
Inhibitors of Discoidin Domain Receptor (DDR) Kinases for Cancer and Inflammation


Abstract
:1. Introduction
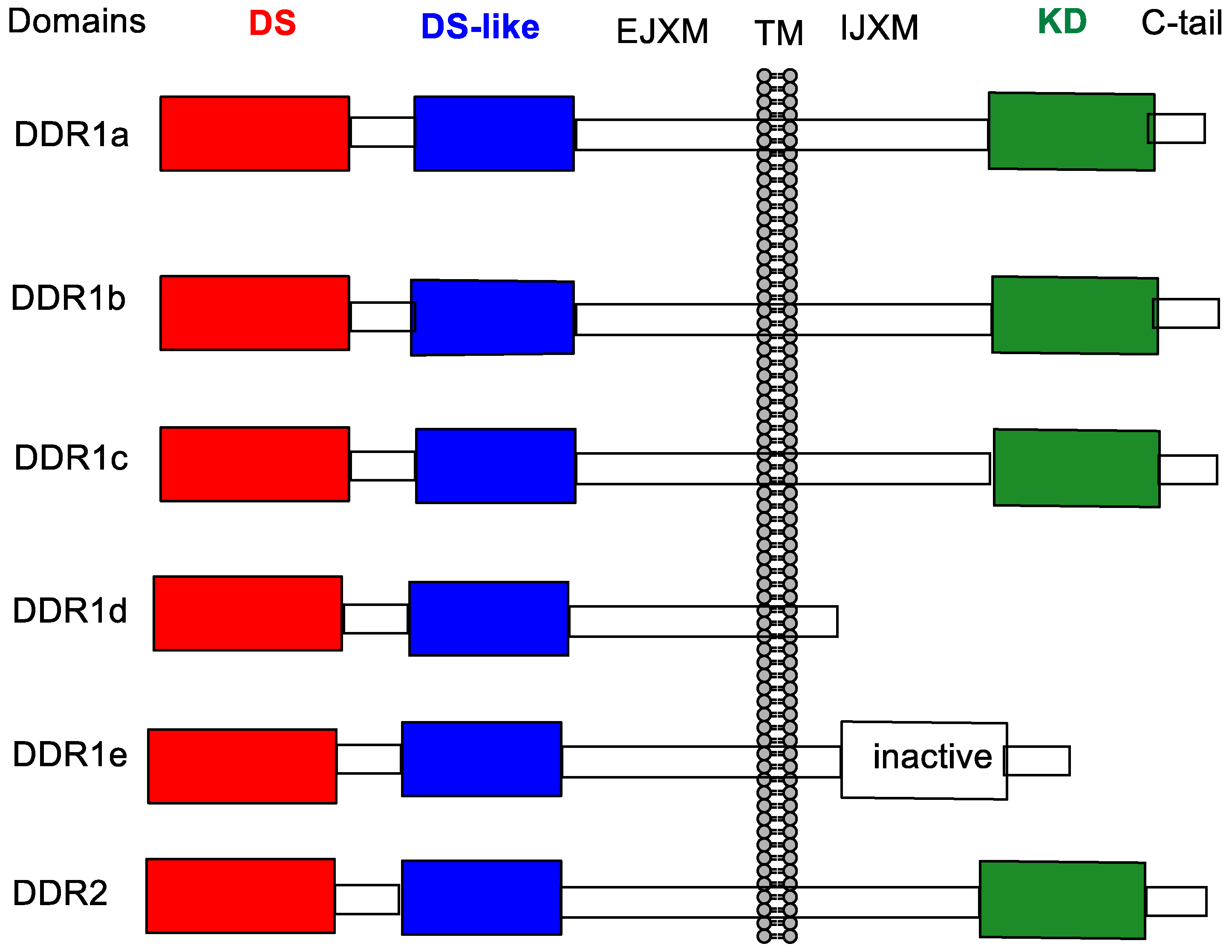
2. Approved Cancer Drugs with Significant DDR1/2 Activity
3. The DDR Kinase Domain
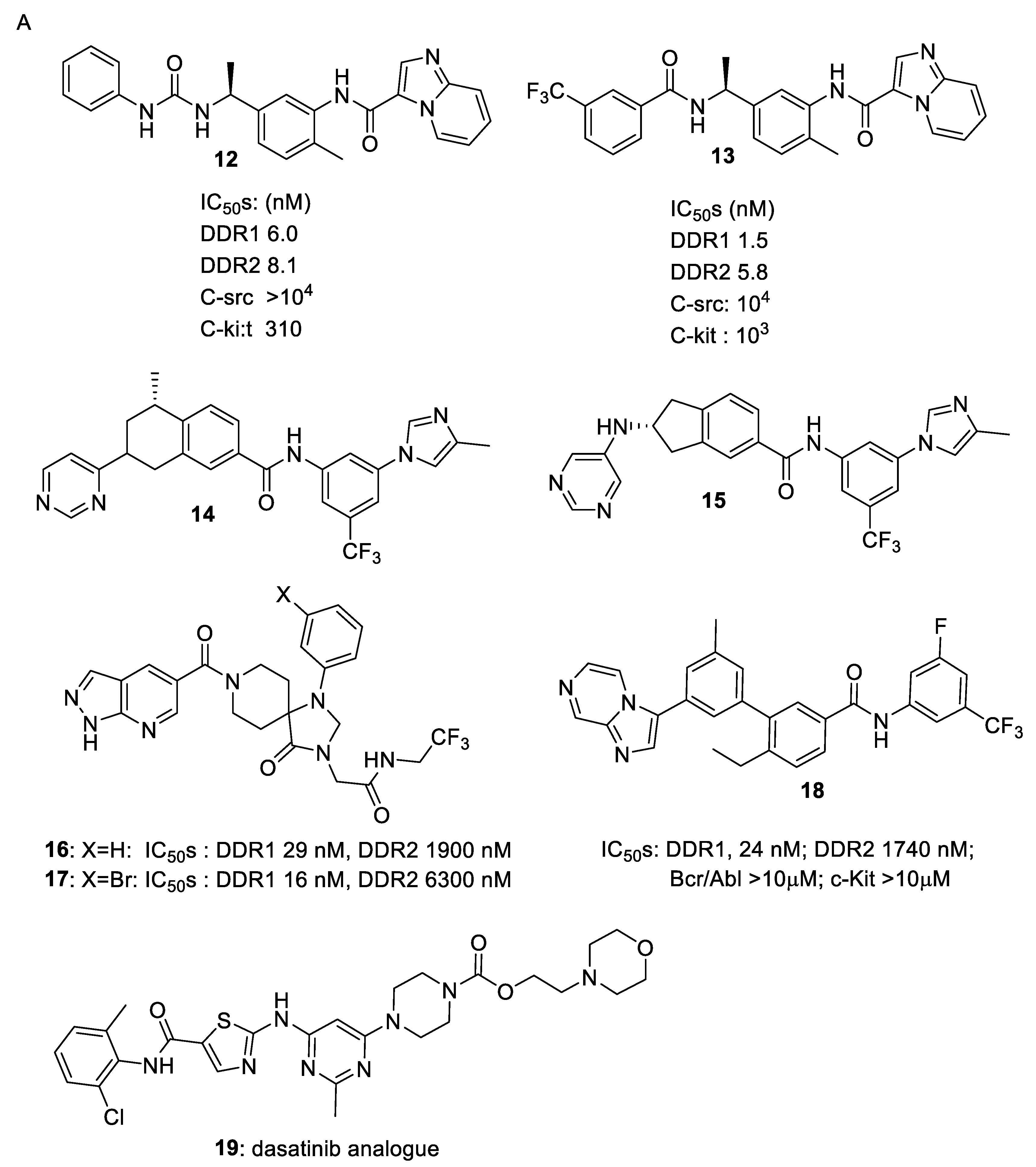
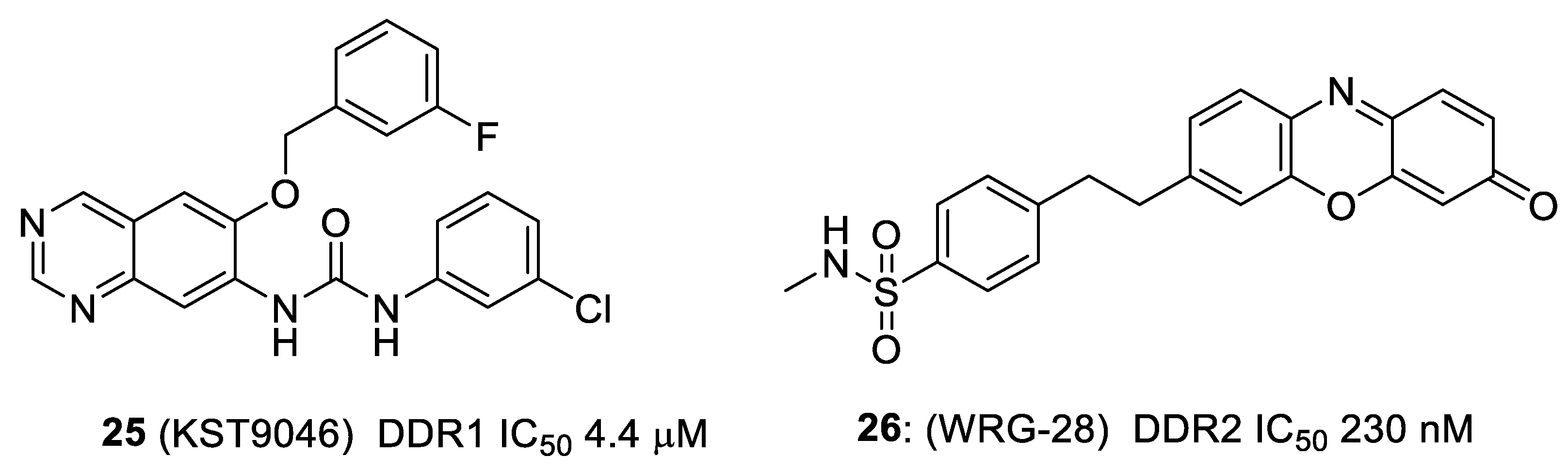
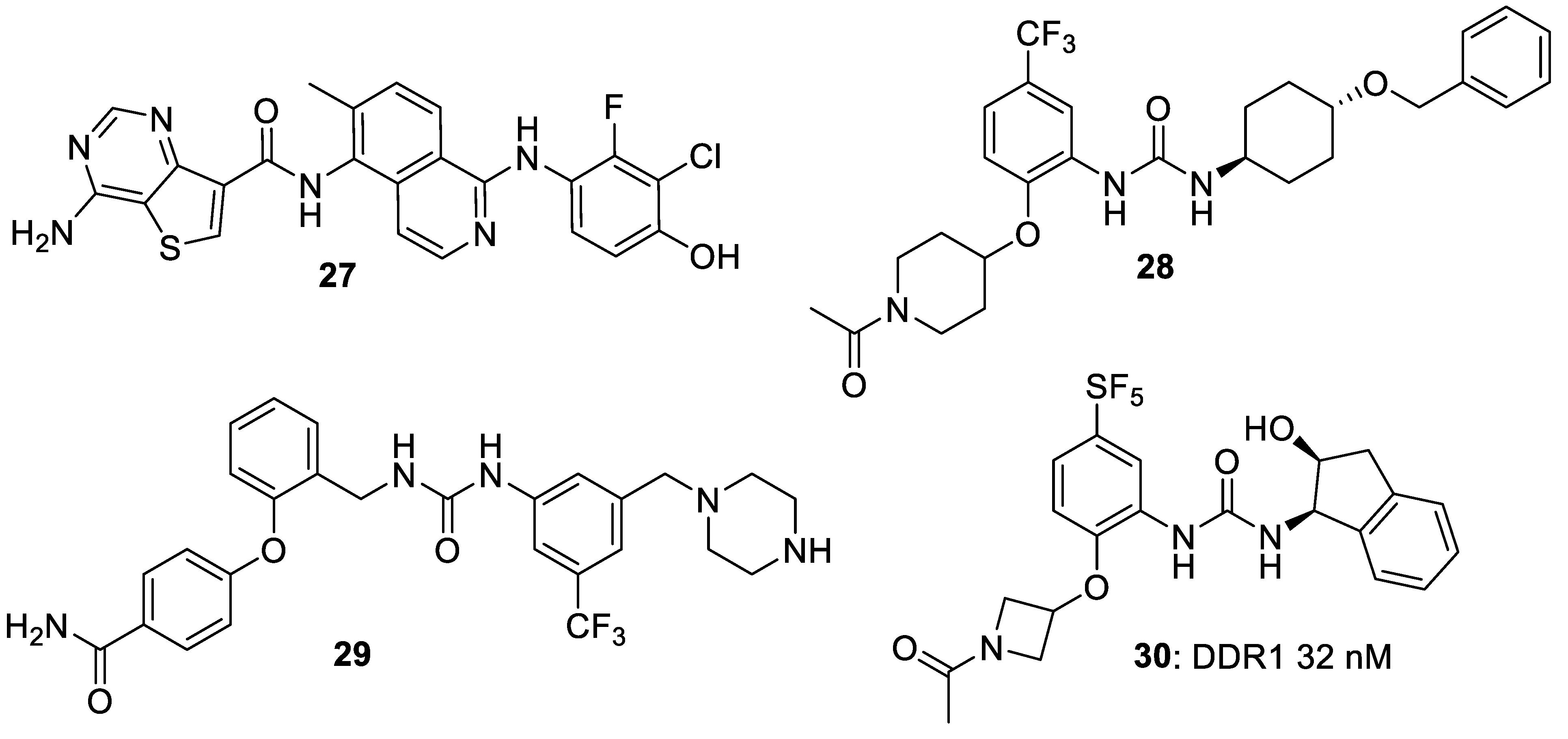
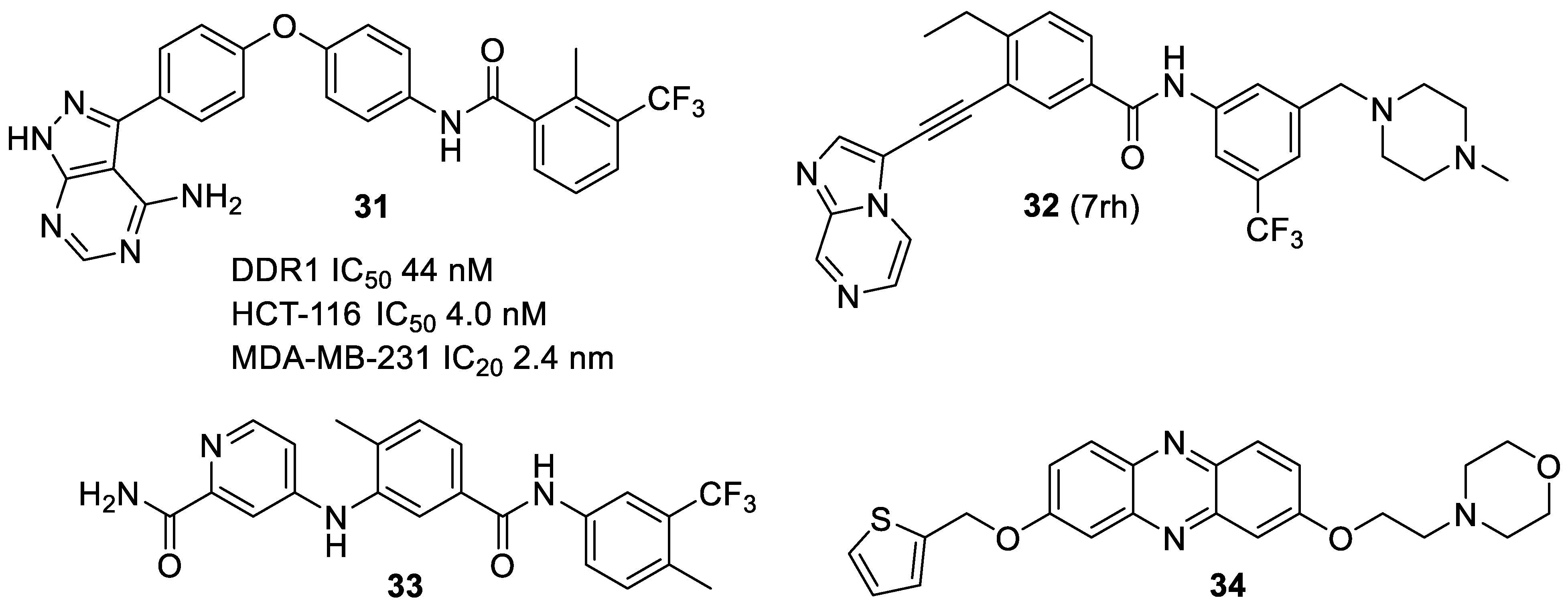
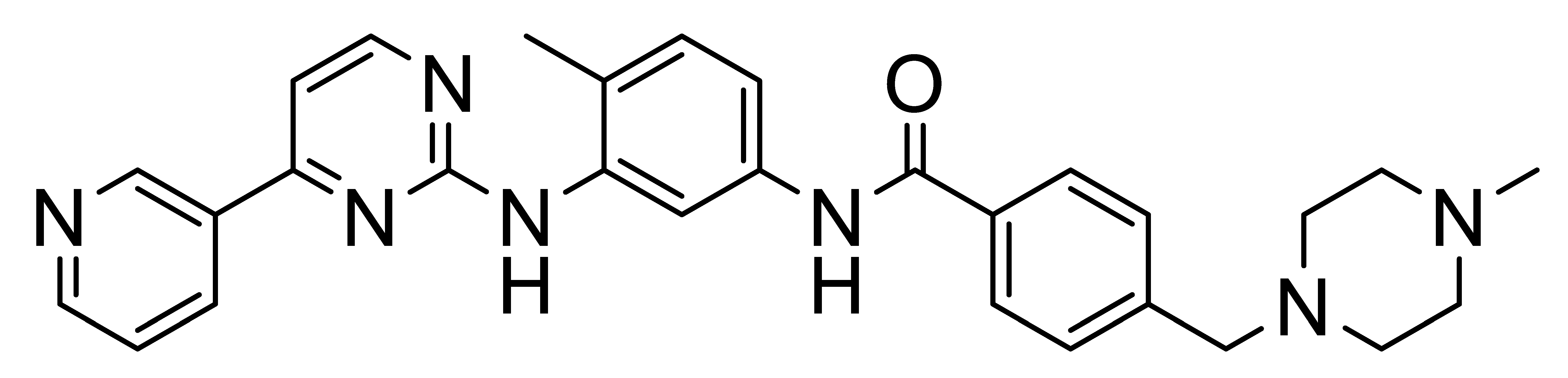
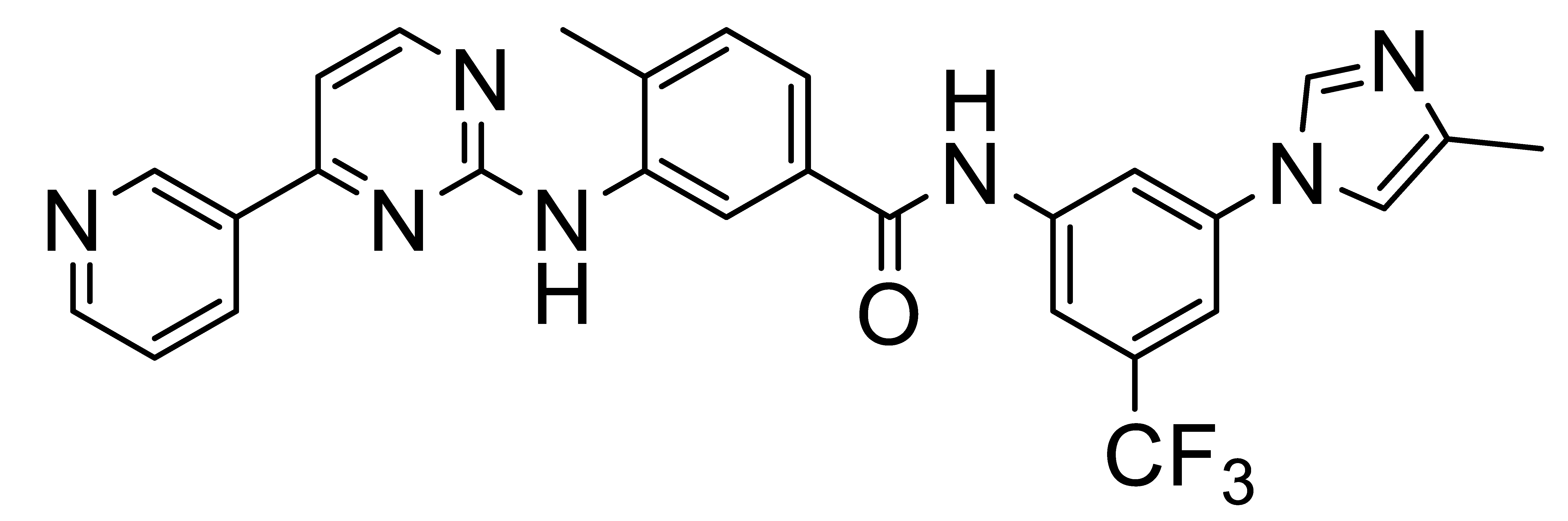
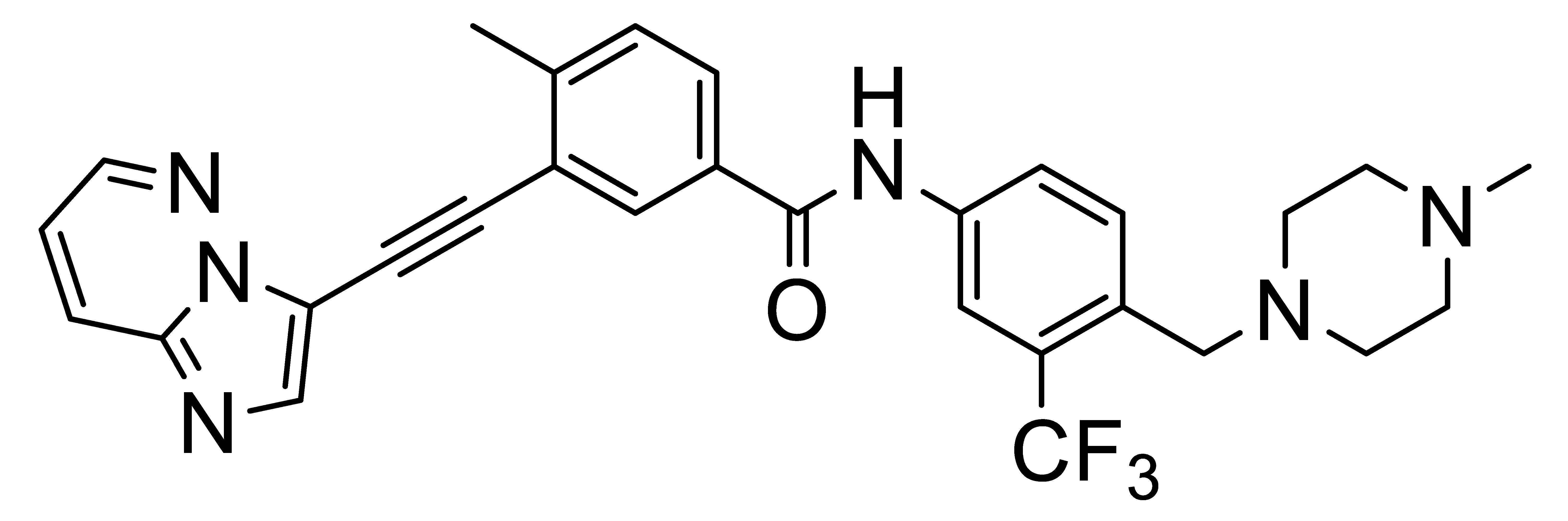
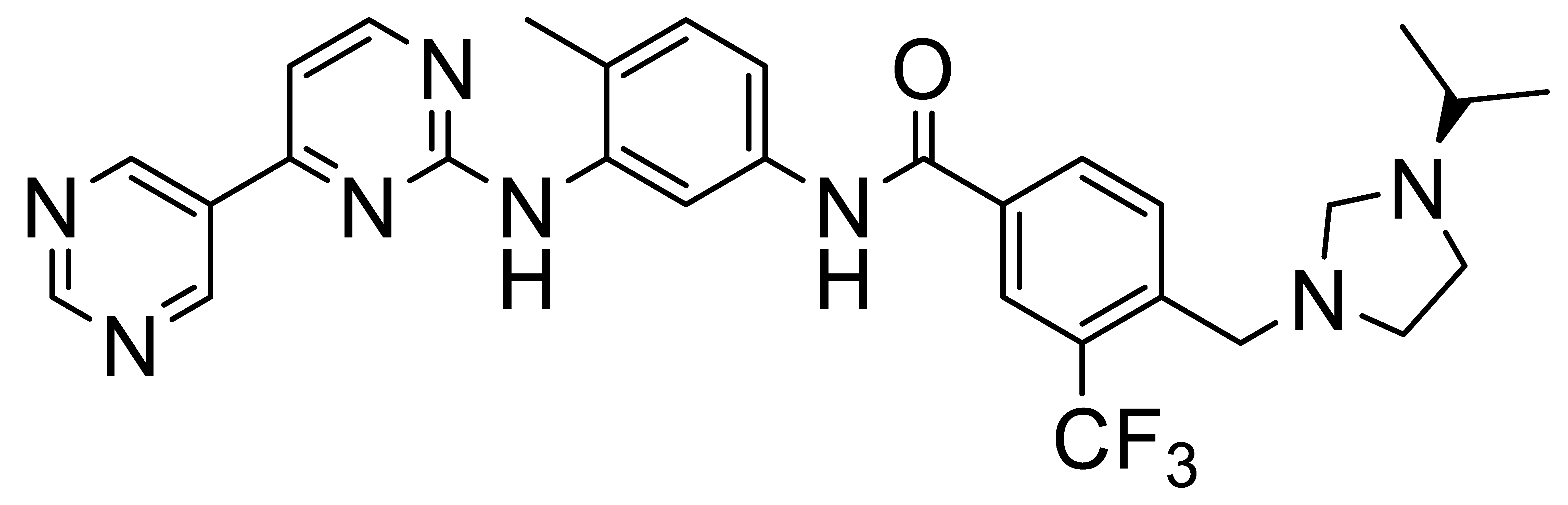
4. Selective DDR1 Inhibitors
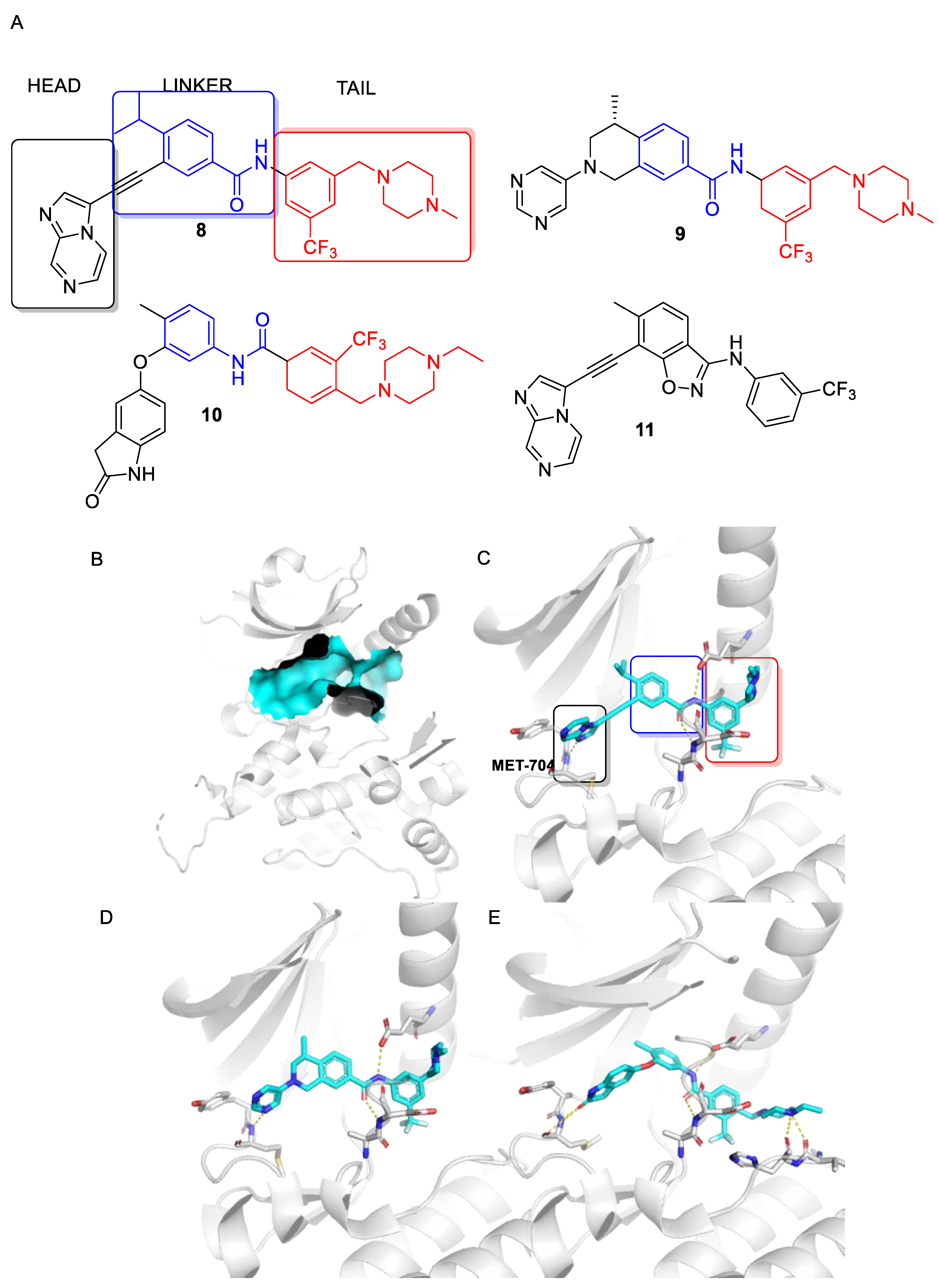
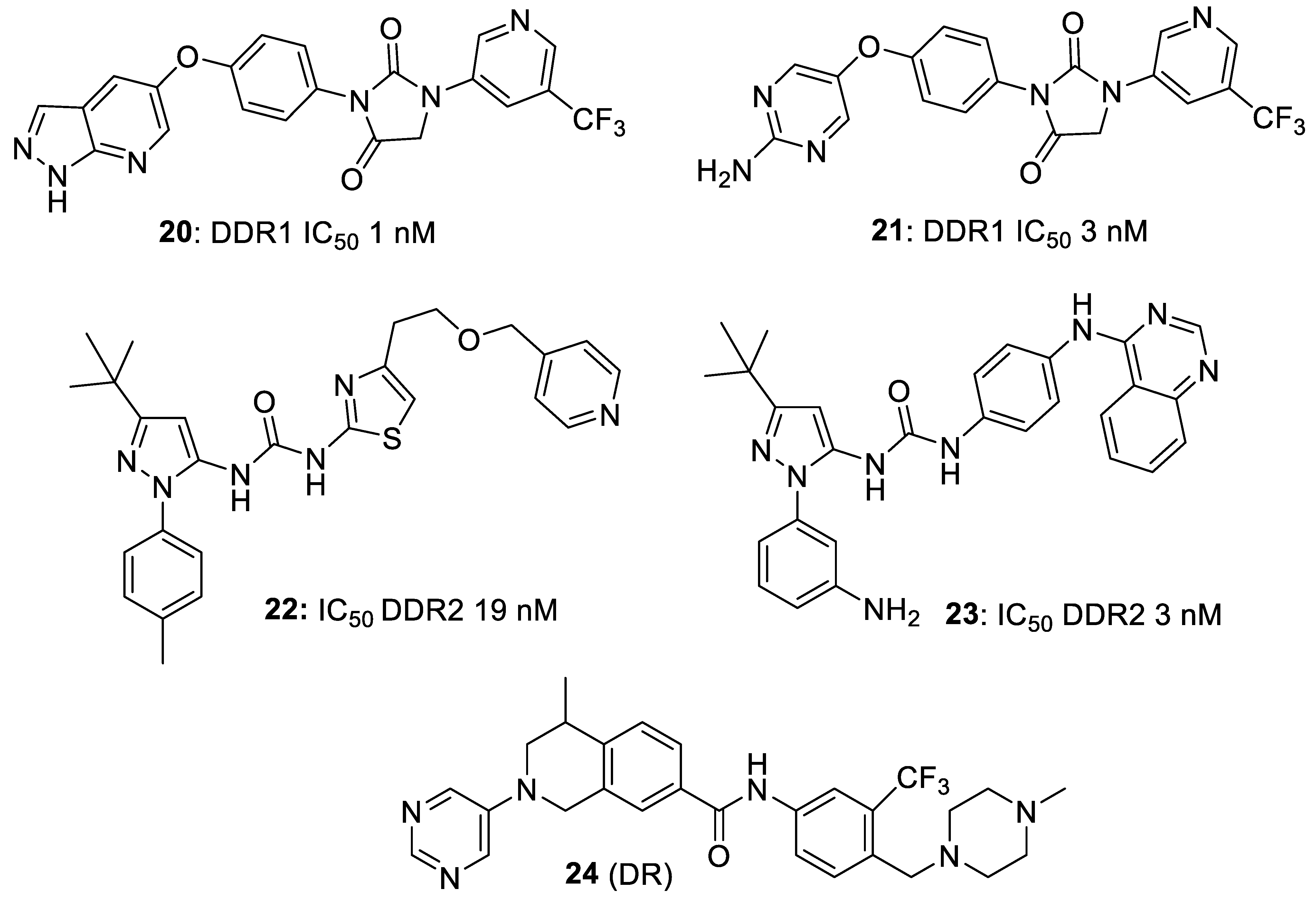
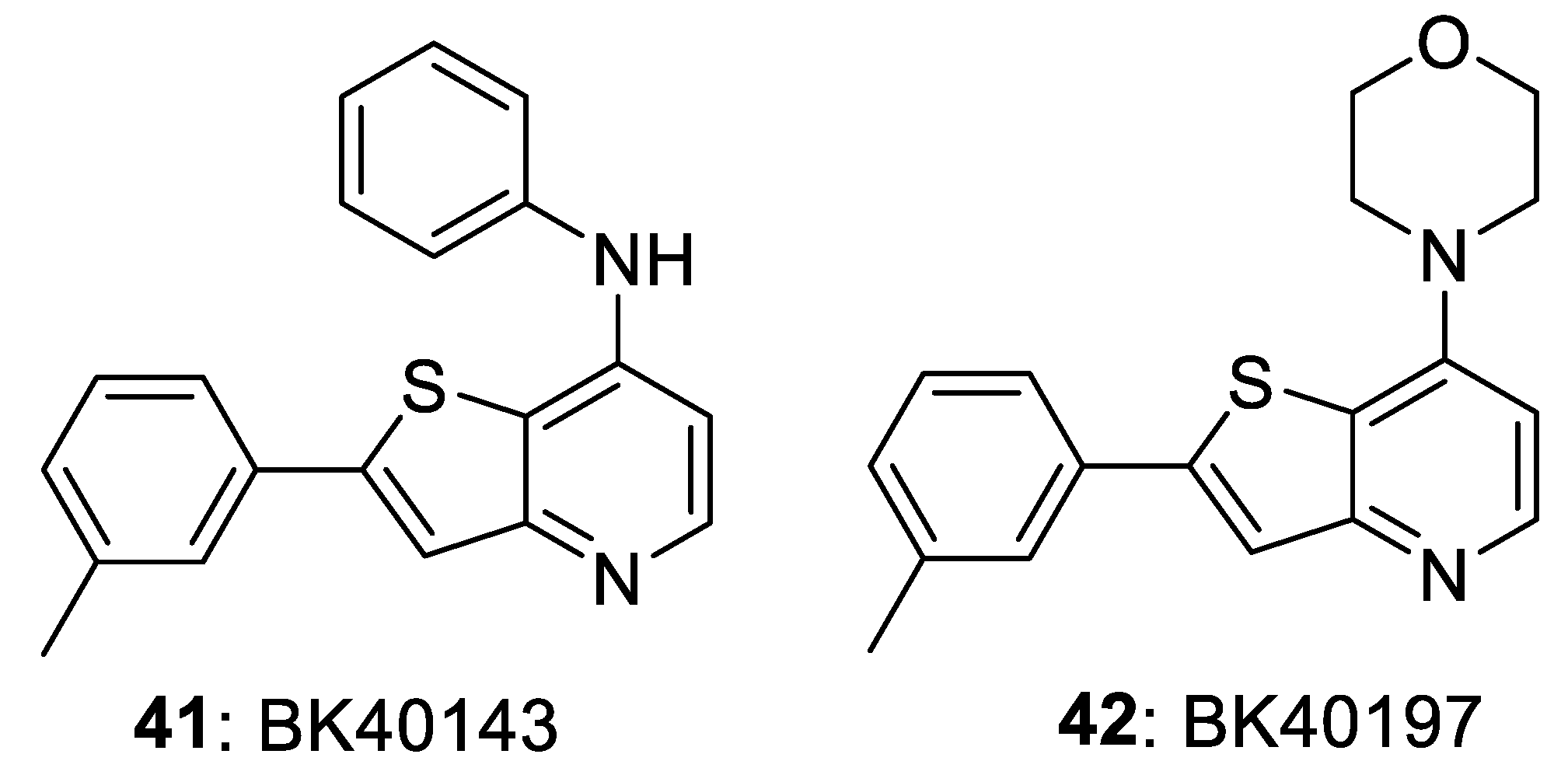
5. Allosteric Inhibitors
6. Other Types of DDR Inhibitors in Patent Claims
7. DDR1 Inhibitors in Cancer Treatment
8. DDR Inhibitors for Inflammatory Conditions
9. DDR Inhibitors in Brain Disease
10. DDR Inhibitors in Renal Disease
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carafoli, F.; Bihan, D.; Stathopoulos, S.; Konitsiotis, A.D.; Kvansakul, M.; Farndale, R.W.; Leitinger, B.; Hohenester, E. Crystallographic insight into collagen recognition by discoidin domain receptor 2. Structure 2009, 17, 1573–1581. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhou, J.; Li, J. Discoidin domain receptors orchestrate cancer progression: A focus on cancer therapies. Cancer Sci. 2021, 112, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, F.; Mayer, M.C.; Shiraishi, K.; Pecheva, M.A.; Chan, L.Y.; Nan, R.; Leitinger, B.; Hofmeister, E.E. Structure of the Discoidin Domain Receptor 1 extracellular region bound to an inhibitory Fab fragment reveals features important for signaling. Structure 2012, 8, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, D.S.; Juskaite, V.; Xu, Y.; Görlitz, F.; Alexandrov, Y.; Dunsby, C.; Paul, M.W.; French, P.W.M.; Leitinger, B. DDR1 autophosphorylation is a result of aggregation into dense clusters. Sci. Rep. 2019, 9, 17104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriet, E.; Sala, M.; Abou Hammoud, A.A.; Tuariihionoa, A.; Di Martino, J.; Ros, M.; Saltel, F. Multitasking discoidin domain receptors are involved in several and specific hallmarks of cancer. Cell Adhes. Migr. 2018, 12, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Dorison, A.; Dussaule, J.