
Changes in Cardiac Metabolism in Prediabetes


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cardiac Fat
3. Adipose Tissue Surrounding the Heart
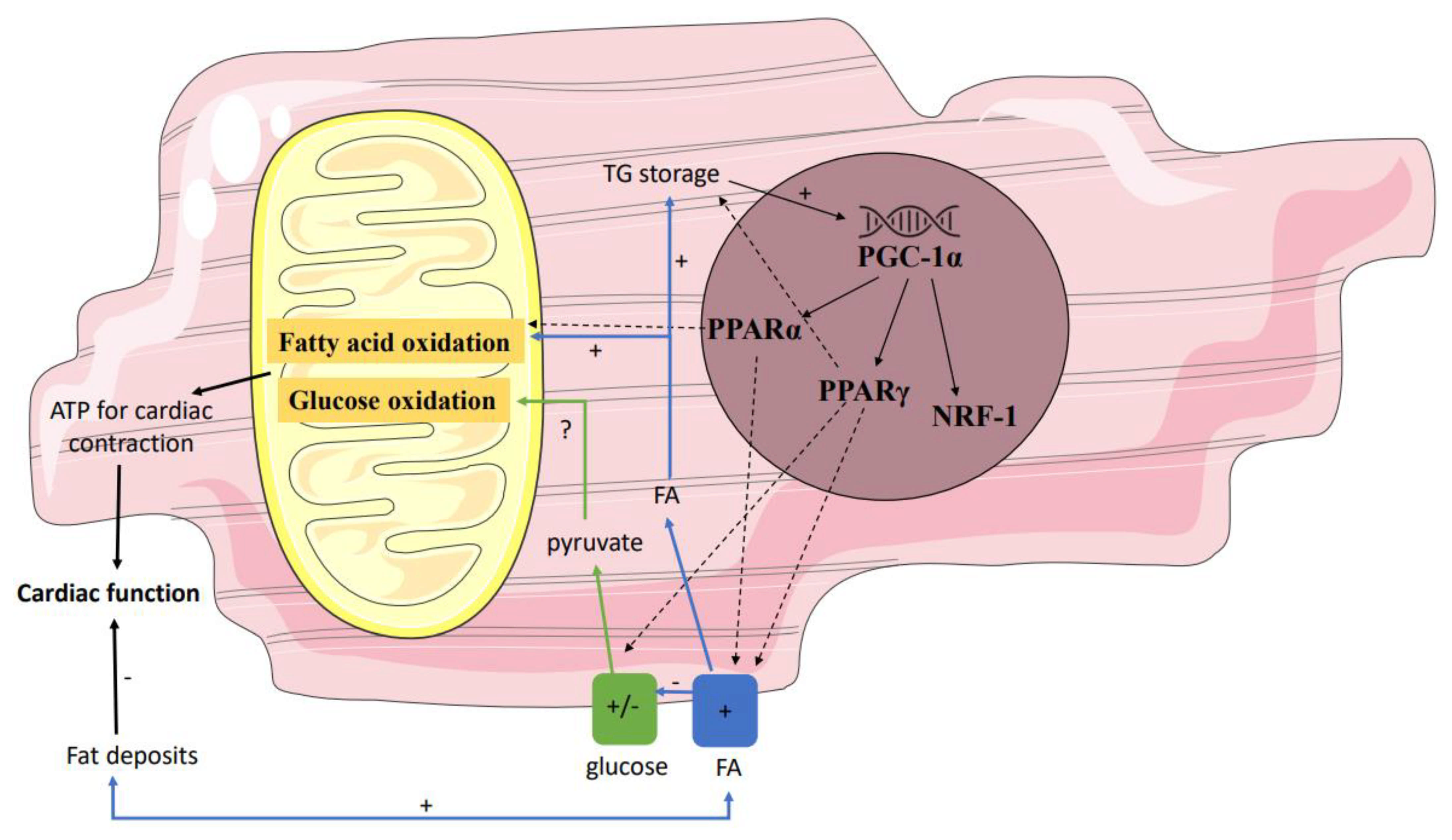
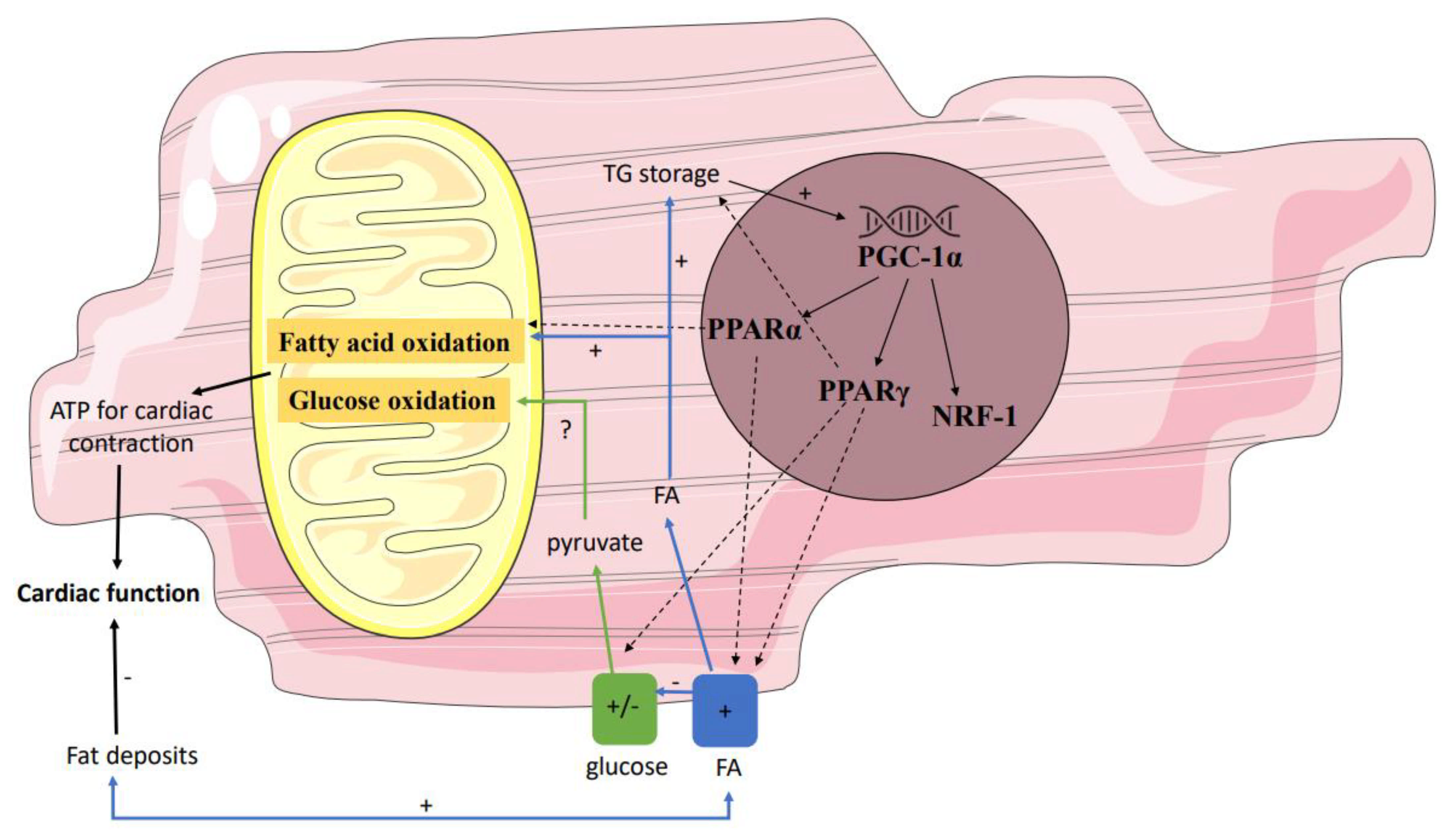
4. Enhanced Cardiac Lipid Metabolism
5. Decreased Cardiac Glucose Metabolism
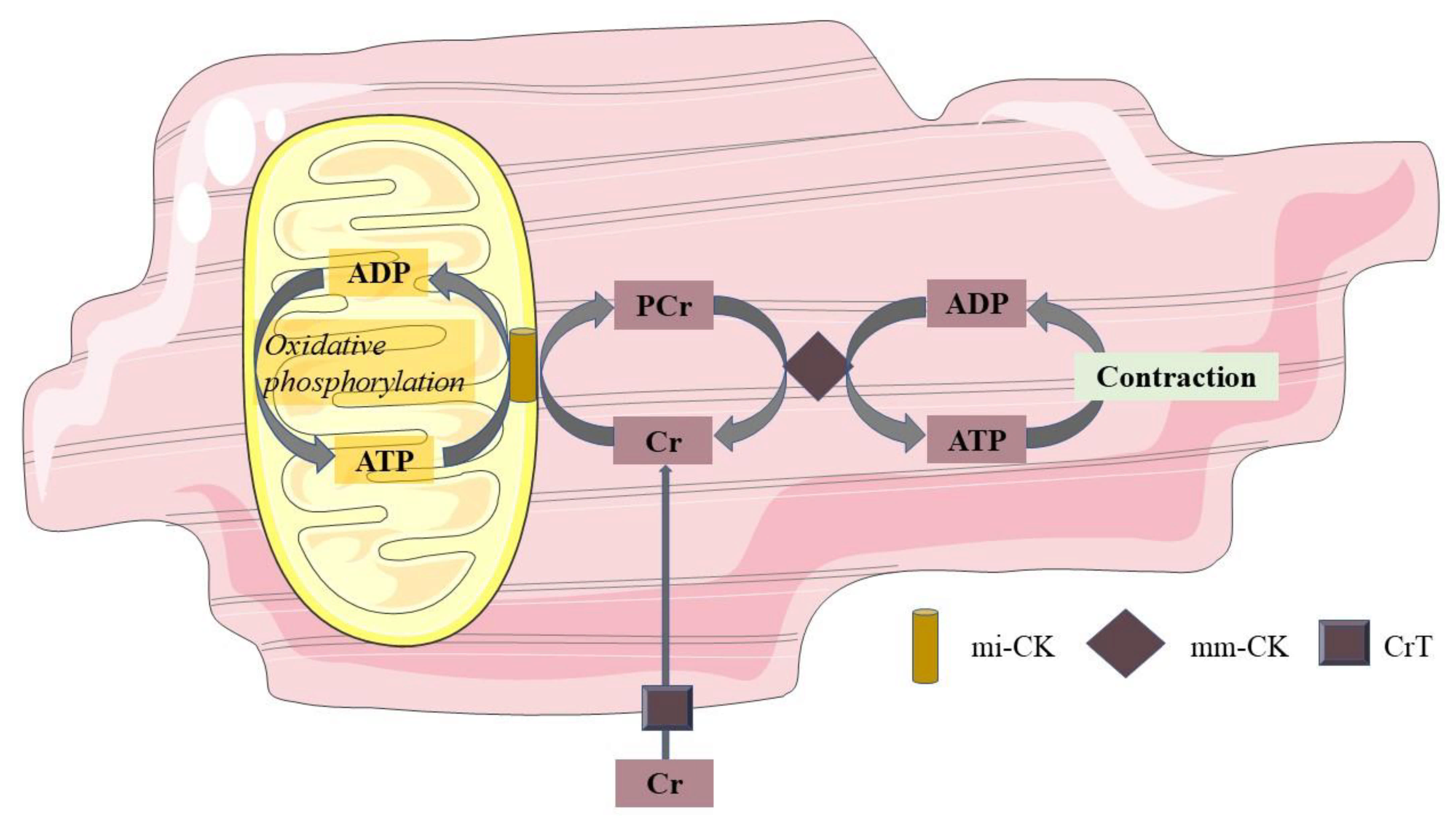
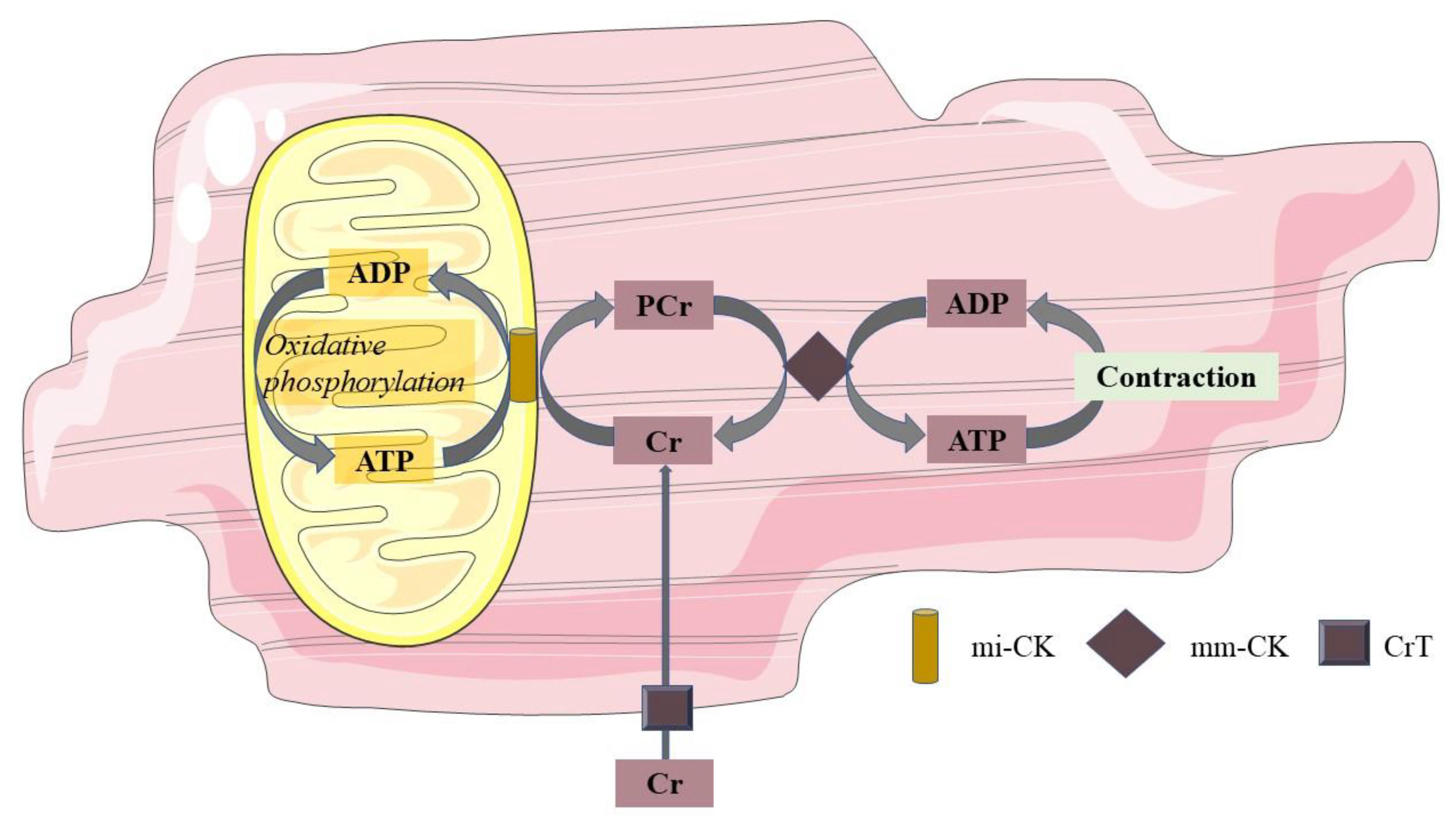
6. Mitochondrial Function
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1H-MRS | proton MRS |
| 18F-FDG | [18F]-fluorodeoxyglucose |
| 18F-FTHA | 18F-fluoro-6-thia-heptadecanoic acid |
| 18F-FTP | 16-[18F]fluoro-4-thiapalmitate |
| A | peak late filling velocity |
| ADP | adenosine diphosphate |
| ATP | adenosine triphosphate |
| CVD | cardiovascular disease |
| Cr | creatine |
| DAG | diacylglycerol |
| DCM | diabetic cardiomyopathy |
| E | peak early filling velocity |
| EAT | epicardial adipose tissue |
| FFA | free fatty acid |
| HSL | hormone-sensitive lipase |
| IRS1 | insulin receptor substrate 1 |
| LVEF | left ventricular ejection fraction |
| LVM | left ventricular mass |
| MRS | magnetic resonance spectroscopy |
| NEFA | nonesterified fatty acid |
| NGM | normal glucose metabolism |
| PCr | phosphocreatine |
| PET | positron emission tomography |
| PI3K | PI 3-kinases |
| PPAR | peroxisome proliferator-activated receptor |
| ROS | reactive oxygen species |
| T2DM | type 2 diabetes mellitus |
| TG | triglyceride |
References
- World Health Organization; International Diabetes Federation. Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycemia: Report of a WHO/IDF Consultation: WHO. 2006. Available online: https://www.who.int/diabetes/publications/Definition%20and%20diagnosis%20of%20diabetes_new.pdf (accessed on 12 October 2021).
- Punthakee, Z.; Goldenberg, R.; Katz, P. Definition, Classification and Diagnosis of Diabetes, Prediabetes and Metabolic Syndrome. Can. J. Diabetes 2018, 42 (Suppl. S1), S8–S11. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Zhang, Y.; Li, M.; Wu, J.H.; Mai, L.; Li, J.; Yang, Y.; Hu, Y.; Huang, Y. Association between prediabetes and risk of all cause mortality and cardiovascular disease: Updated meta-analysis. BMJ 2020, 370, m2297. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Mykkanen, L.; Festa, A.; Burke, J.P.; Stern, M.P. Insulin-resistant prediabetic subjects have more atherogenic risk factors than insulin-sensitive prediabetic subjects: Implications for preventing coronary heart disease during the prediabetic state. Circulation 2000, 101, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [PubMed] [Green Version]
- Grundy, S.M.; Benjamin, I.J.; Burke, G.L.; Chait, A.; Eckel, R.H.; Howard, B.V.; Mitch, W.; Smith, S.C., Jr.; Sowers, J.R. Diabetes and cardiovascular disease: A statement for healthcare professionals from the American Heart Association. Circulation 1999, 100, 1134–1146. [Google Scholar] [CrossRef] [Green Version]
