
Advances in Ophthalmic Optogenetics: Approaches and Applications


Abstract
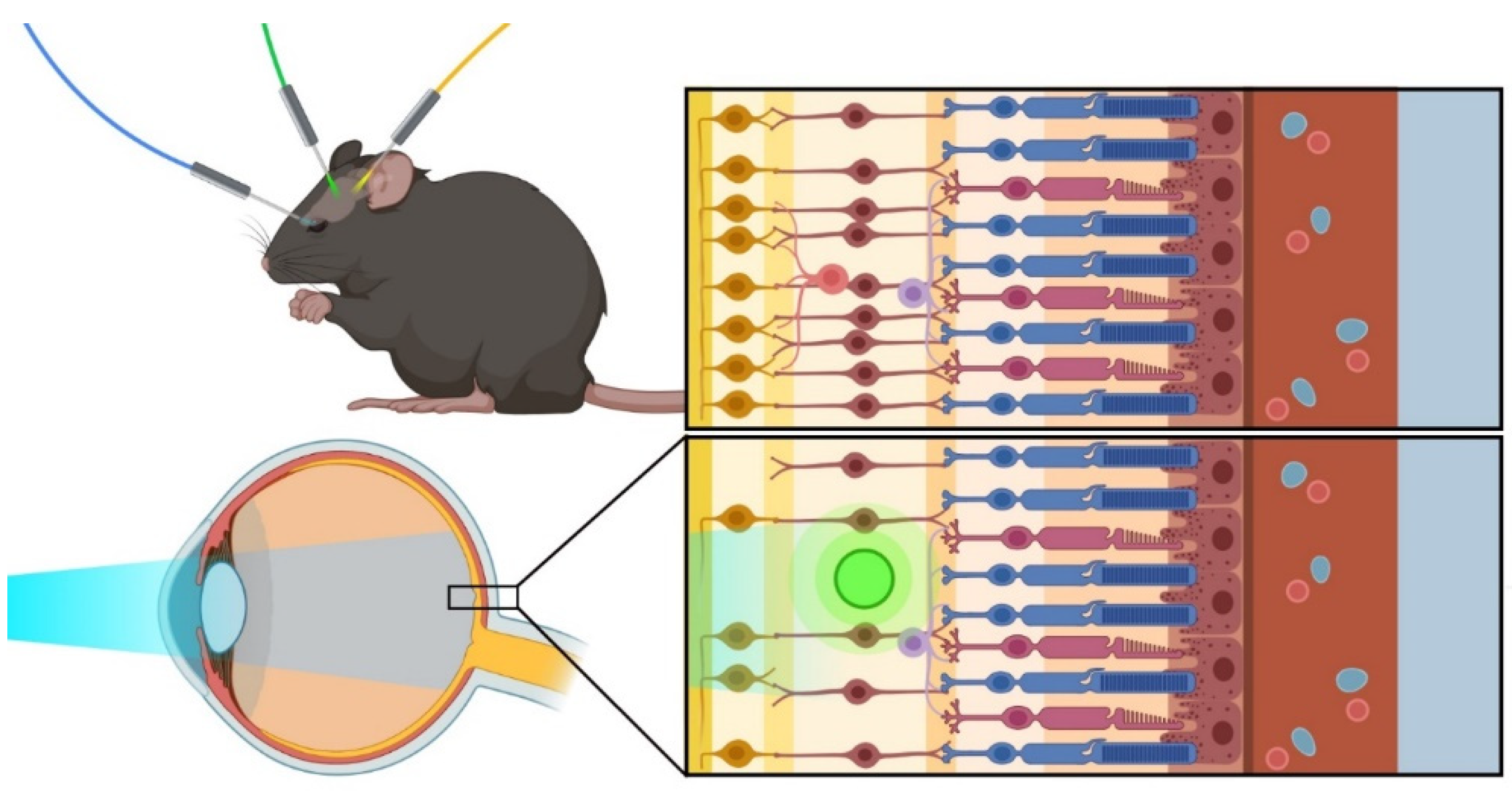
:1. Introduction
2. Opsins Used for Optogenetic Therapy
3. Cryptochrome-Based Dimerizers in Optogenetics
4. Approaches for the Delivery of Optogenetic Therapy in the Eye
5. Using Optogenetics in Retinal Degeneration and Glaucoma
6. Current Clinical Trials
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Deisseroth, K.; Feng, G.; Majewska, A.; Miesenboeck, G.; Ting, A.; Schnitzer, M.J. Next-Generation Optical Technologies for Illuminating Genetically Targeted Brain Circuits. J. Neurosci. 2006, 26, 10380–10386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2000, 17, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef]
- Jeffery, R.C.H.; Mukhtar, S.A.; McAllister, I.L.; Morgan, W.H.; Mackey, D.A.; Chen, F.K. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021, 42, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Prosseda, P.P.; Alvarado, J.A.; Wang, B.; Kowal, T.J.; Ning, K.; Stamer, W.D.; Hu, Y.; Sun, Y. Optogenetic stimulation of phosphoinositides reveals a critical role of primary cilia in eye pressure regulation. Sci. Adv. 2020, 6, eaay8699. [Google Scholar] [CrossRef]
- Kowal, T.J.; Prosseda, P.P.; Ning, K.; Wang, B.; Alvarado, J.; Sendayen, B.E.; Jabbehdari, S.; Stamer, W.D.; Hu, Y.; Sun, Y. Optogenetic Modulation of Intraocular Pressure in a Glucocorticoid-Induced Ocular Hypertension Mouse Model. Transl. Vis. Sci. Technol. 2021, 10, 10. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Xue, K.; de la Camara, C.M.-F.; Nanda, A.; Davies, A.; Wood, L.J.; Salvetti, A.P.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.-A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Degli Esposti, S.; Delaux, A.; de Saint Aubert, J.-B.; de Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Bi, A.; Cui, J.; Ma, Y.-P.; Olshevskaya, E.; Pu, M.; Dizhoor, A.; Pan, Z.-H. Ectopic Expression of a Microbial-Type Rhodopsin Restores Visual Responses in Mice with Photoreceptor Degeneration. Neuron 2006, 50, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Sahel, J.-A.; Roska, B. Gene Therapy for Blindness. Annu. Rev. Neurosci. 