
Pharmacological Inhibition of the VCP/Proteasome Axis Rescues Photoreceptor Degeneration in RHOP23H Rat Retinal Explants

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
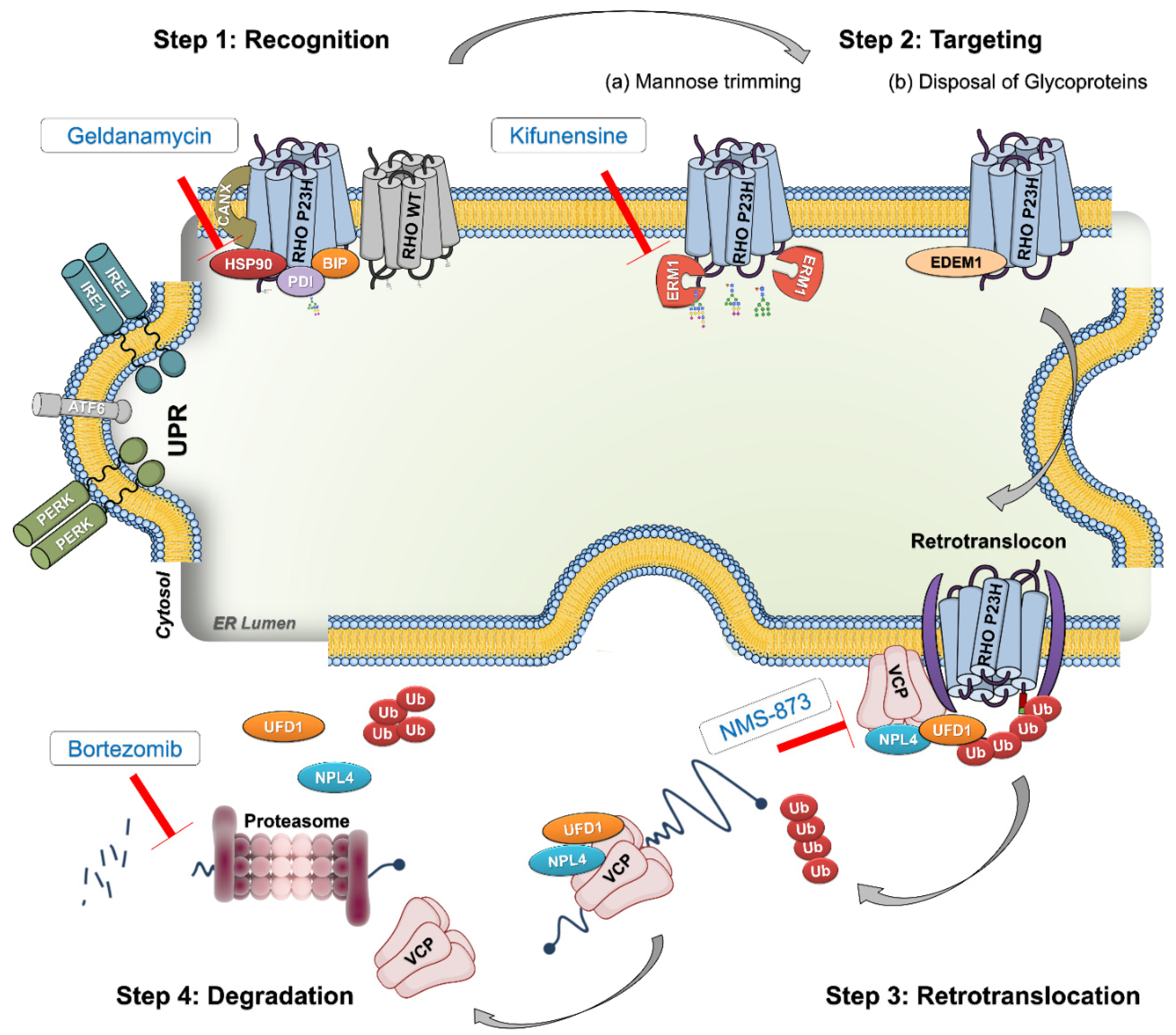
:1. Introduction
2. Results
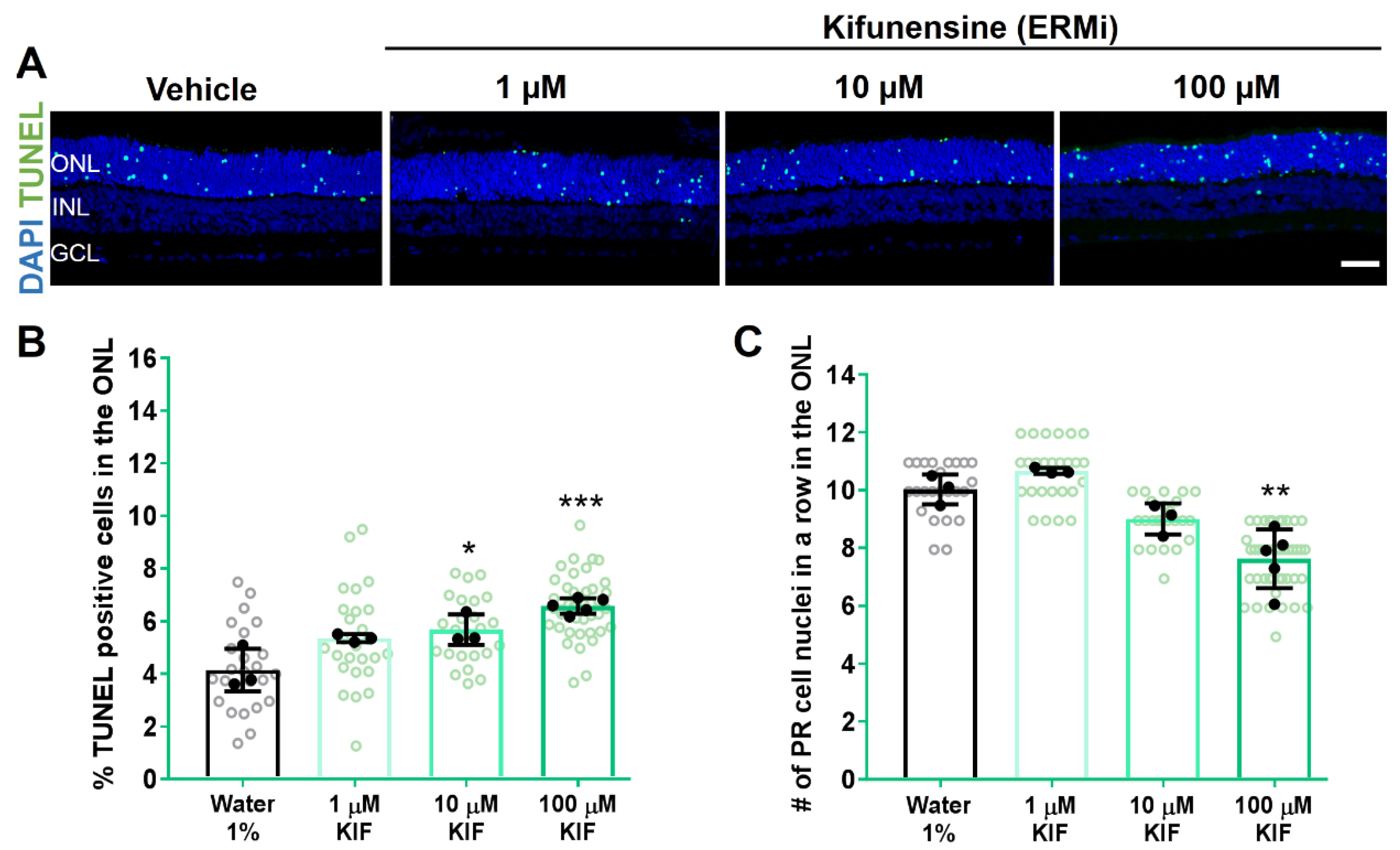
2.1. Hsp90 or ERM1 Inhibition Is Detrimental for RHOP23H Rat Retinae Ex Vivo
2.2. VCP Inhibition Enhances Photoreceptor Cell Survival in RHOP23H Retinal Cultures
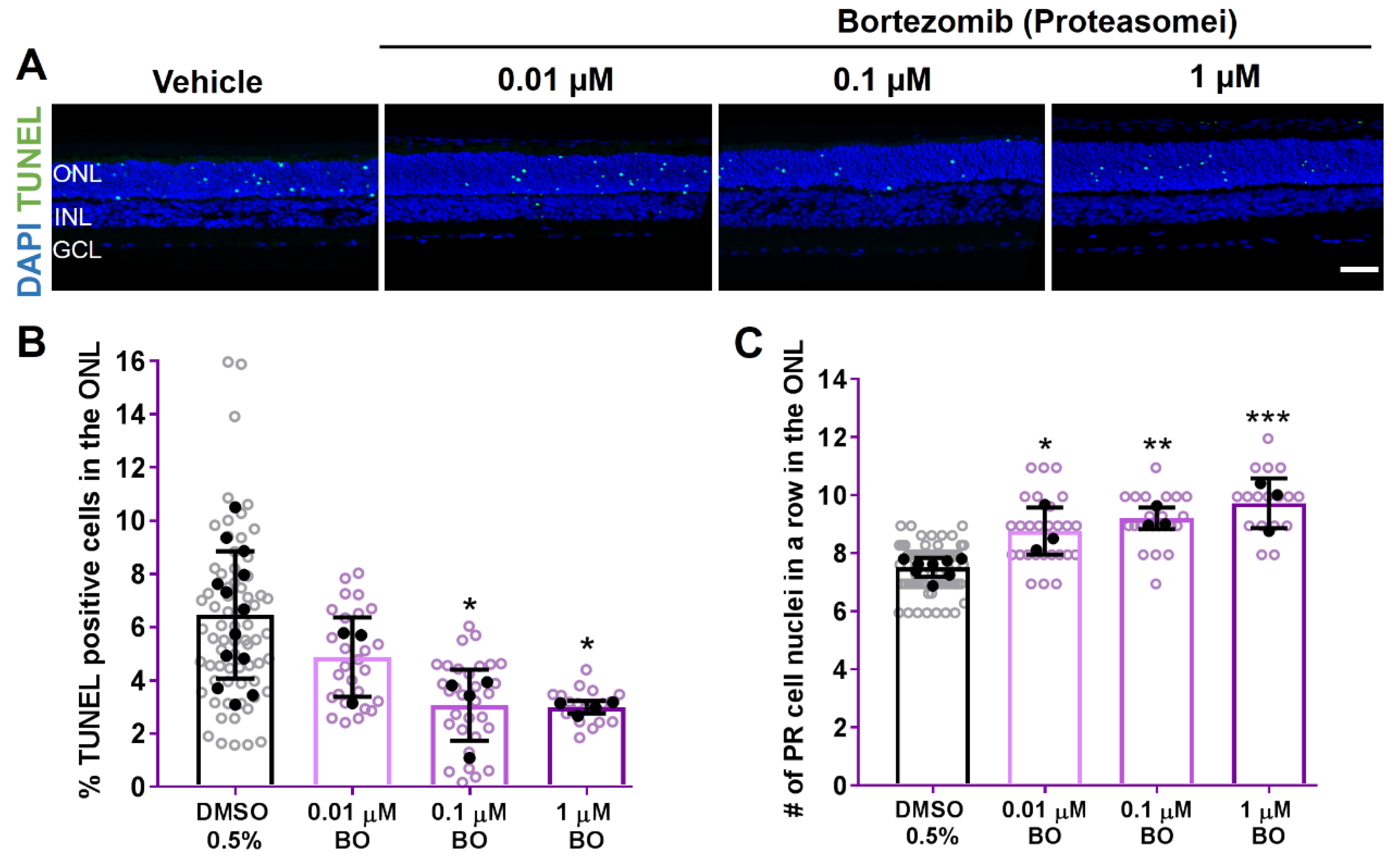
2.3. 26S Proteasome Inhibition Reduces Photoreceptor Degeneration in RHOP23H Rat Explants
2.4. Only VCP Inhibition Enhances RHO Trafficking to the Outer Segment in RHOP23H Rat Retinal Explants
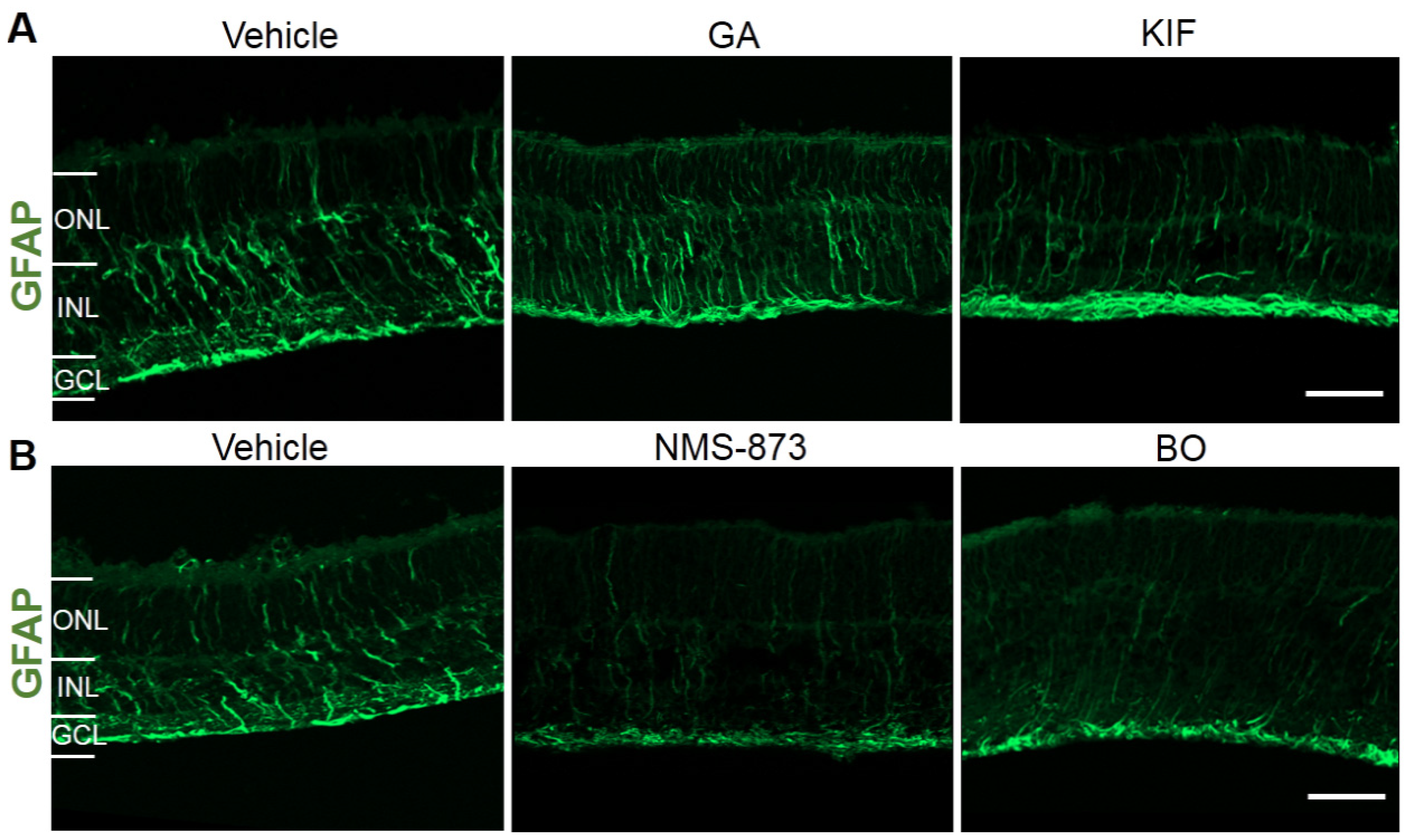
2.5. Effects of Different ERAD Modulators on Retinal Stress and Inflammation in RHOP23H Retinal Organotypic Cultures
3. Discussion
4. Material and Methods
4.1. Study Approval and Animals
4.2. Organotypic Retinal Explant Cultures of P23H Heterozygous Transgenic Rats
4.3. Histology
4.4. TUNEL Assay
4.5. Immunofluorescence Staining
4.6. Hematoxylin and Eosin Staining
4.7. Western Blotting of Retinal Cultures
4.8. Microscopy and Image Analysis
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Lualdi, M.; Alberio, T.; Fasano, M. Proteostasis and proteotoxicity in the network medicine era. Int. J. Mol. Sci. 2020, 21, 6405. [Google Scholar] [CrossRef]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 2007, 125, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Farrar, G.J.; Kenna, P.F.; Humphries, P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 