- Hammoud, T.; Tanguay, J.F.; Bourassa, M.G. Management of coronary artery disease: Therapeutic options in patients with diabetes. J. Am. Coll. Cardiol. 2000, 36, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Taegtmeyer, H.; McNulty, P.; Young, M.E. Adaptation and maladaptation of the heart in diabetes: Part I: General concepts. Circulation 2002, 105, 1727–1733. [Google Scholar] [CrossRef] [Green Version]
- Eckel, R.H.; York, D.A.; Rossner, S.; Hubbard, V.; Caterson, I.; St Jeor, S.T.; Hayman, L.L.; Mullis, R.M.; Blair, S.N. Prevention Conference VII: Obesity, a worldwide epidemic related to heart disease and stroke: Executive summary. Circulation 2004, 110, 2968–2975. [Google Scholar] [CrossRef]
- Calle, E.E.; Thun, M.J.; Petrelli, J.M.; Rodriguez, C.; Heath, C.W., Jr. Body-mass index and mortality in a prospective cohort of U.S. adults. N. Engl. J. Med. 1999, 341, 1097–10105. [Google Scholar] [CrossRef] [PubMed]
- Wolk, R.; Berger, P.; Lennon, R.J.; Brilakis, E.S.; Davison, D.E.; Somers, V.K. Association between plasma adiponectin levels and unstable coronary syndromes. Eur. Heart J. 2007, 28, 292–298. [Google Scholar] [CrossRef]
- Tirosh, A.; Shai, I.; Afek, A.; Dubnov-Raz, G.; Ayalon, N.; Gordon, B.; Derazne, E.; Tzur, D.; Shamis, A.; Vinker, S.; et al. Adolescent BMI trajectory and risk of diabetes versus coronary disease. N. Engl. J. Med. 2011, 364, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Jones, D.; Adams, R.J.; Brown, T.M.; Carnethon, M.; Dai, S.; De Simone, G.; Ferguson, T.B.; Ford, E.; Furie, K.; Gillespie, C.; et al. Executive summary: Heart disease and stroke statistics—2010 update: A report from the American Heart Association. Circulation 2010, 121, 948–954. [Google Scholar] [PubMed]
- Van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- LeWinter, M.M.; Meyer, M. Mechanisms of diastolic dysfunction in heart failure with a preserved ejection fraction: If it’s not one thing it’s another. Circ. Heart Fail. 2013, 6, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Markus, M.R.P.; Rospleszcz, S.; Ittermann, T.; Baumeister, S.E.; Schipf, S.; Siewert-Markus, U.; Lorbeer, R.; Storz, C.; Ptushkina, V.; Peters, A.; et al. Glucose and insulin levels are associated with arterial stiffness and concentric remodeling of the heart. Cardiovasc. Diabetol. 2019, 18, 145. [Google Scholar] [CrossRef]
- Bell, D.S. Diabetic cardiomyopathy. Diabetes Care 2003, 26, 2949–2951. [Google Scholar] [CrossRef] [Green Version]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Atale, N.; Yadav, D.; Rani, V.; Jin, J.O. Pathophysiology, Clinical Characteristics of Diabetic Cardiomyopathy: Therapeutic Potential of Natural Polyphenols. Front. Nutr. 2020, 7, 564352. [Google Scholar] [CrossRef]