-C.; Chatziantoniou, C. The role of discoidin domain receptor 1 in inflammation, fibrosis and renal disease. Nephron 2017, 137, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Ferri, N.; Carragher, N.O.; Raines, E.W. Role of discoidin domain receptors 1 and 2 in human smooth muscle cell-mediated collagen remodeling—Potential implications in atherosclerosis and lymphangioleiomyomatosis. Am. J. Pathol. 2004, 164, 1575–1585. [Google Scholar] [CrossRef]
- Prakoura, N.; Hadchouel, J.; Chatziantoniou, C. Novel targets for therapy of renal fibrosis. J. Histochem. Cytochem. 2019, 67, 701–715. [Google Scholar] [CrossRef]
- Alfieri, C.; Kavvadas, P.; Simonini, P.; Ikehata, M.; Dussaule, J.C.; Chadjichristos, C.E.; Rastaldi, M.P.; Messa, P.; Chatziantoniou, C. Dicoidin domain receptor-1 and periostin: New players in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 1965–1971. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Zhang, Z.; Ding, K. A patent review of discoidin domain receptor 1 (DDR1) modulators (2014-present). Exp. Opin. Ther. Pat. 2020, 30, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Day, E.; Waters, B.; Spiegel, K.; Alnadaf, T.; Manley, P.W.; Buchdunger, E.; Walker, C.; Jarai, G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. [Google Scholar] [CrossRef]
- Fowler, A.J.; Hebron, M.; Missner, A.A.; Wang, R.; Gao, X.; Kurd-Misto, B.T.; Liu, X.; Moussa, E.-H. Multikinase Abl/DDR/Src inhibition produces optimal effects for tyrosine kinase inhibition in neurodegeneration. Drugs R&D 2019, 19, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Jeffries, D.E.; Borza, C.M.; Blobaum, A.L.; Pozzi, A.; Lindsley, C.W. Discovery of VU6015929: A selective discoidin domain receptor 1/2 (DDR1/2) inhibitor to explore the role of DDR1 in antifibrotic therapy. ACS Med. Chem. Lett. 2020, 11, 29–33. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Pinkas, D.M.; Fox, A.E.; Luo, J.; Huang, H.; Cui, S.; Xiang, Q.; Xu, T.; Xun, Q.; et al. Design, synthesis, and biological evaluation of 3-(imidazo[1,2-a]pyrazin-3-ylethynyl)-4-isopropyl-N-(3-((4-methylpiperazin- 1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide as a dual inhibitor of discoidin domain receptors 1 and 2. J. Med. Chem. 2018, 61, 7977–7990. [Google Scholar] [CrossRef]