2002, 21, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Gragg, M.; Park, P.S.H. Detection of misfolded rhodopsin aggregates in cells by Förster resonance energy transfer. In Methods in Cell Biology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 149, pp. 87–105. ISBN 9780128151075. [Google Scholar]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Kisselev, O.G. Focus on Molecules: Rhodopsin. Exp. Eye Res. 2005, 81, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Aron, L.; Piccoli, G.; Ueffing, M. Clearance of RhodopsinP23H aggregates requires the ERAD effector VCP. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 424–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, R.S.; Munro, P.M.G.; Luthert, P.J.; Cheetham, M.E. The cellular fate of mutant rhodopsin: Quality control, degradation and aggresome formation. J. Cell Sci. 2002, 115, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Illing, M.E.; Rajan, R.S.; Bence, N.F.; Kopito, R.R. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 2002, 277, 34150–34160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; LaVail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griciuc, A.; Aron, L.; Roux, M.J.; Klein, R.; Giangrande, A.; Ueffing, M. Inactivation of VCP/ter94 Suppresses Retinal Pathology Caused by Misfolded Rhodopsin in Drosophila. PLoS Genet. 2010, 6, e1001075. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, H.; Chiang, W.C.; Lin, J.H. Endoplasmic Reticulum-Associated Degradation (ERAD) of misfolded glycoproteins and mutant P23H rhodopsin in photoreceptor cells. Adv. Exp. Med. Biol. 2012, 723, 559–565. [Google Scholar]