- Gulsin, G.S.; Athithan, L.; McCann, G.P. Diabetic cardiomyopathy: Prevalence, determinants and potential treatments. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018819834869. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.B. Abnormalities of energy expenditure and the development of obesity. Obes. Res. 1995, 3 (Suppl. S2), 155s–163s. [Google Scholar] [CrossRef]
- Szczepaniak, L.S.; Dobbins, R.L.; Metzger, G.J.; Sartoni-D’Ambrosia, G.; Arbique, D.; Vongpatanasin, W.; Unger, R.; Victor, R.G. Myocardial triglycerides and systolic function in humans: In vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn. Reson. Med. 2003, 49, 417–423. [Google Scholar] [CrossRef]
- McGavock, J.M.; Lingvay, I.; Zib, I.; Tillery, T.; Salas, N.; Unger, R.; Levine, B.D.; Raskin, P.; Victor, R.G.; Szczepaniak, L.S. Cardiac steatosis in diabetes mellitus: A 1H-magnetic resonance spectroscopy study. Circulation 2007, 116, 1170–1175. [Google Scholar] [CrossRef] [Green Version]
- Marfella, R.; Di Filippo, C.; Portoghese, M.; Barbieri, M.; Ferraraccio, F.; Siniscalchi, M.; Cacciapuoti, F.; Rossi, F.; D’Amico, M.; Paolisso, G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J. Lipid Res. 2009, 50, 2314–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meer, R.W.; Hammer, S.; Smit, J.W.; Frolich, M.; Bax, J.J.; Diamant, M.; de Roos, A.; Romijn, J.A.; Lamb, H.J. Short-term caloric restriction induces accumulation of myocardial triglycerides and decreases left ventricular diastolic function in healthy subjects. Diabetes 2007, 56, 2849–2853. [Google Scholar] [CrossRef] [Green Version]
- Malavazos, A.E.; Di Leo, G.; Secchi, F.; Lupo, E.N.; Dogliotti, G.; Coman, C.; Morricone, L.; Corsi, M.M.; Sardanelli, F.; Iacobellis, G. Relation of echocardiographic epicardial fat thickness and myocardial fat. Am. J. Cardiol. 2010, 105, 1831–1835. [Google Scholar] [CrossRef]
- Bakkum, M.J.; Danad, I.; Romijn, M.A.; Stuijfzand, W.J.; Leonora, R.M.; Tulevski, I.I.; Somsen, G.A.; Lammertsma, A.A.; van Kuijk, C.; van Rossum, A.C.; et al. The impact of obesity on the relationship between epicardial adipose tissue, left ventricular mass and coronary microvascular function. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1562–1573. [Google Scholar] [CrossRef] [Green Version]
- Cherian, S.; Lopaschuk, G.D.; Carvalho, E. Cellular cross-talk between epicardial adipose tissue and myocardium in relation to the pathogenesis of cardiovascular disease. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E937–E949. [Google Scholar] [CrossRef] [Green Version]
- Gaborit, B.; Abdesselam, I.; Dutour, A. Epicardial fat: More than just an “epi” phenomenon? Horm. Metab. Res. 2013, 45, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, P. Myocardial, perivascular, and epicardial fat. Diabetes Care 2011, 34 (Suppl. S2), S371–S379. [Google Scholar] [CrossRef] [Green Version]
