
Revealing Topological Barriers against Knot Untying in Thermal and Mechanical Protein Unfolding by Molecular Dynamics Simulations


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Models
2.2. Molecular Dynamics Simulation
2.3. Thermal Denaturation
2.4. Mechanical Denaturation
2.5. Knot Detection Algorithm
3. Results
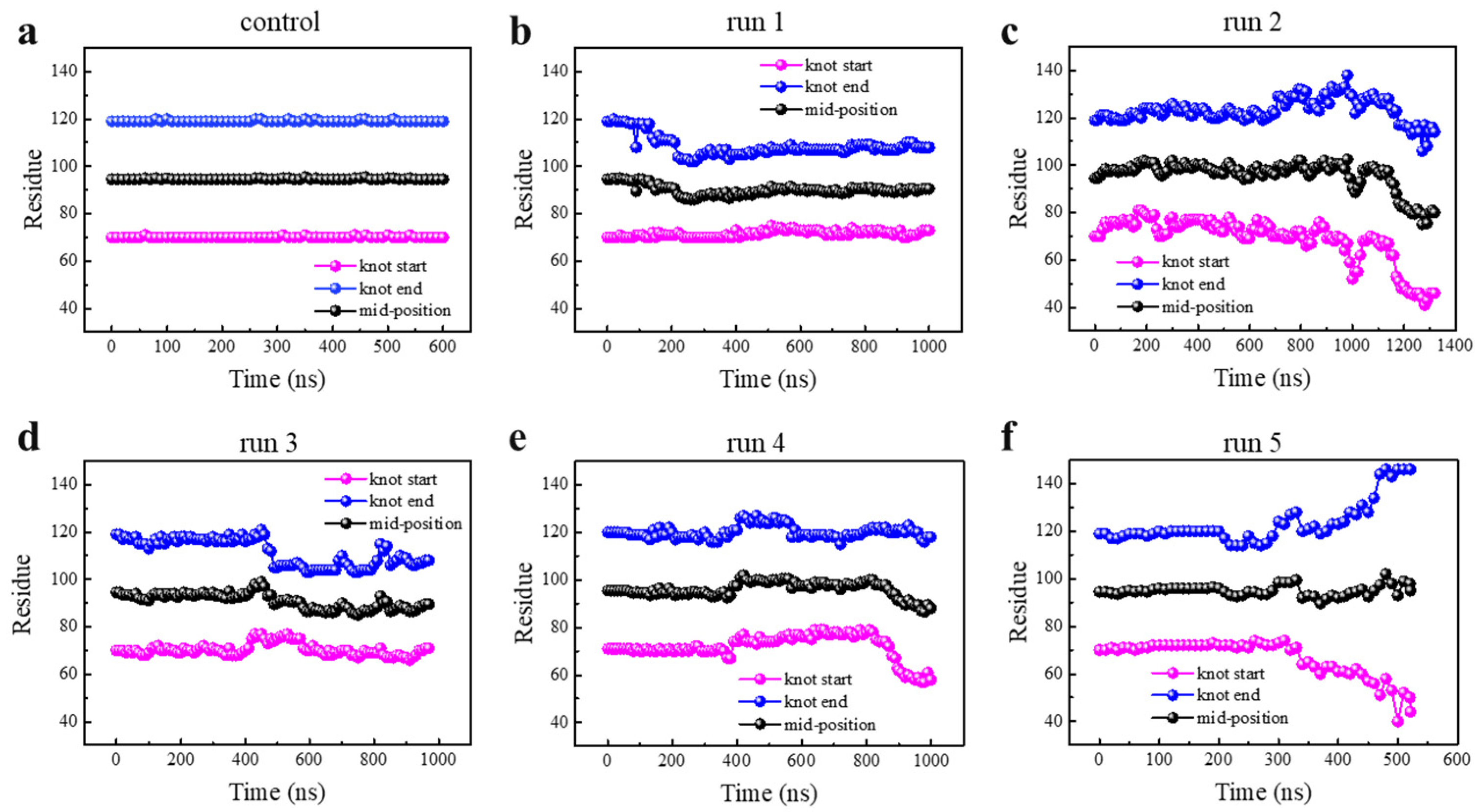
3.1. Knot Untying under Enhanced Thermal Fluctuations
3.2. Knot Maintenance in Thermal Protein Unfolding
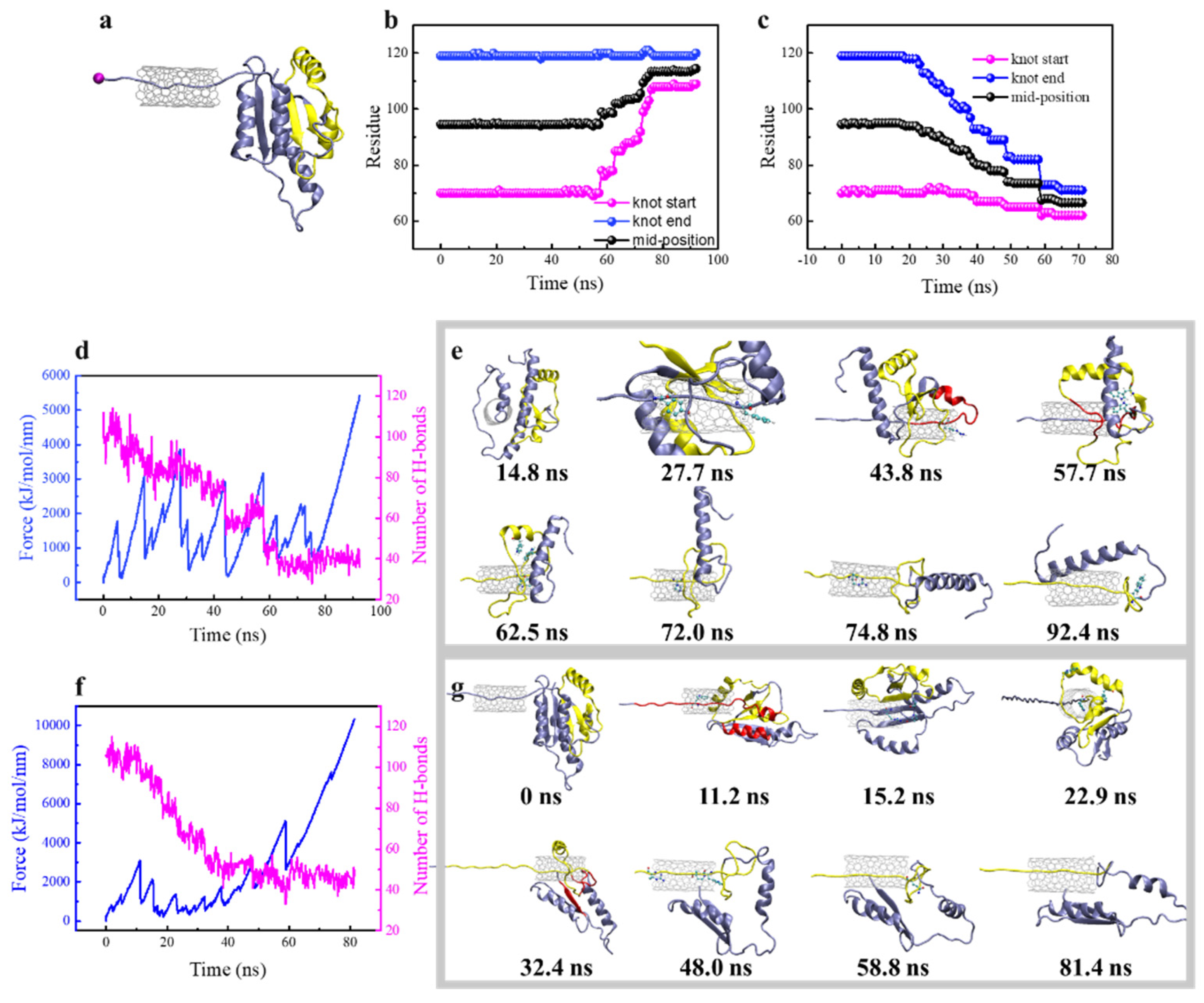
3.3. Steered Translocation of YbeA through SWCNT
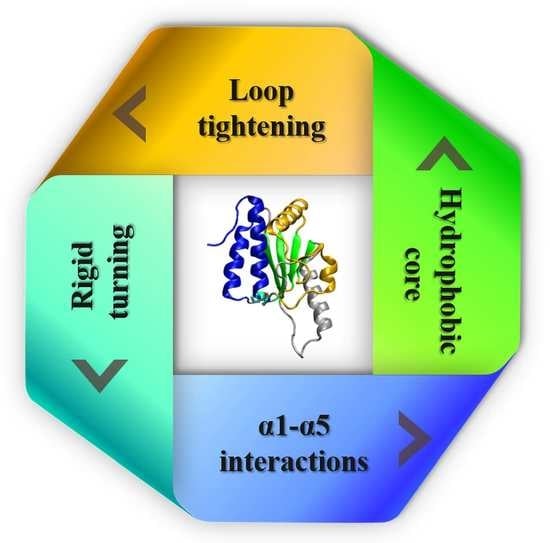
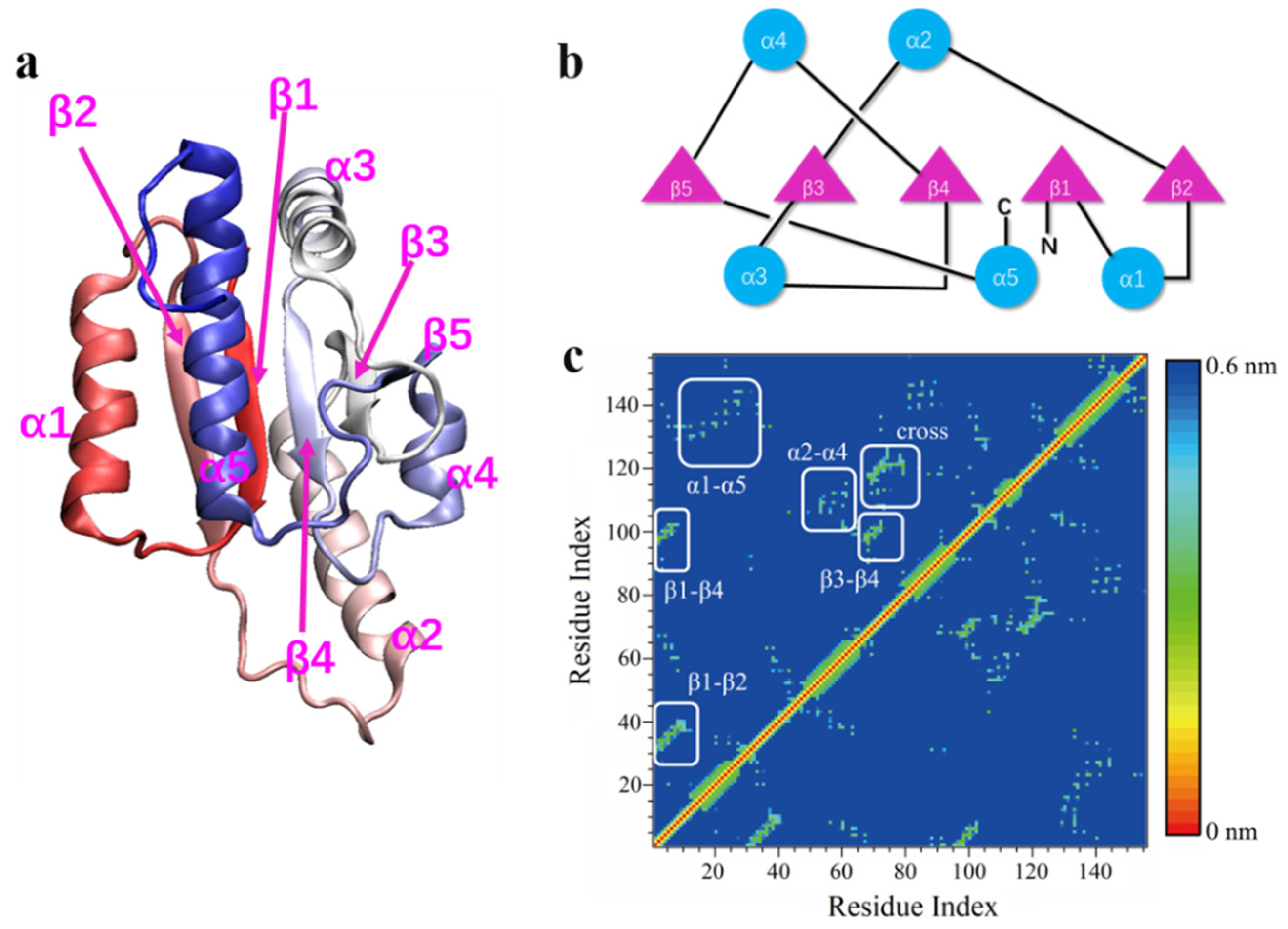
3.4. Four Aspects of Barriers Jointly against Knot Untying in Protein Unfolding
3.5. Site Mutations Manifesting Barriers against Knot Untying
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, E.; Anfinsen, C.B. Regeneration of enzyme activity by air oxidation of reduced subtilisin-modified ribonuclease. J. Biol. Chem. 1961, 236, 422–424. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Dill, K.A.; Ozkan, S.B.; Weikl, T.R.; Chodera, J.D.; Voelz, V.A. The protein folding problem: When will it be solved? Curr. Opin. Struc. Biol. 2007, 17, 342–346. [Google Scholar] [CrossRef]
- Nureki, O.; Shirouzu, M.; Hashimoto, K.; Ishitani, R.; Terada, T.; Tamakoshi, M.; Oshima, T.; Chijimatsu, M.; Takio, K.; Vassylyev, D.G.; et al. An enzyme with a deep trefoil knot for the active-site architecture. Acta Cryst. D 2002, 58, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Taylor, W.R. Protein knots and fold complexity: Some new twists. Comput. Biol. Chem. 2007, 31, 151–162. [Google Scholar] [CrossRef]