2013, 36, 467–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busskamp, V.; Picaud, S.; Sahel, J.A.; Roska, B. Optogenetic therapy for retinitis pigmentosa. Gene Ther. 2012, 19, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.; Bennett, J.; A Wellman, J.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.; Beattie, S.G.; Gardner, P.; et al. Long-Term Effect of Gene Therapy on Leber’s Congenital Amaurosis. New Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.; Jolly, J.K.; Barnard, A.R.; Rudenko, A.; Salvetti, A.P.; Patrício, M.I.; Edwards, T.L.; Groppe, M.; Orlans, H.O.; Tolmachova, T.; et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat. Med. 2018, 24, 1507–1512. [Google Scholar] [CrossRef]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Moritz, O.L. Photoreceptors at a glance. J. Cell Sci. 2015, 128, 4039–4045. [Google Scholar] [CrossRef] [Green Version]
- Ptito, M.; Bleau, M.; Bouskila, J. The Retina: A Window into the Brain. Cells 2021, 10, 3269. [Google Scholar] [CrossRef]
- Murakami, Y.; Notomi, S.; Hisatomi, T.; Nakazawa, T.; Ishibashi, T.; Miller, J.W.; Vavvas, D.G. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog. Retin. Eye Res. 2013, 37, 114–140. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.W.; Watt, C.B.; Frederick, J.M.; Baehr, W.; Chen, C.-K.; Levine, E.M.; Milam, A.H.; Lavail, M.M.; Marc, R.E. Retinal remodeling triggered by photoreceptor degenerations. J. Comp. Neurol. 2003, 464, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Peng, B. Retinal Ganglion Cells are Resistant to Photoreceptor Loss in Retinal Degeneration. PLoS ONE 2013, 8, e68084. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, F.; Novelli, E.; Strettoi, E. Retinal Ganglion Cells Survive and Maintain Normal Dendritic Morphology in a Mouse Model of Inherited Photoreceptor Degeneration. J. Neurosci. 2008, 28, 14282–14292. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Perspective on Genes and Mutations Causing Retinitis Pigmentosa. Arch. Ophthalmol. 2007, 125, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RetNet - Retinal Information Network. Available online: https://sph.uth.edu/retnet/ (accessed on 23 July 2021).
- Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 29 September 2020).
- Simon, C.-J.; Sahel, J.-A.; Duebel, J.; Herlitze, S.; Dalkara, D. Opsins for vision restoration. Biochem. Biophys. Res. Commun. 2020, 527, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 2015, 18, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegemann, P.; Nagel, G. From channelrhodopsins to optogenetics. EMBO Mol. Med. 2013, 5, 173–176. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.; Barnard, A.R.; Hughes, S.; Tam, S.K.E.; Martin, C.; Singh, M.S.; Barnea-Cramer, A.O.; McClements, M.E.; During, M.J.; Peirson, S.N.; et al. Long-term restoration of visual function in end-stage retinal degeneration using subretinal human melanopsin gene therapy. Proc. Natl. Acad. Sci. USA 2017, 114, 11211–11216. [Google Scholar] [CrossRef] [Green Version]
- Cehajic-Kapetanovic, J.; Eleftheriou, C.; Allen, A.; Milosavljevic, N.; Pienaar, A.; Bedford, R.; Davis, K.E.; Bishop, P.N.; Lucas, R. Restoration of Vision with Ectopic Expression of Human Rod Opsin. Curr. Biol. 2015, 25, 2111–2122. [Google Scholar] [CrossRef] [Green Version]
- Busskamp, V.; Duebel, J.; Balya, D.; Fradot, M.; Viney, T.J.; Siegert, S.; Groner, A.C.; Cabuy, E.; Forster, V.; Seeliger, M.; et al. Genetic Reactivation of Cone Photoreceptors Restores Visual Responses in Retinitis Pigmentosa. Science 2010, 329, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Macé, E.; Caplette, R.; Marre, O.; Sengupta, A.; Chaffiol, A.; Barbe, P.; Desrosiers, M.; Bamberg, E.; Sahel, J.-A.; Picaud, S.; et al. Targeting Channelrhodopsin-2 to ON-bipolar Cells With Vitreally Administered AAV Restores ON and OFF Visual Responses in Blind Mice. Mol. Ther. 2015, 23, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Nash, Z.; Conley, S.M.; Fliesler, S.J.; Cooper, M.J.; Naash, M.I. A Partial Structural and Functional Rescue of a Retinitis Pigmentosa Model with Compacted DNA Nanoparticles. PLoS ONE 2009, 4, e5290. [Google Scholar] [CrossRef] [Green Version]
- Lagali, P.S.; Balya, D.; Awatramani, G.B.; Münch, T.; Kim, D.; Busskamp, V.; Cepko, C.L.; Roska, B. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat. Neurosci. 2008, 11, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Doroudchi, M.M.; Greenberg, K.P.; Liu, J.; A Silka, K.; Boyden, E.S.; A Lockridge, J.; Arman, A.C.; Janani, R.; E Boye, S.; Boye, S.L.; et al. Virally delivered Channelrhodopsin-2 Safely and Effectively Restores Visual Function in Multiple Mouse Models of Blindness. Mol. Ther. 2011, 19, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Klapoetke, N.; Murata, Y.; Kim, S.S.; Pulver, S.; Birdsey-Benson, A.; Cho, Y.K.; Morimoto, T.K.; Chuong, A.S.; Carpenter, E.J.; Tian, Z.; et al. Independent optical excitation of distinct neural populations. Nat. Chem. Biol. 2014, 11, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Kim, J.; Tzeng, S.Y.; Ding, K.; Hafiz, Z.; Long, D.; Wang, J.; Green, J.J.; Campochiaro, P.A. Suprachoroidal gene transfer with nonviral nanoparticles. Sci. Adv. 2020, 6, eaba1606. [Google Scholar] [CrossRef] [PubMed]