- Griciuc, A.; Aron, L.; Ueffing, M. ER stress in retinal degeneration: A target for rational therapy? Trends Mol. Med. 2011, 17, 442–451. [Google Scholar] [CrossRef]
- Arango-Gonzalez, B.; Sen, M.; Guarascio, R.; Ziaka, K.; del Amo, E.M.; Hau, K.; Poultney, H.; Asfahani, R.; Urtti, A.; Chou, T.-F.; et al. Inhibition of VCP preserves retinal structure and function in autosomal dominant retinal degeneration. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sen, M.; Bassetto, M.; Poulhes, F.; Zelphati, O.; Ueffing, M.; Arango-Gonzalez, B. Efficient Ocular Delivery of VCP siRNA via Reverse Magnetofection in RHO P23H Rodent Retina Explants. Pharmaceutics 2021, 13, 225. [Google Scholar] [CrossRef]
- Sen, M.; Al-Amin, M.; Kicková, E.; Sadeghi, A.; Puranen, J.; Urtti, A.; Caliceti, P.; Salmaso, S.; Arango-Gonzalez, B.; Ueffing, M. Retinal neuroprotection by controlled release of a VCP inhibitor from self-assembled nanoparticles. J. Control. Release 2021, 339, 307–320. [Google Scholar] [CrossRef]
- Aguilà, M.; Bevilacqua, D.; Mcculley, C.; Schwarz, N.; Athanasiou, D.; Kanuga, N.; Novoselov, S.S.; Lange, C.A.K.; Ali, R.R.; Bainbridge, J.W.; et al. Hsp90 inhibition protects against inherited retinal degeneration. Hum. Mol. Genet. 2014, 23, 2164–2175. [Google Scholar] [CrossRef] [Green Version]
- Arango-Gonzalez, B.; Szabó, A.; Pinzon-Duarte, G.; Lukáts, Á.; Guenther, E.; Kohler, K. In Vivo and In Vitro Development of S- and M-Cones in Rat Retina. Investig. Opthalmology Vis. Sci. 2010, 51, 5320. [Google Scholar] [CrossRef] [Green Version]