- Sułkowska, J.I.; Sułkowski, P.; Szymczak, P.; Cieplak, M. Stabilizing effect of knots on proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19714–19719. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, S.; Yan, Z.; Luo, Z.; Ren, H.; Ge, B.; Huang, F.; Yue, T. Stabilizing Effect of Inherent Knots on Proteins Revealed by Molecular Dynamics Simulations. Biophys. J. 2018, 115, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, S.; Yan, Z.; Ge, B.; Huang, F.; Yue, T. Revealing Cooperation between Knotted Conformation and Dimerization in Protein Stabilization by Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2019, 10, 5815–5822. [Google Scholar] [CrossRef]
- Taylor, W.R. A deeply knotted protein structure and how it might fold. Nature 2000, 406, 916–919. [Google Scholar] [CrossRef]
- Lim, N.C.; Jackson, S.E. Mechanistic insights into the folding of knotted proteins in vitro and in vivo. J. Mol. Biol. 2015, 427, 248–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallam, A.L.; Jackson, S.E. Knot formation in newly translated proteins is spontaneous and accelerated by chaperonins. Nat. Chem. Biol. 2011, 8, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Sułkowska, J.I.; Sułkowski, P.; Onuchic, J. Dodging the crisis of folding proteins with knots. Proc. Natl. Acad. Sci. USA 2009, 106, 3119–3124. [Google Scholar] [CrossRef] [Green Version]
- Noel, J.K.; Sułkowska, J.I.; Onuchic, J.N. Slipknotting upon native-like loop formation in a trefoil knot protein. Proc. Natl. Acad. Sci. USA 2010, 107, 15403–15408. [Google Scholar] [CrossRef] [Green Version]
- a Beccara, S.; Škrbić, T.; Covino, R.; Micheletti, C.; Faccioli, P. Folding Pathways of a Knotted Protein with a Realistic Atomistic Force Field. PLoS Comput. Biol. 2013, 9, e1003002. [Google Scholar] [CrossRef] [PubMed]
- Mallam, A.L.; Rogers, J.M.; Jackson, S.E. Experimental detection of knotted conformations in denatured proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8189–8194. [Google Scholar] [CrossRef] [Green Version]