- Gaub, B.M.; Berry, M.H.; E Holt, A.; Isacoff, E.Y.; Flannery, J.G. Optogenetic Vision Restoration Using Rhodopsin for Enhanced Sensitivity. Mol. Ther. 2015, 23, 1562–1571. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Sugano, E.; Isago, H.; Hiroi, T.; Wang, Z.; Ohta, E.; Tamai, M. Channelrhodopsin-2 gene transduced into retinal ganglion cells restores functional vision in genetically blind rats. Exp. Eye Res. 2010, 90, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Koizumi, A.; Tanaka, N.; Panda, S.; Masland, R.H. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc. Natl. Acad. Sci. USA 2008, 105, 16009–16014. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Liu, H.; Klejnot, J.; Lin, C. The Cryptochrome Blue Light Receptors. Arab. Book 2010, 8, e0135. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, M.J.; Hughes, R.; A Peteya, L.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid blue-light–mediated induction of protein interactions in living cells. Nat. Methods 2010, 7, 973–975. [Google Scholar] [CrossRef] [Green Version]
- Herrou, J.; Crosson, S. Function, structure and mechanism of bacterial photosensory LOV proteins. Nat. Rev. Genet. 2011, 9, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.L.; Zhulin, I.B. PAS Domains: Internal Sensors of Oxygen, Redox Potential, and Light. Microbiol. Mol. Biol. Rev. 1999, 63, 479–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoltowski, B.D.; Gardner, K.H. Tripping the Light Fantastic: Blue-Light Photoreceptors as Examples of Environmentally Modulated Protein−Protein Interactions. Biochemistry 2010, 50, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Huala, E.; Oeller, P.W.; Liscum, E.; Han, I.-S.; Larsen, E.; Briggs, W.R. Arabidopsis NPH1: A Protein Kinase with a Putative Redox-Sensing Domain. Science 1997, 278, 2120–2123. [Google Scholar] [CrossRef]
- Salomon, M.; Christie, J.M.; Knieb, E.; Lempert, U.; Briggs, W.R. Photochemical and Mutational Analysis of the FMN-Binding Domains of the Plant Blue Light Receptor, Phototropin. Biochemistry 2000, 39, 9401–9410. [Google Scholar] [CrossRef]
- Balaggan, K.S.; Ali, R. Ocular gene delivery using lentiviral vectors. Gene Ther. 2011, 19, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Puppo, A.; Cesi, G.; Marrocco, E.; Piccolo, P.; Jacca, S.; Shayakhmetov, D.M.; Parks, R.J.; Davidson, B.; Colloca, S.; Brunetti-Pierri, N.; et al. Retinal transduction profiles by high-capacity viral vectors. Gene Ther. 2014, 21, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, P.C.; De Silva, S.R.; Lipinski, D.M.; Singh, M.S.; Mouravlev, A.; You, Q.S.; Barnard, A.R.; Hankins, M.W.; During, M.J.; MacLaren, R.E. Assessment of Tropism and Effectiveness of New Primate-Derived Hybrid Recombinant AAV Serotypes in the Mouse and Primate Retina. PLoS ONE 2013, 8, e60361. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.M.; Flotte, T.R. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef]
- Nuzbrokh, Y.; Kassotis, A.S.; Ragi, S.D.; Jauregui, R.; Tsang, S.H. Treatment-Emergent Adverse Events in Gene Therapy Trials for Inherited Retinal Diseases: A Narrative Review. Ophthalmol. Ther. 2020, 9, 709–724. [Google Scholar] [CrossRef]
- Fischer, M.D.; McClements, M.E.; de la Camara, C.M.-F.; Bellingrath, J.-S.; Dauletbekov, D.; Ramsden, S.C.; Hickey, D.G.; Barnard, A.R.; MacLaren, R. Codon-Optimized RPGR Improves Stability and Efficacy of AAV8 Gene Therapy in Two Mouse Models of X-Linked Retinitis Pigmentosa. Mol. Ther. 2017, 25, 1854–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., III; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajala, A.; Wang, Y.; Zhu, Y.; Ranjo-Bishop, M.; Ma, J.-X.; Mao, C.; Rajala, R.V.S. Nanoparticle-Assisted Targeted Delivery of Eye-Specific Genes to Eyes Significantly Improves the Vision of Blind Mice In Vivo. Nano Lett. 2014, 14, 5257–5263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Koirala, A.; Makkia, R.; Cooper, M.J.; I Naash, M. Direct gene transfer with compacted DNA nanoparticles in retinal pigment epithelial cells: Expression, repeat delivery and lack of toxicity. Nanomedicine 2012, 7, 521–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimczak, R.R.; Koerber, J.T.; Dalkara, D.; Flannery, J.G.; Schaffer, D.V. A Novel Adeno-Associated Viral Variant for Efficient and Selective Intravitreal Transduction of Rat Müller Cells. PLoS ONE 2009, 4, e7467. [Google Scholar] [CrossRef] [Green Version]