- Arango-Gonzalez, B.; Trifunović, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef]
- Peterson, L.B.; Blagg, B.S. To fold or not to fold: Modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283. [Google Scholar] [CrossRef] [Green Version]
- Hering, K.W.; Karaveg, K.; Moremen, K.W.; Pearson, W.H. A practical synthesis of kifunensine analogues as inhibitors of endoplasmic reticulum α-mannosidase I. J. Org. Chem. 2005, 70, 9892–9904. [Google Scholar] [CrossRef]
- Hosokawa, N.; Tremblay, L.O.; You, Z.; Herscovics, A.; Wada, I.; Nagata, K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded null Hong Kong α1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 2003, 278, 26287–26294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, S.; Rumpf, S. Cdc48 (p97): A “molecular gearbox” in the ubiquitin pathway? Trends Biochem. Sci. 2007, 32, 6–11. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef]
- Wang, F.; Li, S.; Gan, T.; Stott, G.M.; Flint, A.; Chou, T.F. Allosteric p97 Inhibitors Can Overcome Resistance to ATP-Competitive p97 Inhibitors for Potential Anticancer Therapy. ChemMedChem 2020, 15, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Lobanova, E.S.; Finkelstein, S.; Skiba, N.P.; Arshavsky, V.Y. Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9986–9991. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Sakami, S.; Kolesnikov, A.V.; Kefalov, V.J.; Palczewski, K. P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum. Mol. Genet. 