- Mallam, A.L.; Jackson, S.E. Folding studies on a knotted protein. J. Mol. Biol. 2005, 346, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Andrews, B.T.; Capraro, D.T.; Sulkowska, J.I.; Onuchic, J.N.; Jennings, P.A. Hysteresis as a Marker for Complex, Overlapping Landscapes in Proteins. J. Phys. Chem. Lett. 2013, 4, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sułkowska, J.I.; Sułkowski, P.; Szymczak, P.; Cieplak, M. Tightening of knots in proteins. Phys. Rev. Lett. 2008, 100, 058106. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, F.; Lim, N.C.H.; Mandal, S.S.; Pelz, B.; Ng, W.-P.; Schlierf, M.; Jackson, S.E.; Rief, M. Knotting and unknotting of a protein in single molecule experiments. Proc. Natl. Acad. Sci. USA 2016, 113, 7533–7538. [Google Scholar] [CrossRef] [Green Version]
- Bornschlogl, T.; Anstrom, D.M.; Mey, E.; Dzubiella, J.; Rief, M.; Forest, K.T. Tightening the knot in phytochrome by single-molecule atomic force microscopy. Biophys. J. 2009, 96, 1508–1514. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Lamour, G.; Xiao, A.; Gsponer, J.; Li, H. Mechanically tightening a protein slipknot into a trefoil knot. J. Am. Chem. Soc. 2014, 136, 11946–11955. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Li, S.; Gao, X.; Xiao, A.; Hu, C.; Hu, X.; Hu, X.; Li, H. Direct observation of the fast and robust folding of a slipknotted protein by optical tweezers. Nanoscale 2019, 11, 3945–3951. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Genchev, G.Z.; Lu, H.; Li, H. Mechanically untying a protein slipknot: Multiple pathways revealed by force spectroscopy and steered molecular dynamics simulations. J. Am. Chem. Soc. 2012, 134, 10428–10435. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Gao, X.; Hu, X.; Hu, X.; Hu, C.; Li, H. Mechanical Unfolding and Folding of a Complex Slipknot Protein Probed by Using Optical Tweezers. Biochemistry 2019, 58, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, H. Mechanically tightening, untying and retying a protein trefoil knot by single-molecule force spectroscopy. Chem. Sci. 2020, 11, 12512–12521. [Google Scholar] [CrossRef]