- Kim, H.G.; Tan, L.; Weisberg, E.L.; Liu, F.; Canning, P.; Choi, H.G.; Ezell, S.A.; Wu, H.; Zhao, Z.; Wang, J.; et al. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem. Biol. 2013, 8, 2145–2150. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, Y.; Bartual, S.G.; Luo, J.; Xu, T.; Du, W.; Xun, Q.; Tu, Z.; Brekken, R.A.; Ren, X.; et al. Tetrahydroisoquinoline-7-carboxamide derivatives as new selective discoidin domain receptor 1 (DDR1) inhibitors. ACS Med. Chem. Lett. 2017, 8, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Sammon, D.; Hohenester, E.; Leitinger, B. Two-step release of kinase autoinhibition in discoidin domain receptor 1. Proc. Natl. Acad. Sci. USA 2020, 117, 22051–22060. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.J.; Andrews, R.C. Targeting protein multiple conformations: A structure-based strategy for kinase drug design. Curr. Top. Med. Chem. 2007, 7, 1394–1407. [Google Scholar] [CrossRef]
- Van Linden, O.P.; Kooistra, A.J.; Leurs, R.; de Esch, I.J.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef]
- Murray, C.W.; Berdini, V.; Buck, I.M.; Carr, M.E.; Cleasby, A.; Coyle, J.E.; Curry, J.E.; Day, J.E.H.; Day, P.J.; Hearn, K.; et al. Fragment-based discovery of potent and selective DDR1/2 inhibitors. ACS Med. Chem. Lett. 2015, 6, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Bian, H.; Bartual, S.G.; Du, W.; Luo, J.; Hu, Z.; Shasha, Z.; Cheng, Z.; Yang, M.Z.; Yong, X.; et al. Structure-based design of tetrahydroisoquinoline-7-carboxamides as selective discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2016, 59, 5911–5916. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Duan, L.; Luo, J.; Zhang, L.; Lu, X.; Zhang, Y.; Zhang, Z.; Tu, Z.; Xu, Y.; Ren, X.; et al. Discovery and optimization of 3(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2013, 56, 3281–3295. [Google Scholar] [CrossRef]
- Zhu, D.; Huang, H.; Pinkas, D.M.; Luo, J.; Ganguly, D.; Fox, A.E.; Arner, E.; Xiang, Q.; Tu, Z.C.; Bullock, A.N.; et al. 2-Amino-2,3-dihydro-1H-indene-5-carboxamide-based Discoidin Domain receptor 1 (DDR1) inhibitors: Design, synthesis, and in vivo antipancreatic cancer efficacy. J. Med. Chem. 2019, 62, 7431–7444. [Google Scholar] [CrossRef] [PubMed]
- Richters, H.; Satz, A.L.; Bedoucha, M.; Buettelmann, B.; Petersen, A.C.; Harmeier, A.; Hermosilla, R.; Hochstrasser, R.; Burger, D.; Gsell, B. DNA-encoded library-derived DDR1 inhibitor prevents fibrosis and renal function loss in a genetic mouse model of Alport syndrome. ACS Chem. Biol. 2019, 14, 37–49. [Google Scholar] [CrossRef]