2014, 23, 1723–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosche, J.; Grimm, D.; Clemens, N.; Reichenbach, A. Retinal light damage vs. normal aging of rats: Altered morphology, intermediate filament expression, and nuclear organization of Müller (glial) cells. J. Hirnforsch. 1997, 38, 459–470. [Google Scholar]
- Fernández-Sánchez, L.; Lax, P.; Campello, L.; Pinilla, I.; Cuenca, N. Astrocytes and Müller Cell Alterations during Retinal Degeneration in a Transgenic Rat Model of Retinitis Pigmentosa. Front. Cell. Neurosci. 2015, 9, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.; Wagner, F.; Lorenz, B.; Stieger, K. Organotypic cultures of adult mouse retina: Morphologic changes and gene expression. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1930–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, J.; Maruya, M.; Yashiroda, H.; Yahara, I.; Tanaka, K. The molecular chaperone Hsp90 plays a role in the assembly and maintenance of the 26S proteasome. EMBO J. 2003, 22, 3557–3567. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Zhang, X.M.; Wang, X.D.; Wang, B.J.; Wang, W. 17-AAG, a Hsp90 inhibitor, attenuates the hypoxia-induced expression of SDF-1α and ILK in mouse RPE cells. Mol. Biol. Rep. 2010, 37, 1203–1209. [Google Scholar] [CrossRef]
- Wu, W.C.; Wu, M.H.; Chang, Y.C.; Hsieh, M.C.; Wu, H.J.; Cheng, K.C.; Lai, Y.H.; Kao, Y.H. Geldanamycin and its analog induce cytotoxicity in cultured human retinal pigment epithelial cells. Exp. Eye Res. 2010, 91, 211–219. [Google Scholar] [CrossRef]