- Noel, J.K.; Onuchic, J.N.; Sulkowska, J.I. Knotting a Protein in Explicit Solvent. J. Phys. Chem. Lett. 2013, 4, 3570–3573. [Google Scholar] [CrossRef]
- Wang, P.; Yang, L.; Liu, P.; Gao, Y.Q.; Zhao, X.S. Single-molecule detection reveals knot sliding in TrmD denaturation. Chemistry 2013, 19, 5909–5916. [Google Scholar] [CrossRef]
- Zou, A.; Xiu, P.; Ou, X.; Zhou, R. Spontaneous Translocation of Single-Stranded DNA in Graphene-MoS2 Heterostructure Nanopores: Shape Effect. J. Phys. Chem. B. 2020, 124, 9490–9496. [Google Scholar] [CrossRef]
- Gu, Z.; Zhang, Y.; Luan, B.; Zhou, R. DNA translocation through single-layer boron nitride nanopores. Soft Matter 2016, 12, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.K.; Gordon, R.K.; Tal, J.; Zeng, G.C.; Doctor, B.P.; Pardhasaradhi, K.; McCann, P.P. S-Adenosylmethionine and methylation. FASEB J. 1996, 10, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Levitt, M.; Warshel, A. Computer simulation of protein folding. Nature 1975, 253, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.C.; Jacobson, D.; Yatsenko, K.; Sritharan, D.; Weinreich, T.M.; Shaw, D.E. Atomic-level characterization of protein–protein association. Proc. Natl. Acad. Sci. USA 2019, 116, 4244–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arantes, P.R.; Saha, A.; Palermo, G. Fighting COVID-19 Using Molecular Dynamics Simulations. ACS Central Sci. 2020, 6, 1654–1656. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhou, R. Planar graphene/h-BN/graphene heterostructures for protein stretching and confinement. Nanoscale 2020, 12, 13822–13828. [Google Scholar] [CrossRef]