- GBD 2019 Blindness; Vision Impairment Collaborators; the Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Wiggs, J.L.; Pasquale, L.R. Genetics of glaucoma. Hum. Mol. Genet. 2017, 26, R21–R27. [Google Scholar] [CrossRef]
- McClements, M.E.; Staurenghi, F.; MacLaren, R.E.; Cehajic-Kapetanovic, J. Optogenetic Gene Therapy for the Degenerate Retina: Recent Advances. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Geeraerts, E.; Claes, M.; Dekeyster, E.; Salinas-Navarro, M.; De Groef, L.; Haute, C.V.D.; Scheyltjens, I.; Baekelandt, V.; Arckens, L.; Moons, L. Optogenetic Stimulation of the Superior Colliculus Confers Retinal Neuroprotection in a Mouse Glaucoma Model. J. Neurosci. 2019, 39, 2313–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreb, R.N.; Khaw, P.T. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef]
- Gordon, M.O.; Beiser, J.A.; Brandt, J.D.; Heuer, D.K.; Higginbotham, E.J.; Johnson, C.; Keltner, J.L.; Miller, J.P.; Parrish, R.K.; Wilson, M.R.; et al. The Ocular Hypertension Treatment Study. Arch. Ophthalmol. 2002, 120, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Agis Investigators. The advanced glaucoma intervention study (AGIS): 7. the relationship between control of intraocular pressure and visual field deterioration. Am. J. Ophthalmol. 2000, 130, 429–440. [Google Scholar] [CrossRef]
- Lee, R.K.; Liu, Y. Cell transplantation to replace retinal ganglion cells faces challenges – the Switchboard Dilemma. Neural Regen. Res. 2021, 16, 1138–1143. [Google Scholar] [CrossRef]
- Youssef, P.N.; Sheibani, N.; Albert, D.M. Retinal light toxicity. Eye 2010, 25, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Van Norren, D.; Vos, J.J. Light damage to the retina: An historical approach. Eye 2015, 30, 169–172. [Google Scholar] [CrossRef] [Green Version]
- ANSI Z136.1-2014 - American National Standard for Safe Use of Lasers. Available online: https://webstore.ansi.org/standards/lia/ansiz1362014 (accessed on 1 January 2022).
- International Commission on Non-Ionizing Radiation Protection. ICNIRP Guidelines on Limits of Exposure to Laser Radiation of Wavelengths between 180 nm and 1,000 μm. Heal. Phys. 2013, 105, 271–295. [Google Scholar] [CrossRef] [Green Version]
- Delori, F.C.; Webb, R.H.; Sliney, D.H. Maximum permissible exposures for ocular safety (ANSI 2000), with emphasis on ophthalmic devices. J. Opt. Soc. Am. A 2007, 24, 1250–1265. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, S.; Van Wyk, M.; Lehmann, K.; Löwel, S.; Feng, G.; Wässle, H. Visual Function in Mice with Photoreceptor Degeneration and Transgenic Expression of Channelrhodopsin 2 in Ganglion Cells. J. Neurosci. 2010, 30, 8745–8758. [Google Scholar] [CrossRef]
- Ham, W.T.; Ruffolo, J.J.; A Mueller, H.; Clarke, A.M.; E Moon, M. Histologic analysis of photochemical lesions produced in rhesus retina by short-wave-length light. Investig. Ophthalmol. Vis. Sci. 1978, 17, 1029–1035. [Google Scholar]
- Chen, E. Inhibition of cytochrome oxidase and blue-light damage in rat retina. Graefe’s Arch. Clin. Exp. Ophthalmol. 1993, 231, 416–423. [Google Scholar] [CrossRef]
- Wu, J.; Seregard, S.; Algvere, P.V. Photochemical Damage of the Retina. Surv. Ophthalmol. 2006, 51, 461–481. [Google Scholar] [CrossRef] [PubMed]
