calmodulin Controls Surface Retention of the L-type Ca2+ Channel CaV1.2. Neuron 2013, 78, 483–497. [Google Scholar] [CrossRef]
- Zamponi, G.; Bourinet, E.; Nelson, D.; Nargeot, J.; Snutch, T.P. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature 1997, 385, 442–446. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Li, Z.Q.; Qian, X.Y.; Lu, S.; Dong, M.Q.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature. 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Li, Z.Q.; Yan, C.; Lu, S.; Dong, M.Q.; Yan, N. Structure of the voltage-gated calcium channel Cav1.1 complex. Science 2015, 350. [Google Scholar] [CrossRef]
- Peterson, B.Z.; DeMaria, C.D.; Yue, D.T. Calmodulin Is the Ca2+ Sensor for Ca2+-Dependent Inactivation of L-Type Calcium Channels. Neuron 1999, 22, 549–558. [Google Scholar] [CrossRef]
- Tippens, A.L.; Lee, A. Caldendrin, a Neuron-specific Modulator of Cav/1.2 (L-type) Ca2+ Channels. J. Biol. Chem. 2007, 282, 8464–8473. [Google Scholar] [CrossRef]
- Zhou, H.; Kim, S.-A.; Kirk, E.A.; Tippens, A.L.; Sun, H.; Haeseleer, F.; Lee, A. Ca2+-Binding Protein-1 Facilitates and Forms a Postsynaptic Complex with Cav1.2 (L-Type) Ca2+ Channels. J. Neurosci. 2004, 24, 4698–4708. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.G.; Liang, H.; Mori, M.X.; Yue, D.T. FRET Two-Hybrid Mapping Reveals Function and Location of L-Type Ca2+ Channel CaM Preassociation. Neuron 2003, 39, 97–107. [Google Scholar] [CrossRef]
- Lian, L.-Y.; Myatt, D.; Kitmitto, A. Apo calmodulin binding to the L-type voltage-gated calcium channel Cav1.2 IQ peptide. Biochem. Biophys. Res. Commun. 2007, 353, 565–570. [Google Scholar] [CrossRef]
- Xiong, L.; Kleerekoper, Q.K.; He, R.; Putkey, J.A.; Hamilton, S.L. Sites on Calmodulin That Interact with the C-terminal Tail of Cav1.2 Channel. J. Biol. Chem. 2005, 280, 7070–7079. [Google Scholar] [CrossRef] [PubMed]
- Johny, M.; Yang, P.S.; Bazzazi, H.; Yue, D.T. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat. Commun. 2013, 4, 1717. [Google Scholar] [CrossRef]
- Erickson, M.G.; Alseikhan, B.A.; Peterson, B.Z.; Yue, D.T. Preassociation of Calmodulin with Voltage-Gated Ca2+ Channels Revealed by FRET in Single Living Cells. Neuron 2001, 31, 973–985. [Google Scholar] [CrossRef]
- Peterson, B.Z.; Lee, J.S.; Mulle, J.G.; Wang, Y.; de Leon, M.; Yue, D.T. Critical Determinants of Ca2+-Dependent Inactivation within an EF-Hand Motif of L-Type Ca2+ Channels. Biophys. J. 2000, 78, 1906–1920. [Google Scholar] [CrossRef]
- Findeisen, F.; Rumpf, C.H.; Minor, D.L. Apo States of Calmodulin and CaBP1 Control CaV1 Voltage-Gated Calcium Channel Function through Direct Competition for the IQ Domain. J. Mol. Biol. 2013, 425, 3217–3234. [Google Scholar] [CrossRef] [PubMed]
- Moncrief, N.; Kretsinger, R.H.; Goodman, M. Evolution of EF-hand calcium-modulated proteins. I. Relationships based on amino acid sequences. J. Mol. Evol. 1990, 30, 522–562. [Google Scholar] [CrossRef]
- Babu, Y.; Bugg, C.E.; Cook, W.J. Structure of calmodulin refined at 2.2 Å resolution. J. Mol. Biol. 1988, 204, 191–204. [Google Scholar] [CrossRef]
- Gilli, R.; Lafitte, D.; Lopez, C.; Kilhoffer, M.-C.; Makarov, A.; Briand, A.C.; Haiech, J. Thermodynamic Analysis of Calcium and Magnesium Binding to Calmodulin. Biochemistry 1998, 37, 5450–5456. [Google Scholar] [CrossRef]
- Ikura, M. Calcium binding and conformational response in EF-hand proteins. Trends Biochem. Sci. 1996, 21, 14–17. [Google Scholar] [CrossRef]
- Evans, T.I.A.; Hell, J.W.; Shea, M.A. Thermodynamic linkage between calmodulin domains binding calcium and contiguous sites in the C-terminal tail of CaV1.2. Biophys. Chem. 2011, 159, 172–187. [Google Scholar] [CrossRef][Green Version]
- Turner, M.; Anderson, D.E.; Bartels, P.; Nieves, C.; Andrea, M.C.; Henderson, P.B.; Man, K.N.M.; Tseng, P.-Y.; Yarov-Yarovoy, V.; Bers, D.M.; et al. α-Actinin-1 promotes gating of the L-type Ca2+ Channel CaV1.2. EMBO J. 2020, 39, e102622. [Google Scholar] [CrossRef] [PubMed]
- Zühlke, R.D.; Pitt, G.S.; Tsien, R.W.; Reuter, H. Ca2+-sensitive Inactivation and Facilitation of L-type Ca2+ Channels Both Depend on Specific Amino Acid Residues in a Consensus Calmodulin-binding Motif in theα1C subunit. J. Biol. Chem. 2000, 275, 21121–21129. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ghosh, S.; A Nunziato, D.; Pitt, G.S. Identification of the Components Controlling Inactivation of Voltage-Gated Ca2+ Channels. Neuron 2004, 41, 745–754. [Google Scholar] [CrossRef]