- Ponomarev, S.Y.; Thayer, K.M.; Beveridge, D.L. Ion motions in molecular dynamics simulations on DNA. Proc. Natl. Acad. Sci. USA 2004, 101, 14771–14775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, Y.; Huynh, T.; Yang, Y.; Yang, X.; Zhou, R. Directional extraction and penetration of phosphorene nanosheets to cell membranes. Nanoscale 2020, 12, 2810–2819. [Google Scholar] [CrossRef]
- Meng, X.Y.; Liu, S.; Cui, M.; Zhou, R.; Logothetis, D.E. The Molecular Mechanism of Opening the Helix Bundle Crossing (HBC) Gate of a Kir Channel. Sci. Rep. 2016, 6, 29399. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.Y.; Kang, S.G.; Zhou, R. Molecular mechanism of phosphoinositides’ specificity for the inwardly rectifying potassium channel Kir2.2. Chem. Sci. 2018, 9, 8352–8362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutzner, C.; Pall, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmuller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wallin, S.; Zeldovich, K.B.; Shakhnovich, E.I. The folding mechanics of a knotted protein. J. Mol. Biol. 2007, 368, 884–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosé, S.; Klein, M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wang, Q.; Christiansen, A.; Samiotakis, A.; Wittung-Stafshede, P.; Cheung, M.S. Comparison of chemical and thermal protein denaturation by combination of computational and experimental approaches. II. J. Chem. Phys. 2011, 135, 11B604. [Google Scholar] [CrossRef]
- Rocco, A.G.; Mollica, L.; Ricchiuto, P.; Baptista, A.M.; Gianazza, E.; Eberini, I. Characterization of the protein unfolding processes induced by urea and temperature. Biophys. J. 2008, 94, 2241–2251. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Liu, S.; Yin, X.; Li, Z.; Yang, Z.; Zhou, R. Molecular Origin of the Stability Difference in Four Shark IgNAR Constant Domains. Biophys. J. 2019, 116, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, P. Ions at aqueous interfaces. Faraday Discuss. 