- Gauvain, G.; Akolkar, H.; Chaffiol, A.; Arcizet, F.; Khoei, M.A.; Desrosiers, M.; Jaillard, C.; Caplette, R.; Marre, O.; Bertin, S.; et al. Optogenetic therapy: High spatiotemporal resolution and pattern discrimination compatible with vision restoration in non-human primates. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef]
- Sengupta, A.; Chaffiol, A.; Macé, E.; Caplette, R.; Desrosiers, M.; Lampič, M.; Forster, V.; Marre, O.; Lin, J.; Sahel, J.-A.; et al. Red-shifted channelrhodopsin stimulation restores light responses in blind mice, macaque retina, and human retina. EMBO Mol. Med. 2016, 8, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.N.; Boulanger-Scemama, E.; Degli Esposti, S.; Galluppi, F.; Blouin, L.; Taiel, M.; Sahel, J.A. Optogenetic GS030 Therapy in Subjects with Retinitis Pigmentosa: Safety and Tolerability Up to Two Years After Treatment Administration in the Phase 1/2a PIONEER Study. Investigative Ophthalmology & Visual Science 2021, 62, 3236. [Google Scholar]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.A.; Wright, J.F. Challenges Posed by Immune Responses to AAV Vectors: Addressing Root Causes. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Yin, L.; Strazzeri, J.M.; Flannery, J.; Merigan, W.H.; Schaffer, D.V. Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther. 2014, 22, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Dick, A.D.; Hall, S.M.; Langmann, T.; Scribner, C.L.; Mansfield, B.C. For the Ocular Gene Therapy Inflammation Working Group Inflammation in Viral Vector-Mediated Ocular Gene Therapy: A Review and Report From a Workshop Hosted by the Foundation Fighting Blindness, 9/2020. Transl. Vis. Sci. Technol. 2021, 10, 3. [Google Scholar] [CrossRef]
- Tummala, G.; Crain, A.; Rowlan, J.; Pepple, K.L. Characterization of Gene Therapy Associated Uveitis Following Intravitreal Adeno-Associated Virus Injection in Mice. Investig. Opthalmology Vis. Sci. 2021, 62, 41. [Google Scholar] [CrossRef]
- Simunovic, M.; Shen, W.; Lin, J.; Protti, D.; Lisowski, L.; Gillies, M. Optogenetic approaches to vision restoration. Exp. Eye Res. 2018, 178, 15–26. [Google Scholar] [CrossRef]
- Jabs, D.A.; Nussenblatt, R.B.; Rosenbaum, J.T. Standardization of Uveitis Nomenclature for Reporting Clinical Data. Results of the First International Workshop. Am. J. Ophthalmol. 2005, 140, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, D.; Byrne, L.C.; Klimczak, R.R.; Visel, M.; Yin, L.; Merigan, W.H.; Flannery, J.G.; Schaffer, D.V. In Vivo–Directed Evolution of a New Adeno-Associated Virus for Therapeutic Outer Retinal Gene Delivery from the Vitreous. Sci. Transl. Med. 2013, 5, 189ra76. [Google Scholar] [CrossRef]
- Posch, C.; Matolin, D.; Wohlgenannt, R. A QVGA 143 dB Dynamic Range Frame-Free PWM Image Sensor With Lossless Pixel-Level Video Compression and Time-Domain CDS. IEEE J. Solid-state Circuits 2010, 46, 259–275. [Google Scholar] [CrossRef]
- Nirenberg, S.; Pandarinath, C. Retinal prosthetic strategy with the capacity to restore normal vision. Proc. Natl. Acad. Sci. USA 2012, 109, 15012–15017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batabyal, S.; Gajjeraman, S.; Pradhan, S.; Bhattacharya, S.; Wright, W.; Mohanty, S. Sensitization of ON-bipolar cells with ambient light activatable multi-characteristic opsin rescues vision in mice. Gene Ther. 2020, 28, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.W.; Gajjeraman, S.; Batabyal, S.; Pradhan, S.; Bhattacharya, S.; Mahapatra, V.; Tripathy, A.; Mohanty, S.K. Restoring vision in mice with retinal degeneration using