- Mo, C.; Zhang, Z.; Li, Y.; Huang, M.; Zou, J.; Luo, J.; Tu, Z.-C.; Xu, Y.; Ren, X.; Ding, K.; et al. Design and optimization of 3’-(imidazo[1,2-a]pyrazin-3-yl)-[1,1’-biphenyl]-3-carboxamides as selective DDR1 inhibitors. ACS Med. Chem. Lett. 2020, 11, 379–384. [Google Scholar] [CrossRef]
- Reger de Moura, C.; Battistella, M.; Sohail, A.; Caudron, A.; Feugeas, J.P.; Podgorniak, M.-P.; Pages, C.; Dorval, M.; Oren, M.; Menashi, S.; et al. Discoidin domain receptors: A promising target in melanoma et al. Pigm. Cell Melan. Res. 2019, 32, 697–707. [Google Scholar] [CrossRef]
- Liu, L.; Hussain, M.; Luo, J.; Duan, A.; Chen, C.; Tu, Z.; Zhang, J. Synthesis and biological evaluation of novel dasatinib analogues as potent DDR1 and DDR2 kinase inhibitors. Chem. Biol. Drug Des. 2017, 89, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Nishiota, Y.; Kurono, M.; Nishizawa, R.; Bandodkar, B.; Gao, X.; Wan, Z.; Lv, R.; Doyle, K.; Goldsmith, M. Preparation of Hydantoin Derivatives As Selective DDR1 Inhibitors. WO 2021043245, 11 March 2020. [Google Scholar]
- Richters, A.; Nguyen, H.D.; Phan, T.; Simard, J.R.; Grutter, C.; Engel, J.; Rauh, D. Identification of type II and III DDR2 inhibitors. J. Med. Chem. 2014, 57, 4252–4262. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Z.; Lin, L.N.; Sun, H.; White, L.V.; Ding, K.; Li, Z. Targeting discoidin domain receptor 1 (DDR1) signaling and its crosstalk with β1-integrin emerges as a key factor for breast cancer chemosensitization upon collagen type 1 binding 16. Quantitative proteomics reveals cellular off-targets of a DDR1 inhibitor. ACS Med. Chem. Lett. 2020, 11, 535–540. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Park, J.-E.; Cho, N.-C.; Sim, T.; Pae, A.N.; Roh, E.J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Grither, W.R.; Longmore, G.D. Inhibition of tumor-microenvironment interaction and tumor invasion by small-molecule allosteric inhibitor of DDR2 extracellular domain. Proc. Natl. Acad. Sci. USA 2018, 115, E7786–E7794. [Google Scholar] [CrossRef] [Green Version]
- Bae, I.H.; Jung, S.H.; Ahn, Y.G.; Suh, K.H. Preparation of thieno[3,2-d]pyrimidine Compound with Inhibitory Activity for Protein Kinases Useful in Treatment and Prevention of Diseases. WO 2019107987, 6 June 2019. [Google Scholar]