- Henrichot, E.; Juge-Aubry, C.E.; Pernin, A.; Pache, J.C.; Velebit, V.; Dayer, J.M.; Meda, P.; Chizzolini, C.; Meier, C.A. Production of chemokines by perivascular adipose tissue: A role in the pathogenesis of atherosclerosis? Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2594–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibasaki, I.; Nishikimi, T.; Mochizuki, Y.; Yamada, Y.; Yoshitatsu, M.; Inoue, Y.; Kuwata, T.; Ogawa, H.; Tsuchiya, G.; Ishimitsu, T.; et al. Greater expression of inflammatory cytokines, adrenomedullin, and natriuretic peptide receptor-C in epicardial adipose tissue in coronary artery disease. Regul. Pept. 2010, 165, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat. Rev. Endocrinol. 2015, 11, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Willens, H.J. Echocardiographic epicardial fat: A review of research and clinical applications. J. Am. Soc. Echocardiogr. 2009, 22, 1311–1319. [Google Scholar] [CrossRef]
- Dabbah, S.; Komarov, H.; Marmor, A.; Assy, N. Epicardial fat, rather than pericardial fat, is independently associated with diastolic filling in subjects without apparent heart disease. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 877–882. [Google Scholar] [CrossRef]
- Hua, N.; Chen, Z.; Phinikaridou, A.; Pham, T.; Qiao, Y.; LaValley, M.P.; Bigornia, S.J.; Ruth, M.R.; Apovian, C.M.; Ruberg, F.L.; et al. The influence of pericardial fat upon left ventricular function in obese females: Evidence of a site-specific effect. J. Cardiovasc. Magn. Reson. 2014, 16, 37. [Google Scholar] [CrossRef] [Green Version]
- Konishi, M.; Sugiyama, S.; Sugamura, K.; Nozaki, T.; Matsubara, J.; Akiyama, E.; Utsunomiya, D.; Matsuzawa, Y.; Yamashita, Y.; Kimura, K.; et al. Accumulation of pericardial fat correlates with left ventricular diastolic dysfunction in patients with normal ejection fraction. J. Cardiol. 2012, 59, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Leonetti, F.; Singh, N.; Sharma, A.M. Relationship of epicardial adipose tissue with atrial dimensions and diastolic function in morbidly obese subjects. Int. J. Cardiol. 2007, 115, 272–273. [Google Scholar] [CrossRef]
- De Wit-Verheggen, V.H.W.; Altintas, S.; Spee, R.J.M.; Mihl, C.; van Kuijk, S.M.J.; Wildberger, J.E.; Schrauwen-Hinderling, V.B.; Kietselaer, B.; van de Weijer, T. Pericardial fat and its influence on cardiac diastolic function. Cardiovasc. Diabetol. 2020, 19, 129. [Google Scholar] [CrossRef] [PubMed]
