
DispHScan: A Multi-Sequence Web Tool for Predicting Protein Disorder as a Function of pH

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Methods
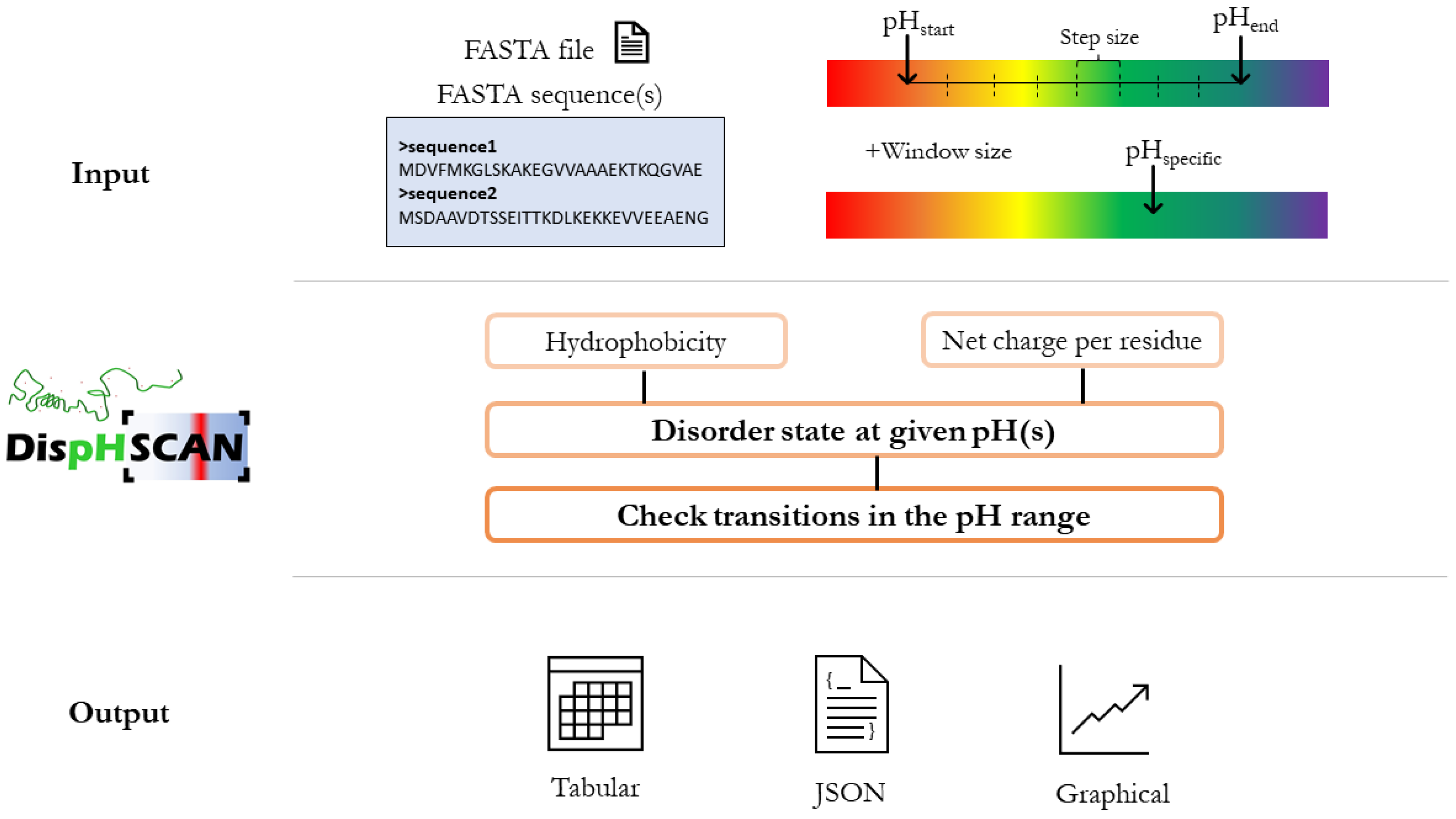
2.1. DispHScan Pipeline
2.2. Server Implementation
2.3. Gene Ontology Annotation
3. Results
3.1. Performance
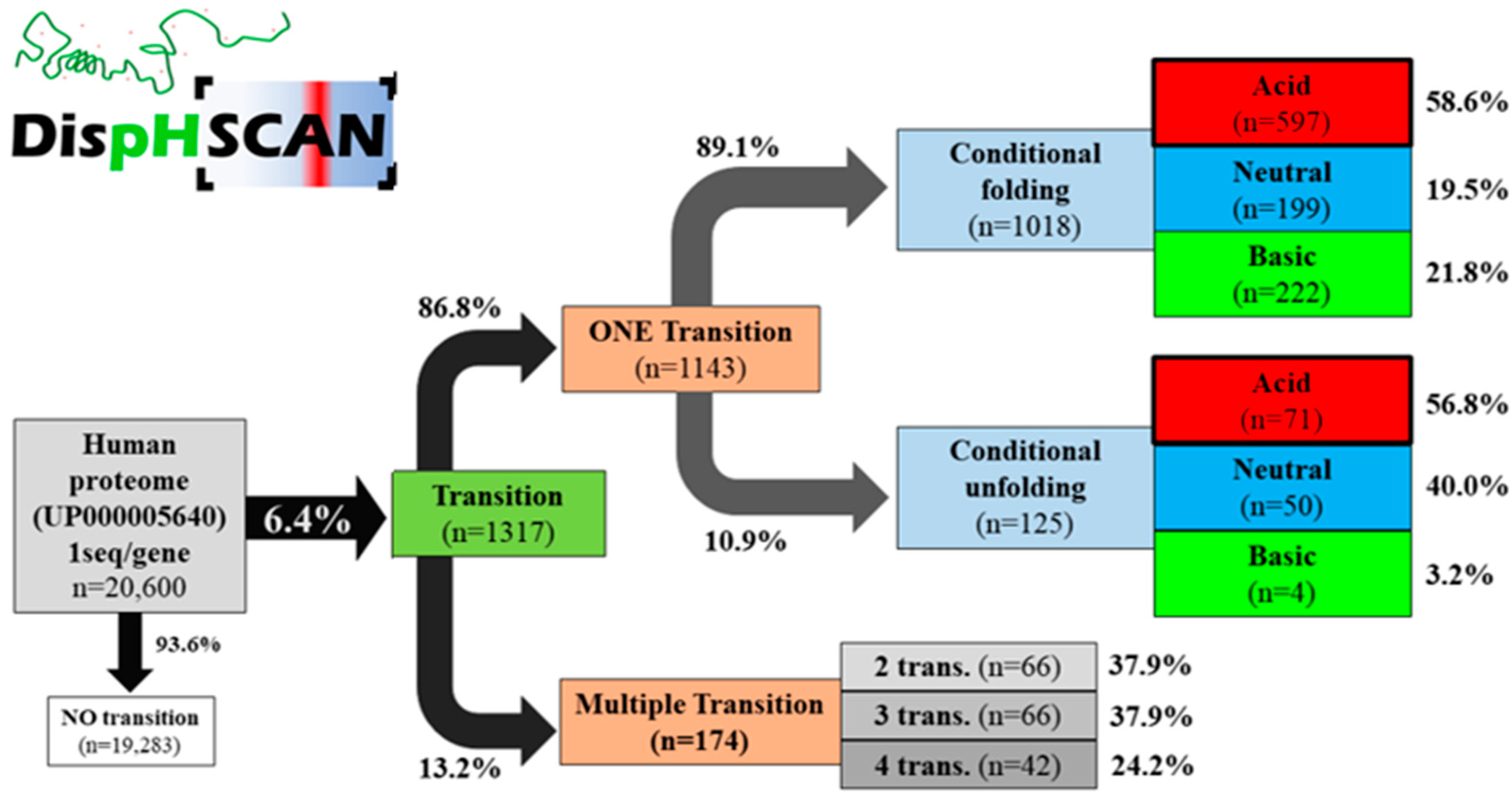
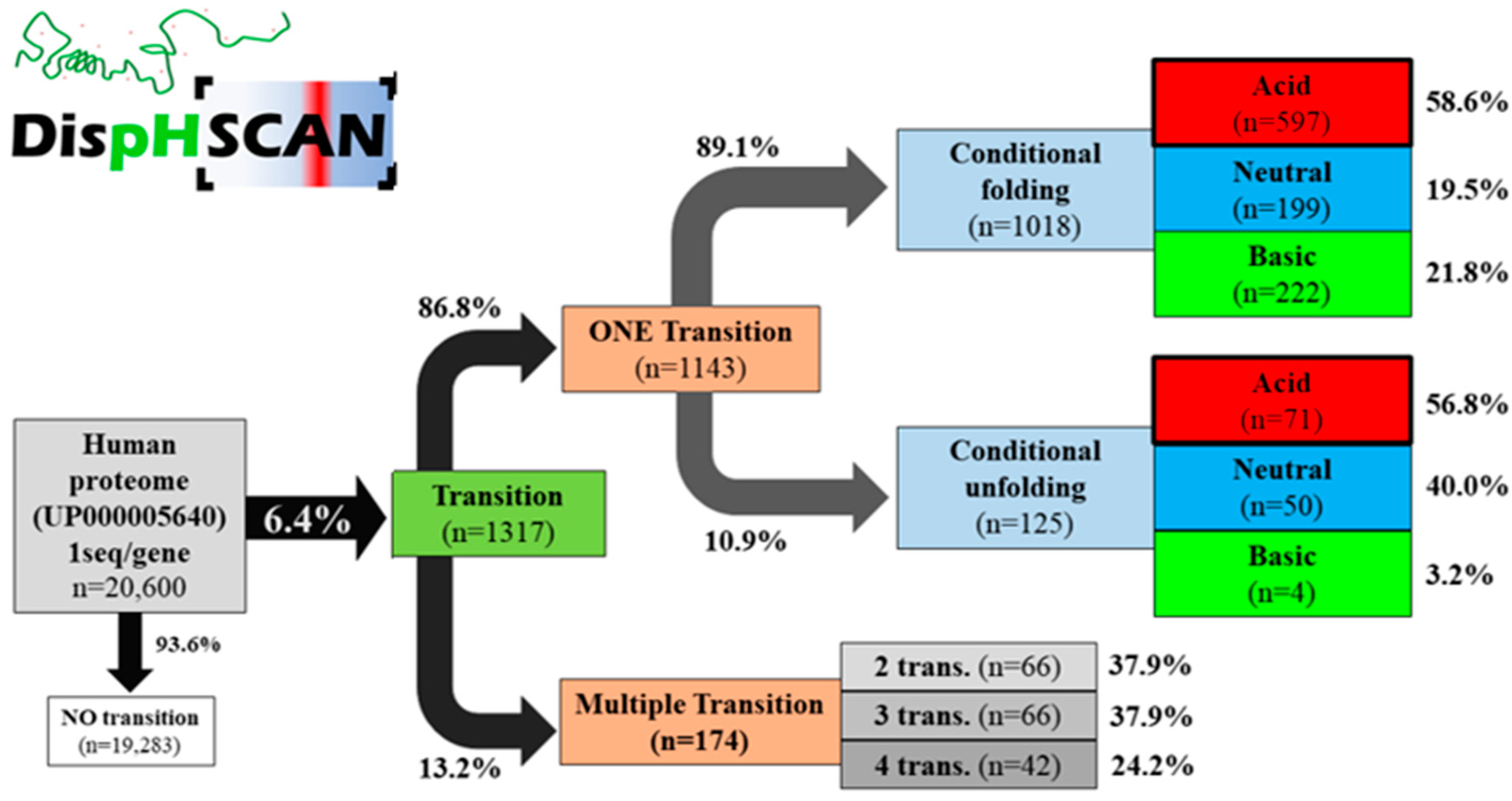
3.2. Analysis of Model Organisms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Minde, D.P.; Halff, E.F.; Tans, S. Designing disorder: Tales of the unexpected tails. Intrinsically Disord. Proteins 2013, 1, e26790. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Predictors, C.; DisProt, C.; Tosatto, S.C.E. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and their environment: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009, 28, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Jakob, U.; Kriwacki, R.; Uversky, V.N. Conditionally and transiently disordered proteins: Awakening cryptic disorder to regulate protein function. Chem. Rev. 2014, 114, 