- Nishio, Y.; Kubota, Y.; Yamamoto, M.; Nishimura, Y.; Masuda, T.; Tsutsui, H.; Okimura, K.; Udagawa, S.; Kaino, M.; Meguro, H.; et al. Preparation of Urea Derivatives for Inhibiting Discoidin Domain Receptor 1. WO 2017038873 A1, 9 March 2017. [Google Scholar]
- Nishio, Y.; Yamamoto, M.; Kubota, Y.; Tsutsui, H.; Masuda, T.; Okimura, K.; Udagawa, S.; Kaino, M.; Meguro, H.; Sekiya, Y. Preparation of Urea Derivatives for Inhibiting Discoidin Domain Receptor 1. WO 2017038871, 9 March 2017. [Google Scholar]
- Tsutsui, H.; Okimura, K.; Udagawa, S.; Kaino, M.; Meguro, H.; Sekiya, Y. Preparation of N-(2,3-dihydro-1H-inden-1-yl)-N’-[3-(pentafluorosulfanyl)phenyl]urea Derivatives as Inhibitors of Discoidin Domain Receptor Kinase 1 (DDR1). WO 2017038870, 3 March 2017. [Google Scholar]
- Dong, R.; Zhou, X.; Wang, M.; Li, W.; Zhang, J.-Y.; Zheng, X.; Tang, K.-X.; Sun, L.-P. Discovery of 4-amino-1H-pyrazolo[3,4-d]pyrimidin derivatives as novel discoidin domain receptor 1 (DDR1) inhibitors. Bioorg. Med. Chem. 2021, 29, 115876. [Google Scholar] [CrossRef]
- Vehlow, A.; Klapproth, E.; Jin, S.; Hannen, R.; Hauswald, M.; Bartsch, J.-W.; Nimsky, C.; Temme, A.; Leitinger, B.; Cordes, N. Interaction of discoidin domain receptor 1 with a 14-3-3-beclin-1-Akt1 complex modulates glioblastoma therapy sensitivity. Cell Rept. 2019, 26, 3672–3683. [Google Scholar] [CrossRef] [Green Version]
- Romayor, I.; Badiola, I.; Benedicto, A.; Marquez, J.; Herrero, A.; Arteta, B.; Olaso, E. Silencing of sinusoidal DDR1 reduces murine liver metastasis by colon carcinoma. Sci. Rep. 2020, 10, 18398. [Google Scholar] [CrossRef] [PubMed]
- Baltes, F.; Caspers, J.; Henze, S.; Schlesinger, M.; Bendas, G. Targeting discoidin domain receptor 1 (DDR1) signaling and its crosstalk with β1-integrin emerges as a key factor for breast cancer chemosensitization upon collagen type 1 binding. Int. J. Mol. Sci. 2020, 21, 4956. [Google Scholar] [CrossRef] [PubMed]
- Hur, H.; Ham, I.-H.; Lee, D.; Jin, H.; Aguilera, K.Y.; Oh, H.J.; Han, S.-U.; Kwon, J.E.; Kim, Y.-B.; Ding, K.; et al. Discoidin domain receptor 1 activity drives an aggressive phenotype in gastric carcinoma. BMC Cancer 2017, 17, 87/1–87/11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, K.Y.; Huang, H.; Du, W.; Hagopian, M.; Wang, Z.; Hinz, S.; Hwang, T.H.; Wang, H.; Fleming, J.B.; Castrillon, D.H.; et al. Inhibition of discoidin domain receptor 1 reduces collagen-mediated tumorigenicity in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2017, 16, 2473–2485. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.-P.; Guan, Z.; Xu, Y.-D.; Zhang, J.-L.; Cai, Q.; Peng, J.-R.; Feng, G.-K. Effects of DDR1 inhibitor and its possible mechanism. Zhongshan Daxue Xuebao Yixue Kexueban 2015, 36, 827–832. [Google Scholar]