- Van Petegem, F.; Chatelain, F.C.; Minor, J.D.L. Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain–Ca2+/calmodulin complex. Nat. Struct. Mol. Biol. 2005, 12, 1108–1115. [Google Scholar] [CrossRef]
- Findeisen, F.; Minor, D.L. Structural Basis for the Differential Effects of CaBP1 and Calmodulin on CaV1.2 Calcium-Dependent Inactivation. Structure 2010, 18, 1617–1631. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F.; Sokal, I.; Verlinde, C.; Erdjument-Bromage, H.; Tempst, P.; Pronin, A.N.; Benovic, J.L.; Fariss, R.; Palczewski, K. Five Members of a Novel Ca2+-binding Protein (CABP) Subfamily with Similarity to Calmodulin. J. Biol. Chem. 2000, 275, 1247–1260. [Google Scholar] [CrossRef]
- Haynes, L.P.; Tepikin, A.; Burgoyne, R.D. Calcium-binding Protein 1 Is an Inhibitor of Agonist-evoked, Inositol 1,4,5-Trisphosphate-mediated Calcium Signaling. J. Biol. Chem. 2004, 279, 547–555. [Google Scholar] [CrossRef]
- Menger, N.; Seidenbecher, C.I.; Gundelfinger, E.D.; Kreutz, M. The cytoskeleton-associated neuronal calcium-binding protein caldendrin is expressed in a subset of amacrine, bipolar and ganglion cells of the rat retina. Cell Tissue Res. 1999, 298, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Seidenbecher, C.I.; Reissner, C.; Kreutz, M.R. Caldendrins in the Inner Retina. Photorecept. Calcium 2002, 514, 451–463. [Google Scholar] [CrossRef]
- Laube, G.; Seidenbecher, C.; Richter, K.; Dieterich, D.; Hoffmann, B.; Landwehr, M.; Smalla, K.; Winter, C.; Böckersbc, T.M.; Wolf, G.; et al. The Neuron-Specific Ca2+-Binding Protein Caldendrin: Gene Structure, Splice Isoforms, and Expression in the Rat Central Nervous System. Mol. Cell. Neurosci. 2002, 19, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, K.; McCoy, K.L.; Lee, A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein 1. J. Biol. Chem. 2005, 280, 29612–29619. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita-Kawada, M.; Tang, J.; Xiao, R.; Kaneko, S.; Foskett, J.K.; Zhu, M.X. Inhibition of TRPC5 channels by Ca2+-binding protein 1 in Xenopus oocytes. Pflug. Arch. 2005, 450, 345–354. [Google Scholar] [CrossRef]
- Wingard, J.N.; Chan, J.; Bosanac, I.; Haeseleer, F.; Palczewski, K.; Ikura, M.; Ames, J.B. Structural Analysis of Mg2+ and Ca2+ Binding to CaBP1, a Neuron-specific Regulator of Calcium Channels. J. Biol. Chem. 2005, 280, 37461–37470. [Google Scholar] [CrossRef] [PubMed]
- Oz, S.; Tsemakhovich, V.; Christel, C.J.; Lee, A.; Nathan, D. CaBP1 regulates voltage-dependent inactivation and activation of Ca(V)1.2 (L-type) calcium channels. J. Biol. Chem. 2011, 286, 13945–13953. [Google Scholar] [CrossRef] [PubMed]
- Oz, S.; Benmocha, A.; Sasson, Y.; Sachyani, D.; Almagor, L.; Lee, A.; Hirsch, J.A.; Dascal, N. Competitive and Non-competitive Regulation of Calcium-dependent Inactivation in CaV1.2 L-type Ca2+ Channels by Calmodulin and Ca2+-binding Protein 1. J. Biol. Chem. 2013, 288, 12680–12691. [Google Scholar] [CrossRef]
- Adams, P.J.; Johny, M.; Dick, I.; Inoue, T.; Yue, D.T. Apocalmodulin Itself Promotes Ion Channel Opening and Ca2+ Regulation. Cell 2014, 159, 608–622. [Google Scholar] [CrossRef]
- Liu, Z.; Vogel, H.J. Structural basis for the regulation of L-type voltage-gated calcium channels: Interactions between the N-terminal cytoplasmic domain and Ca2+-calmodulin. Front. Mol. Neurosci. 2012, 5, 38. [Google Scholar] [CrossRef]
- Dixon, R.; Yuan, C.; Cheng, E.P.; Navedo, M.F.; Santana, L.F. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc. Natl. Acad. Sci. USA 2012, 109, 1749–1754. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ames, J.B. L-Type Ca2+ Channel Regulation by Calmodulin and CaBP1. Biomolecules 2021, 11, 1811. https://doi.org/10.3390/biom11121811
Ames JB. L-Type Ca2+ Channel Regulation by Calmodulin and CaBP1. Biomolecules. 2021; 11(12):1811. https://doi.org/10.3390/biom11121811
Chicago/Turabian StyleAmes, James B. 2021. "L-Type Ca2+ Channel Regulation by Calmodulin and CaBP1" Biomolecules 11, no. 12: 1811. https://doi.org/10.3390/biom11121811
APA StyleAmes, J. B. (2021). L-Type Ca2+ Channel Regulation by Calmodulin and CaBP1. Biomolecules, 11(12), 1811. https://doi.org/10.3390/biom11121811