multicharacteristic opsin. Neurophotonics 2017, 4, 041505. [Google Scholar] [CrossRef]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and Efficacy of Gene Transfer for Leber’s Congenital Amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Seitz, I.P.; Michalakis, S.; Wilhelm, B.; Reichel, F.F.; Ochakovski, G.A.; Zrenner, E.; Ueffing, M.; Biel, M.; Wissinger, B.; Bartz-Schmidt, K.U.; et al. Superior Retinal Gene Transfer and Biodistribution Profile of Subretinal Versus Intravitreal Delivery of AAV8 in Nonhuman Primates. Investig. Opthalmology Vis. Sci. 2017, 58, 5792–5801. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Le Goff, M.M.; Allen, A.; Lucas, R.J.; Bishop, P.N. Glycosidic enzymes enhance retinal transduction following intravitreal delivery of AAV2. Mol. Vis. 2011, 17, 1771–1783. [Google Scholar]
- Cehajic-Kapetanovic, J.; Milosavljevic, N.; Bedford, R.A.; Lucas, R.; Bishop, P.N. Efficacy and Safety of Glycosidic Enzymes for Improved Gene Delivery to the Retina following Intravitreal Injection in Mice. Mol. Ther. - Methods Clin. Dev. 2018, 9, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Miller, R.; Han, P.-Y.; Pang, J.; Dinculescu, A.; Chiodo, V.; Hauswirth, W.W. Intraocular route of AAV2 vector administration defines humoral immune response and therapeutic potential. Mol. Vis. 2008, 14, 1760–1769. [Google Scholar] [PubMed]
- Teo, K.Y.C.; Lee, S.Y.; Barathi, A.V.; Tun, S.B.B.; Tan, L.; Constable, I.J. Surgical Removal of Internal Limiting Membrane and Layering of AAV Vector on the Retina Under Air Enhances Gene Transfection in a Nonhuman Primate. Investig. Opthalmology Vis. Sci. 2018, 59, 3574–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiu, G.; Chung, S.H.; Mollhoff, I.N.; Nguyen, U.T.; Thomasy, S.M.; Yoo, J.; Taraborelli, D.; Noronha, G. Suprachoroidal and Subretinal Injections of AAV Using Transscleral Microneedles for Retinal Gene Delivery in Nonhuman Primates. Mol. Ther. - Methods Clin. Dev. 2020, 16, 179–191. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Company | Disease | Intervention | Opsin | Viral Vector | Delivery Route | Trial Stage | Clinical Trial Identifier |
|---|---|---|---|---|---|---|---|
| GenSight Biologics | Retinitis Pigmentosa | Drug: GS030-DP Medical device: GS030-MD | ChrimsonR | rAAV2.7m8 | Intravitreal injection | Phase 1/2a | NCT03326336 |
| Allergan | Advanced Retinitis Pigmentosa | Drug: RST-001 | ChR2 | rAAV2.7m8 | Intravitreal injection | Phase 1/2a | NCT02556736 |
| Bionic Sight LLC | Retinitis Pigmentosa | Drug: BS01 | ChronosFP | rAAV2 | Intravitreal injection | Phase 1/2 | NCT04278131 |
| Nanoscope Therapeutics Inc. | Retinitis Pigmentosa | Drug: vMCO-010 | MCO1 | rAAV2 | Intravitreal injection | Phase 2b | NCT04945772 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prosseda, P.P.; Tran, M.; Kowal, T.; Wang, B.; Sun, Y. Advances in Ophthalmic Optogenetics: Approaches and Applications. Biomolecules 2022, 12, 269. https://doi.org/10.3390/biom12020269
Prosseda PP, Tran M, Kowal T, Wang B, Sun Y. Advances in Ophthalmic Optogenetics: Approaches and Applications. Biomolecules. 2022; 12(2):269. https://doi.org/10.3390/biom12020269
Chicago/Turabian StyleProsseda, Philipp P., Matthew Tran, Tia Kowal, Biao Wang, and Yang Sun. 2022. "Advances in Ophthalmic Optogenetics: Approaches and Applications" Biomolecules 12, no. 2: 269. https://doi.org/10.3390/biom12020269
APA StyleProsseda, P. P., Tran, M., Kowal, T., Wang, B., & Sun, Y. (2022). Advances in Ophthalmic Optogenetics: Approaches and Applications. Biomolecules, 12(2), 269. https://doi.org/10.3390/biom12020269