- Hannukainen, J.C.; Lautamäki, R.; Pärkkä, J.; Strandberg, M.; Saunavaara, V.; Hurme, S.; Soinio, M.; Dadson, P.; Virtanen, K.A.; Grönroos, T.; et al. Reversibility of Myocardial Metabolism and Remodeling in Morbidly Obese Patients Six Months after Bariatric Surgery. Diabetes Obes. Metab. 2018, 20, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Brassard, P.; Frisch, F.; Lavoie, F.; Cyr, D.; Bourbonnais, A.; Cunnane, S.C.; Soinio, M.; Dadson, P.; Virtanen, K.A.; Gronroos, T.; et al. Impaired plasma nonesterified fatty acid tolerance is an early defect in the natural history of type 2 diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 837–844. [Google Scholar] [CrossRef]
- Labbe, S.M.; Grenier-Larouche, T.; Noll, C.; Phoenix, S.; Guerin, B.; Turcotte, E.E.; Carpentier, A.C. Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes 2012, 61, 2701–2710. [Google Scholar] [CrossRef] [Green Version]
- Mather, K.J.; Hutchins, G.D.; Perry, K.; Territo, W.; Chisholm, R.; Acton, A.; Glick-Wilson, B.; Considine, R.V.; Moberly, S.; DeGrado, T.R. Assessment of myocardial metabolic flexibility and work efficiency in human type 2 diabetes using 16-[18F]fluoro-4-thiapalmitate, a novel PET fatty acid tracer. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E452–E460. [Google Scholar] [CrossRef] [Green Version]
- Labbe, S.M.; Noll, C.; Grenier-Larouche, T.; Kunach, M.; Bouffard, L.; Phoenix, S.; Guerin, B.; Baillargeon, J.P.; Langlois, M.F.; Turcotte, E.E.; et al. Improved cardiac function and dietary fatty acid metabolism after modest weight loss in subjects with impaired glucose tolerance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1388–E1396. [Google Scholar] [CrossRef] [PubMed]
- Noll, C.; Kunach, M.; Frisch, F.; Bouffard, L.; Dubreuil, S.; Jean-Denis, F.; Phoenix, S.; Cunnane, S.C.; Guerin, B.; Turcotte, E.E.; et al. Seven-Day Caloric and Saturated Fat Restriction Increases Myocardial Dietary Fatty Acid Partitioning in Impaired Glucose-Tolerant Subjects. Diabetes 2015, 64, 3690–3699. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin resistance: Metabolic mechanisms and consequences in the heart. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2068–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Jo, K.; Kim, K.J.; Lee, Y.H.; Han, E.; Yoon, H.J.; Wang, H.J.; Kang, E.S.; Yun, M. Visceral adiposity is associated with altered myocardial glucose uptake measured by (18)FDG-PET in 346 subjects with normal glucose tolerance, prediabetes, and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 148. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Qiu, C.; Wang, X.; Xu, M.; Shao, X.; Wang, Y. The association between diabetes mellitus and reduction in myocardial glucose uptake: A population-based 18F-FDG PET/CT study. BMC Cardiovasc. Disord. 2018, 18, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Brom, C.E.; Huisman, M.C.; Vlasblom, R.; Boontje, N.M.; Duijst, S.; Lubberink, M.; Molthoff, C.F.; Lammertsma, A.A.; van der Velden, J.; Boer, C.; et al. Altered myocardial substrate metabolism is associated with myocardial dysfunction in early diabetic cardiomyopathy in rats: Studies using positron emission tomography. Cardiovasc. Diabetol. 