- Borza, C.M.; Su, Y.; Tran, T.L.; Yu, L.; Steyns, N.; Temple, K.J.; Skwark, M.J.; Meiler, J.; Lindsley, C.W.; Hicks, B.R.; et al. Discoidin domain receptor 1 kinase activity is required for regulating collagen IV synthesis. Matrix Biol. 2017, 57–58, 258–271. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Ham, I.-H.; Oh, H.J.; Bae, C.A.; Lee, D.; Kim, Y.-B.; Son, S.-Y.; Chwae, Y.-J.; Han, S.-U.; Brekken, R.A.; et al. Inhibition of discoidin domain receptor 1 prevents stroma-induced peritoneal metastasis in gastric carcinoma. Mol. Cancer Res. 2018, 16, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Rovati, L.C.; Pilkington-Miksa, M.; Bovino, C.; Mandelli, S.; Zanelli, T. Preparation of Phenazines as Inhibitors of Discoidin Domain Receptors 2 (DDR2). WO 2020207611A1, 15 October 2020. [Google Scholar]
- Vehlow, A.; Cordes, N. DDR1 (discoidin domain receptor tyrosine kinase 1) drives glioblastoma therapy resistance by modulating autophagy. Autophagy 2019, 15, 1487–1488. [Google Scholar] [CrossRef]
- Yuge, R.; Kitadai, Y.; Takigawa, H.; Naito, T.; Oue, N.; Wataru, Y.; Tanaka, S.; Chayama, K. Silencing of discoidin domain receptor-1 (DDR1) concurrently inhibits multiple steps of metastasis cascade in gastric cancer. Trans. Oncol. 2018, 11, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Zhang, W.; Sun, T. DDR1 promotes breast tumor growth by suppressing antitumor immunity. Oncol. Repts. 2019, 42, 2844–2854. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, A.; Asawa, Y.; Kawasaki, E.; Tasaka, T.; Matsuda, S.; Sekikawa, T.; Tanabe, S.; Neya, M.; Natsugari, H.; Kanai, C. Design and synthesis of DDR1 inhibitors with a desired pharmacophore using deep generative models. ChemMedChem 2021, 16, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Atsushi, Y.; Yoshimori, A.; Kawasaki, E.; Kanai, C.; Tasaka, T. Strategies for design of molecular structures with a desired pharmacophore using deep reinforcement learning. Chem. Pharm. Bull. 2020, 68, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Lino, M.; Ngai, D.; Liu, A.; Mohabeer, A.; Harper, C.; Caruso, L.-L.; Schroer, S.A.; Fu, F.; McKee, T.; Giacca, A.; et al. Discoidin domain receptor 1-deletion ameliorates fibrosis and promotes adipose tissue beiging, brown fat activity, and increased metabolic rate in a mouse model of cardiometabolic disease. Mol. Metab. 2020, 39, 101006. [Google Scholar] [CrossRef]