6779–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.; Iglesias, V.; Pintado, C.; Santos-Suarez, J.; Ventura, S. DispHred: A Server to Predict pH-Dependent Order-Disorder Transitions in Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2020, 21, 5814. [Google Scholar] [CrossRef] [PubMed]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- Zamora, W.J.; Campanera, J.M.; Luque, F.J. Development of a Structure-Based, pH-Dependent Lipophilicity Scale of Amino Acids from Continuum Solvation Calculations. J. Phys. Chem. Lett. 2019, 10, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Zambrano, R.; Conchillo-Sole, O.; Iglesias, V.; Illa, R.; Rousseau, F.; Schymkowitz, J.; Sabate, R.; Daura, X.; Ventura, S. PrionW: A server to identify proteins containing glutamine/asparagine rich prion-like domains and their amyloid cores. Nucleic Acids Res. 2015, 43, W331–W337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognesi, B.; Lorenzo Gotor, N.; Dhar, R.; Cirillo, D.; Baldrighi, M.; Tartaglia, G.G.; Lehner, B. A Concentration-Dependent Liquid Phase Separation Can Cause Toxicity upon Increased Protein Expression. Cell Rep. 2016, 16, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toombs, J.A.; Petri, M.; Paul, K.R.; Kan, G.Y.; Ben-Hur, A.; Ross, E.D. De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. USA 2012, 109, 6519–6524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lindt, J.; Bratek-Skicki, A.; Nguyen, P.N.; Pakravan, D.; Duran-Armenta, L.F.; Tantos, A.; Pancsa, R.; Van Den Bosch, L.; Maes, D.; Tompa, P. A generic approach to study the kinetics of liquid-liquid phase separation under near-native conditions. Commun. Biol. 2021, 4, 77. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Organism | n | No Transition (n) | Transition (n) | No Transition (%) | Transition (%) |
|---|---|---|---|---|---|
| H. sapiens | 20,600 | 19,283 | 1317 | 93.6 | 6.4 |
| E. coli | 5062 | 4932 | 130 | 97.5 | 2.6 |
| S. cerevisiae | 6050 | 5661 | 389 | 93.6 | 6.4 |
| C. elegans | 19,813 | 18,735 | 1078 | 94.6 | 5.4 |
| Organism | Single Transition (%) | Multitransition (%) | Conditional Folding (%) | Conditional Unfolding (%) |
|---|---|---|---|---|
| H. sapiens | 86.8 | 13.2 | 89.1 | 10.9 |
| E. coli | 94.6 | 5.4 | 88.6 | 11.4 |