2009, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, R.; Jorsal, A.; Iversen, P.; Tolbod, L.; Bouchelouche, K.; Sorensen, J.; Harms, H.J.; Flyvbjerg, A.; Botker, H.E.; Wiggers, H. Heart failure patients with prediabetes and newly diagnosed diabetes display abnormalities in myocardial metabolism. J. Nucl. Cardiol. 2018, 25, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, T.; Yokoyama, I.; Watanabe, T.; Momose, T.; Serezawa, T.; Nishikawa, J.; Sasaki, Y. Myocardial glucose metabolism in noninsulin-dependent diabetes mellitus patients evaluated by FDG-PET. J. Nucl. Med. 1995, 36, 456–463. [Google Scholar] [PubMed]
- Eriksson, J.W.; Visvanathar, R.; Kullberg, J.; Strand, R.; Skrtic, S.; Ekström, S.; Lubberink, M.; Lundqvist, M.H.; Katsogiannos, P.; Pereira, M.J.; et al. Tissue-specific glucose partitioning and fat content in prediabetes and type 2 diabetes: Whole-body PET/MRI during hyperinsulinemia. Eur. J. Endocrinol. 2021, 184, 879–889. [Google Scholar] [CrossRef]
- Yokoyama, I.; Yonekura, K.; Ohtake, T.; Kawamura, H.; Matsumoto, A.; Inoue, Y.; Aoyagi, T.; Sugiura, S.; Omata, M.; Ohtomo, K.; et al. Role of insulin resistance in heart and skeletal muscle F-18 fluorodeoxyglucose uptake in patients with non-insulin-dependent diabetes mellitus. J. Nucl. Cardiol. 2000, 7, 242–248. [Google Scholar] [CrossRef]
- Cook, S.A.; Varela-Carver, A.; Mongillo, M.; Kleinert, C.; Khan, M.T.; Leccisotti, L.; Strickland, N.; Matsui, T.; Das, S.; Rosenzweig, A.; et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur. Heart J. 2010, 31, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.H.; Lu, K.Y.; Chiu, S.C.; Lo, C.J.; Hung, L.M.; Huang, J.P.; Cheng, M.L.; Wang, C.H.; Tsai, C.K.; Lin, Y.C.; et al. Early Imaging Biomarker of Myocardial Glucose Adaptations in High-Fat-Diet-Induced Insulin Resistance Model by Using (18)F-FDG PET and [U-(13)C]glucose Nuclear Magnetic Resonance Tracer. Contrast Media Mol. Imaging 2018, 2018, 8751267. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.H.; Lau, J.Y.; Chen, A.P.; Geraghty, B.J.; Perks, W.J.; Roifman, I.; Wright, G.A.; Connelly, K.A. Hyperpolarized 13C Metabolic MRI of the Human Heart: Initial Experience. Circ. Res. 2016, 119, 1177–1182. [Google Scholar] [CrossRef]
- Rider, O.J.; Apps, A.; Miller, J.; Lau, J.Y.C.; Lewis, A.J.M.; Peterzan, M.A.; Dodd, M.S.; Lau, A.Z.; Trumper, C.; Gallagher, F.A.; et al. Noninvasive In Vivo Assessment of Cardiac Metabolism in the Healthy and Diabetic Human Heart Using Hyperpolarized (13)C MRI. Circ. Res. 2020, 126, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Chatham, J.C.; Seymour, A.M. Cardiac carbohydrate metabolism in Zucker diabetic fatty rats. Cardiovasc. Res. 2002, 55, 104–112. [Google Scholar] [CrossRef]