- Kanamaru, C.; Yamada, Y.; Hayashi, S.; Matsushita, T.; Suda, A.; Nagayasu, M.; Kimura, K.; Chiba, S. Retinal toxicity induced by small-molecule Hsp90 inhibitors in beagle dogs. J. Toxicol. Sci. 2014, 39, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 inhibition as a means to inhibit activation of the NLRP3 inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfitt, D.A.; Aguila, M.; McCulley, C.H.; Bevilacqua, D.; Mendes, H.F.; Athanasiou, D.; Novoselov, S.S.; Kanuga, N.; Munro, P.M.; Coffey, P.J.; et al. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014, 5, e1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmaoglou, M.; Kanuga, N.; Aguilà, M.; Garriga, P.; Cheetham, M.E. A dual role for EDEM1 in the processing of rod opsin. J. Cell Sci. 2009, 122, 4465–4472. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Song, C.; Li, C.C.H. Molecular perspectives on p97-VCP: Progress in understanding its structure and diverse biological functions. J. Struct. Biol. 2004, 146, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Roux, M.J.; Merl, J.; Giangrande, A.; Hauck, S.M.; Aron, L.; Ueffing, M. Proteomic Survey Reveals Altered Energetic Patterns and Metabolic Failure Prior to Retinal Degeneration. J. Neurosci. 2014, 34, 2797–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasiou, D.; Aguila, M.; Opefi, C.A.; South, K.; Bellingham, J.; Bevilacqua, D.; Munro, P.M.; Kanuga, N.; Mackenzie, F.E.; Dubis, A.M.; et al. Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum. Mol. Genet. 2017, 26, 305–319. [Google Scholar] [CrossRef]