| S. cerevisiae | 85.6 | 14.4 | 82.9 | 17.1 |
| C. elegans | 80.1 | 19.9 | 86.2 | 13.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pintado-Grima, C.; Iglesias, V.; Santos, J.; Uversky, V.N.; Ventura, S. DispHScan: A Multi-Sequence Web Tool for Predicting Protein Disorder as a Function of pH. Biomolecules 2021, 11, 1596. https://doi.org/10.3390/biom11111596
Pintado-Grima C, Iglesias V, Santos J, Uversky VN, Ventura S. DispHScan: A Multi-Sequence Web Tool for Predicting Protein Disorder as a Function of pH. Biomolecules. 2021; 11(11):1596. https://doi.org/10.3390/biom11111596
Chicago/Turabian StylePintado-Grima, Carlos, Valentín Iglesias, Jaime Santos, Vladimir N. Uversky, and Salvador Ventura. 2021. "DispHScan: A Multi-Sequence Web Tool for Predicting Protein Disorder as a Function of pH" Biomolecules 11, no. 11: 1596. https://doi.org/10.3390/biom11111596
APA StylePintado-Grima, C., Iglesias, V., Santos, J., Uversky, V. N., & Ventura, S. (2021). DispHScan: A Multi-Sequence Web Tool for Predicting Protein Disorder as a Function of pH. Biomolecules, 11(11), 1596. https://doi.org/10.3390/biom11111596