- Chatham, J.C.; Forder, J.R. A 13C-NMR study of glucose oxidation in the intact functioning rat heart following diabetes-induced cardiomyopathy. J. Mol. Cell. Cardiol. 1993, 25, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Le Page, L.M.; Rider, O.J.; Lewis, A.J.; Ball, V.; Clarke, K.; Johansson, E.; Carr, C.A.; Heather, L.C.; Tyler, D.J. Increasing Pyruvate Dehydrogenase Flux as a Treatment for Diabetic Cardiomyopathy: A Combined 13C Hyperpolarized Magnetic Resonance and Echocardiography Study. Diabetes 2015, 64, 2735–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morbelli, S.; Marini, C.; Adami, G.F.; Kudomi, N.; Camerini, G.; Iozzo, P.; Massollo, M.; Capitanio, S.; Bodrato, S.; Verardi, M.T.; et al. Tissue specificity in fasting glucose utilization in slightly obese diabetic patients submitted to bariatric surgery. Obesity 2013, 21, E175–E181. [Google Scholar] [CrossRef] [Green Version]
- Lautamäki, R.; Airaksinen, K.E.J.; Seppänen, M.; Toikka, J.; Luotolahti, M.; Ball, E.; Borra, R.; Härkönen, R.; Iozzo, P.; Stewart, M.; et al. Rosiglitazone Improves Myocardial Glucose Uptake in Patients with Type 2 Diabetes and Coronary Artery Disease. A 16-Week Randomized, Double-Blind, Placebo-Controlled Study. Diabetes 2005, 54, 2787–2794. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Koncsos, G.; Varga, Z.V.; Baranyai, T.; Boengler, K.; Rohrbach, S.; Li, L.; Borra, R.; Härkönen, R.; Iozzo, P.; Stewart, M.; et al. Diastolic dysfunction in prediabetic male rats: Role of mitochondrial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H927–H943. [Google Scholar] [CrossRef] [Green Version]
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.M.; Mouton, S.; Sebti, Y.; et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 2014, 130, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Niemann, B.; Chen, Y.; Teschner, M.; Li, L.; Silber, R.E.; Rohrbach, S. Obesity induces signs of premature cardiac aging in younger patients: The role of mitochondria. J. Am. Coll. Cardiol. 2011, 57, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Van de Weijer, T.; Paiman, E.H.M.; Lamb, H.J. Mini-Review on Cardiac Metabolic Imaging: Current imaging modalities and future perspectives. J. Appl. Physiol. 2018, 124, 168–181. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; Jenkins, D. Factors affecting the rate of phosphocreatine resynthesis following intense exercise. Sports Med. 2002, 32, 761–784. [Google Scholar] [CrossRef] [PubMed]
- Diamant, M.; Lamb, H.J.; Groeneveld, Y.; Endert, E.L.; Smit, J.W.; Bax, J.J.; Romijn, J.A.; de Roos, A.; Radder, J.K. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2003, 42, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Scheuermann-Freestone, M.; Madsen, P.L.; Manners, D.; Blamire, A.M.; Buckingham, R.E.; Styles, P.; Radda, G.K.; Neubauer, S.; Clarke, K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation 2003, 107, 3040–3046. [Google Scholar] [CrossRef]