- Krebs, M.P.; Holden, D.C.; Joshi, P.; Clark, C.L.; Lee, A.H.; Kaushal, S. Molecular Mechanisms of Rhodopsin Retinitis Pigmentosa and the Efficacy of Pharmacological Rescue. J. Mol. Biol. 2010, 395, 1063–1078. [Google Scholar] [CrossRef]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [Green Version]
- Kosmaoglou, M.; Cheetham, M.E. Calnexin is not essential for mammalian rod opsin biogenesis. Mol. Vis. 2008, 14, 2466–2474. [Google Scholar]
- Gorbatyuk, M.S.; Knox, T.; LaVail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, M.; Kutsyr, O.; Cao, B.; Bolz, S.; Arango-Gonzalez, B.; Ueffing, M. Pharmacological Inhibition of the VCP/Proteasome Axis Rescues Photoreceptor Degeneration in RHOP23H Rat Retinal Explants. Biomolecules 2021, 11, 1528. https://doi.org/10.3390/biom11101528
Sen M, Kutsyr O, Cao B, Bolz S, Arango-Gonzalez B, Ueffing M. Pharmacological Inhibition of the VCP/Proteasome Axis Rescues Photoreceptor Degeneration in RHOP23H Rat Retinal Explants. Biomolecules. 2021; 11(10):1528. https://doi.org/10.3390/biom11101528
Chicago/Turabian StyleSen, Merve, Oksana Kutsyr, Bowen Cao, Sylvia Bolz, Blanca Arango-Gonzalez, and Marius Ueffing. 2021. "Pharmacological Inhibition of the VCP/Proteasome Axis Rescues Photoreceptor Degeneration in RHOP23H Rat Retinal Explants" Biomolecules 11, no. 10: 1528. https://doi.org/10.3390/biom11101528
APA StyleSen, M., Kutsyr, O., Cao, B., Bolz, S., Arango-Gonzalez, B., & Ueffing, M. (2021). Pharmacological Inhibition of the VCP/Proteasome Axis Rescues Photoreceptor Degeneration in RHOP23H Rat Retinal Explants. Biomolecules, 11(10), 1528. https://doi.org/10.3390/biom11101528