2009, 141, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Alexiadis, A.; Kassinos, S. Molecular Simulation of Water in Carbon Nanotubes. Chem. Rev. 2008, 108, 5014–5034. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Huynh, T.; Li, J.; Zhou, R. Nanomechanics of Protein Unfolding Outside a Generic Nanopore. ACS Nano 2016, 10, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Zhou, R. Single-File Protein Translocations through Graphene-MoS2 Heterostructure Nanopores. J. Phys. Chem. Lett. 2018, 9, 3409–3415. [Google Scholar] [CrossRef]
- Szymczak, P. Periodic forces trigger knot untying during translocation of knotted proteins. Sci. Rep. 2016, 6, 21702. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zhang, S.; Weber, J.K.; Luan, B.; Zhou, R.; Li, J. Sequential protein unfolding through a carbon nanotube pore. Nanoscale 2016, 8, 12143–12151. [Google Scholar] [CrossRef]
- Jamroz, M.; Niemyska, W.; Rawdon, E.J.; Stasiak, A.; Millett, K.C.; Sulkowski, P.; Sulkowska, J.I. KnotProt: A database of proteins with knots and slipknots. Nucleic Acids Res. 2015, 43, D306–D314. [Google Scholar] [CrossRef]
- Dabrowski-Tumanski, P.; Rubach, P.; Goundaroulis, D.; Dorier, J.; Sulkowski, P.; Millett, K.C.; Rawdon, E.J.; Stasiak, A.; Sulkowska, J.I. KnotProt 2.0: A database of proteins with knots and other entangled structures. Nucleic Acids Res. 2019, 47, D367–D375. [Google Scholar] [CrossRef] [Green Version]
- Sulkowska, J.I.; Rawdon, E.J.; Millett, K.C.; Onuchic, J.N.; Stasiak, A. Conservation of complex knotting and slipknotting patterns in proteins. Proc. Natl. Acad. Sci. USA 2012, 109, E1715–E1723. [Google Scholar] [CrossRef] [Green Version]
- Koniaris, K.; Muthukumar, M. Selfentanglement in ring polymers. J. Chem. Phys. 1991, 95, 2873–2881. [Google Scholar] [CrossRef]