- Rijzewijk, L.J.; van der Meer, R.W.; Lamb, H.J.; de Jong, H.W.; Lubberink, M.; Romijn, J.A.; Bax, J.J.; de Roos, A.; Twisk, J.W.; Heine, R.J.; et al. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: Studies with cardiac positron emission tomography and magnetic resonance imaging. J. Am. Coll. Cardiol. 2009, 54, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Tocchetti, C.G.; Bhatt, N.; Paolocci, N.; Cortassa, S. Protective mechanisms of mitochondria and heart function in diabetes. Antioxid. Redox Signal. 2015, 22, 1563–1586. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Gilde, A.; Fruchart, J.C.; Staels, B. PPAR receptors at the crossroads of obesity, diabetes and cardiovascular diseases. Journ. Annu. Diabetol. Hotel Dieu 2007, 21–38. [Google Scholar]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Yue, T.L.; Bao, W.; Jucker, B.M.; Gu, J.L.; Romanic, A.M.; Brown, P.J.; Cui, J.; Thudium, D.T.; Boyce, R.; Burns-Kurtis, C.L.; et al. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation 2003, 108, 2393–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerre-Millo, M.; Gervois, P.; Raspé, E.; Madsen, L.; Poulain, P.; Derudas, B.; Herbert, J.M.; Winegar, D.A.; Willson, T.M.; Fruchart, J.C.; et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J. Biol. Chem. 2000, 275, 16638–16642. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Han, X.; Courtois, M.; Aimond, F.; Nerbonne, J.M.; Kovacs, A.; Gross, R.W.; Kelly, D.P. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc. Natl. Acad. Sci. USA 2003, 100, 1226–1231. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sambandam, N.; Morabito, D.; Wagg, C.; Finck, B.N.; Kelly, D.P.; Lopaschuk, G.D. Chronic activation of PPARalpha is detrimental to cardiac recovery after ischemia. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H87–H95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Wit-Verheggen, V.H.W.; van de Weijer, T. Changes in Cardiac Metabolism in Prediabetes. Biomolecules 2021, 11, 1680. https://doi.org/10.3390/biom11111680
de Wit-Verheggen VHW, van de Weijer T. Changes in Cardiac Metabolism in Prediabetes. Biomolecules. 2021; 11(11):1680. https://doi.org/10.3390/biom11111680
Chicago/Turabian Stylede Wit-Verheggen, Vera H. W., and Tineke van de Weijer. 2021. "Changes in Cardiac Metabolism in Prediabetes" Biomolecules 11, no. 11: 1680. https://doi.org/10.3390/biom11111680
APA Stylede Wit-Verheggen, V. H. W., & van de Weijer, T. (2021). Changes in Cardiac Metabolism in Prediabetes. Biomolecules, 11(11), 1680. https://doi.org/10.3390/biom11111680