- Ushkamary, M.G.; Wang, M.; Harikrishnan, V.; Titus, A.S.; Zhang, J.; Liu, L.; Monticone, R.; Wang, Y.; Mattison, J.A.; de Cabo, R.; et al. Discoidin domain Receptor 2: A determinant of metabolic syndrome-associated arterial fibrosis in non-human primates. PLoS ONE 2019, 14, e0225911. [Google Scholar] [CrossRef] [Green Version]
- Mattison, J.A.; Wang, M.; Bernier, M.; Zhang, J.; Park, S.-S.; Maudsley, S. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. 2014, 20, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Phan, T.N.; Jung, S.H.; Kim, S.Y.; Cho, J.U.; Lee, H.; Woo, S.H.; Park, T.K.; Yang, B.-S. LCB 03-0110, a novel pan-discoidin domain receptor/c-Src family tyrosine kinase inhibitor, suppresses scar formation by inhibiting fibroblast and macrophage activation. J. Pharmacol. Exp. Ther. 2012, 340, 510–519. [Google Scholar] [CrossRef]
- Zhao, H.; Bian, H.; Bu, X.; Zhang, S.; Zhang, P.; Yu, J.; Lai, X.; Li, D.; Zhu, C.; Yao, L.; et al. Targeting of discoidin domain receptor 2 (DDR2) prevents myofibroblast activation and neovessel formation during pulmonary fibrosis. Mol. Ther. 2016, 24, 1734–1744. [Google Scholar] [CrossRef] [Green Version]
- Terai, H.; Tan, L.; Beauchamp, E.M.; Hatcher, J.M.; Liu, Q.; Meyerson, M.; Gray, N.S.; Hammerman, P.S. Characterization of DDR2 inhibitors for the treatment of DDR2 mutated non-small-cell lung cancer. ACS Chem. Biol. 2015, 10, 2687–2696. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Deng, J.; Yu, X.; Wu, F.; Men, K.; Yang, Q.; Zhu, Y.; Liu, X.; Jiang, Q. Identification of novel inhibitors of DDR1 against idiopathic pulmonary fibrosis by integrative transcriptome meta-analysis, computational and experimental screening. Mol. BioSys. 2016, 12, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of PAINS (Pan Assay Interference Compounds) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Chou, H.-C.; Chen, C.-H.; Chou, L.-Y.; Cheng, T.-L.; Kang, L.; Chuang, S.-C.; Lin, Y.-S.; Ho, M.-L.; Wang, Y.-H.; Lin, S.-Y.; et al. Discoidin domain receptors 1 inhibition alleviates osteoarthritis via enhancing autophagy. Int. J. Mol. Sci. 2020, 21, 6991. [Google Scholar] [CrossRef] [PubMed]