- Day, R.; Bennion, B.J.; Ham, S.; Daggett, V. Increasing Temperature Accelerates Protein Unfolding Without Changing the Pathway of Unfolding. J. Mol. Biol. 2002, 322, 189–203. [Google Scholar] [CrossRef]
- Hespenheide, B.M.; Rader, A.; Thorpe, M.F.; Kuhn, L.A. Identifying protein folding cores from the evolution of flexible regions during unfolding. J. Mol. Graph. Model. 2002, 21, 195–207. [Google Scholar] [CrossRef]
- Hutchinson, E.G.; Thornton, J.M. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 1994, 3, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Burban, D.J.; Haglund, E.; Capraro, D.T.; Jennings, P.A. Heterogeneous side chain conformation highlights a network of interactions implicated in hysteresis of the knotted protein, minimal tied trefoil. J. Phys. Condens. Matter 2015, 27, 354108. [Google Scholar] [CrossRef] [Green Version]
- Shih, P.M.; Wang, I.; Lee, Y.T.; Hsieh, S.J.; Chen, S.Y.; Wang, L.W.; Huang, C.T.; Chien, C.T.; Chang, C.Y.; Hsu, S.T. Random-Coil Behavior of Chemically Denatured Topologically Knotted Proteins Revealed by Small-Angle X-ray Scattering. J. Phys. Chem. B 2015, 119, 5437–5443. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Kang, R.; Ren, L.; Yang, L.; Yue, T. Revealing Topological Barriers against Knot Untying in Thermal and Mechanical Protein Unfolding by Molecular Dynamics Simulations. Biomolecules 2021, 11, 1688. https://doi.org/10.3390/biom11111688
Xu Y, Kang R, Ren L, Yang L, Yue T. Revealing Topological Barriers against Knot Untying in Thermal and Mechanical Protein Unfolding by Molecular Dynamics Simulations. Biomolecules. 2021; 11(11):1688. https://doi.org/10.3390/biom11111688
Chicago/Turabian StyleXu, Yan, Runshan Kang, Luyao Ren, Lin Yang, and Tongtao Yue. 2021. "Revealing Topological Barriers against Knot Untying in Thermal and Mechanical Protein Unfolding by Molecular Dynamics Simulations" Biomolecules 11, no. 11: 1688. https://doi.org/10.3390/biom11111688