- Wucherer-Plietker, M.; Werkmann, D.; Gigout, A.; Kuhn, D.; Sawatzky, E. DDR2 Inhibitors for the Treatment of Osteoarthritis. WO 2014032755A2, 17 July 2014. [Google Scholar]
- Murata, T.; Niizuma, S.; Hara, S.; Kawada, H.; Hada, K.; Shimada, H.; Tanaka, H.; Nakanishi, Y. Preparation of Benzamide Derivatives as Discoidin Domain Receptor 1 (DDR1) Inhibitors. WO 2013161851A1, 21 October 2013. [Google Scholar]
- Murata, T.; Kawada, H.; Niizuma, S.; Hara, S.; Hada, K.; Shimada, H.; Tanaka, H.; Mio, T. Preparation of Quinazolinedione Derivatives as Discoidin Domain Receptor 1 (DDR1) Inhibitors. WO 2013161853 A1, 31 October 2013. [Google Scholar]
- Fowler, A.J.; Hebron, M.; Balaraman, K.; Wangke Shi, W.; Missner, A.A.; Greenzaid, J.D.; Chiu, T.L.; Ullman, C.; Weatherdon, E.; Duka, V.; et al. Discoidin Domain Receptor 1 is a therapeutic target for neurodegenerative diseases. Hum. Mol. Gen. 2020, 29, 2882–2898. [Google Scholar] [CrossRef]
- Flamant, M.; Placier, S.; Rodenas, A.; Curat, C.A.; Vogel, W.F.; Chatziantoniou, C.; Dussaule, J.C. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J. Am. Soc. Nephrol. 2006, 17, 3374–3381. [Google Scholar] [CrossRef]
- Kerroch, M.; Guerrot, D.; Vandermeersch, S.; Placier, S.; Mesnard, L.; Jouanneau, C.; Rondeau, E.; Ronco, P.; Boffa, J.-J.; Chatziantoniou, C.; et al. Genetic inhibition of discoidin domain receptor 1 protects mice against crescentic glomerulonephritis. FASEB J. 2012, 26, 4079–4091. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denny, W.A.; Flanagan, J.U. Inhibitors of Discoidin Domain Receptor (DDR) Kinases for Cancer and Inflammation. Biomolecules 2021, 11, 1671. https://doi.org/10.3390/biom11111671
Denny WA, Flanagan JU. Inhibitors of Discoidin Domain Receptor (DDR) Kinases for Cancer and Inflammation. Biomolecules. 2021; 11(11):1671. https://doi.org/10.3390/biom11111671
Chicago/Turabian StyleDenny, William A., and Jack U. Flanagan. 2021. "Inhibitors of Discoidin Domain Receptor (DDR) Kinases for Cancer and Inflammation" Biomolecules 11, no. 11: 1671. https://doi.org/10.3390/biom11111671
APA StyleDenny, W. A., & Flanagan, J. U. (2021). Inhibitors of Discoidin Domain Receptor (DDR) Kinases for Cancer and Inflammation. Biomolecules, 11(11), 1671. https://doi.org/10.3390/biom11111671