
Alternative Pre-mRNA Splicing of the Mu Opioid Receptor Gene, OPRM1: Insight into Complex Mu Opioid Actions

 ,
, 
Abstract
1. Introduction
2. Mechanisms and Functions of Alternative Pre-mRNA Splicing
3. Alternative Splicing in GPCRs
4. Alternative Splicing of the Mu Opioid Receptor Gene, OPRM1
4.1. Concept of Multiple Mu Opioid Receptors
4.2. Evolution of OPRM1 Gene
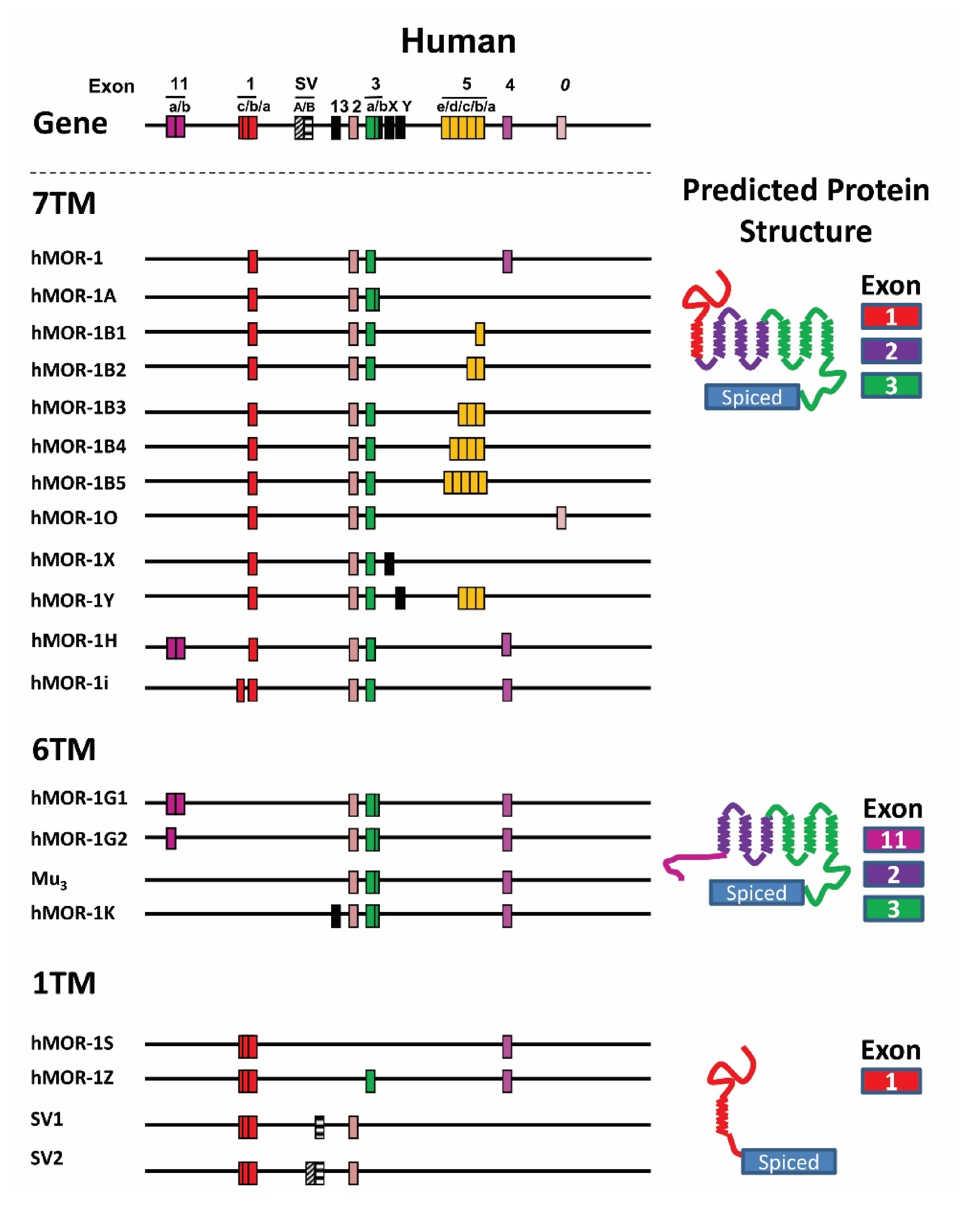
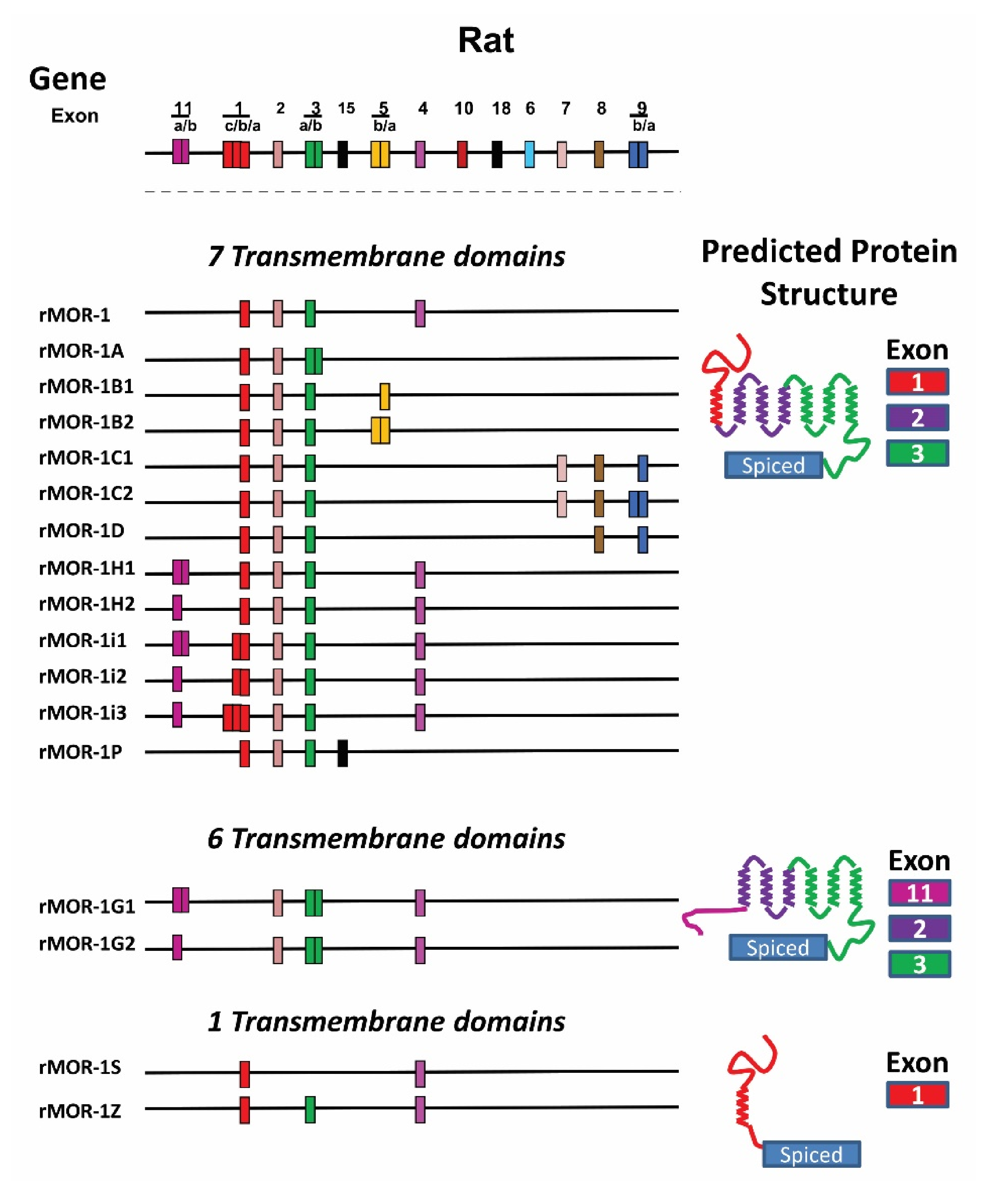
4.3. OPRM1 Gene Structure
4.4. Alternatively Spliced OPRM1 Variants
4.4.1. Full-Length 7TM C-Terminal Splice Variants
4.4.2. Truncated 6TM Variants
4.4.3. Single TM Variants
5. Expression and Function of the OPRM1 Variants
5.1. Expression of the OPRM1 Variant mRNAs and Proteins
5.1.1. Region-Specific and Strain-Specific Expression of the OPRM1 Variant mRNAs
5.1.2. Region-Specific and Cell-Specific Expression of the OPRM1 Variant Proteins
5.2. The Function of Full-Length 7TM C-Terminal Variants
5.2.1. Opioid Receptor Binding Affinity
5.2.2. Mu Agonist-Induced G Protein Coupling, Internalization, Phosphorylation, and Post-Endocytic Sorting
5.2.3. Mu Agonist-Induced Biased Signaling at 7TM C-Terminal Variants
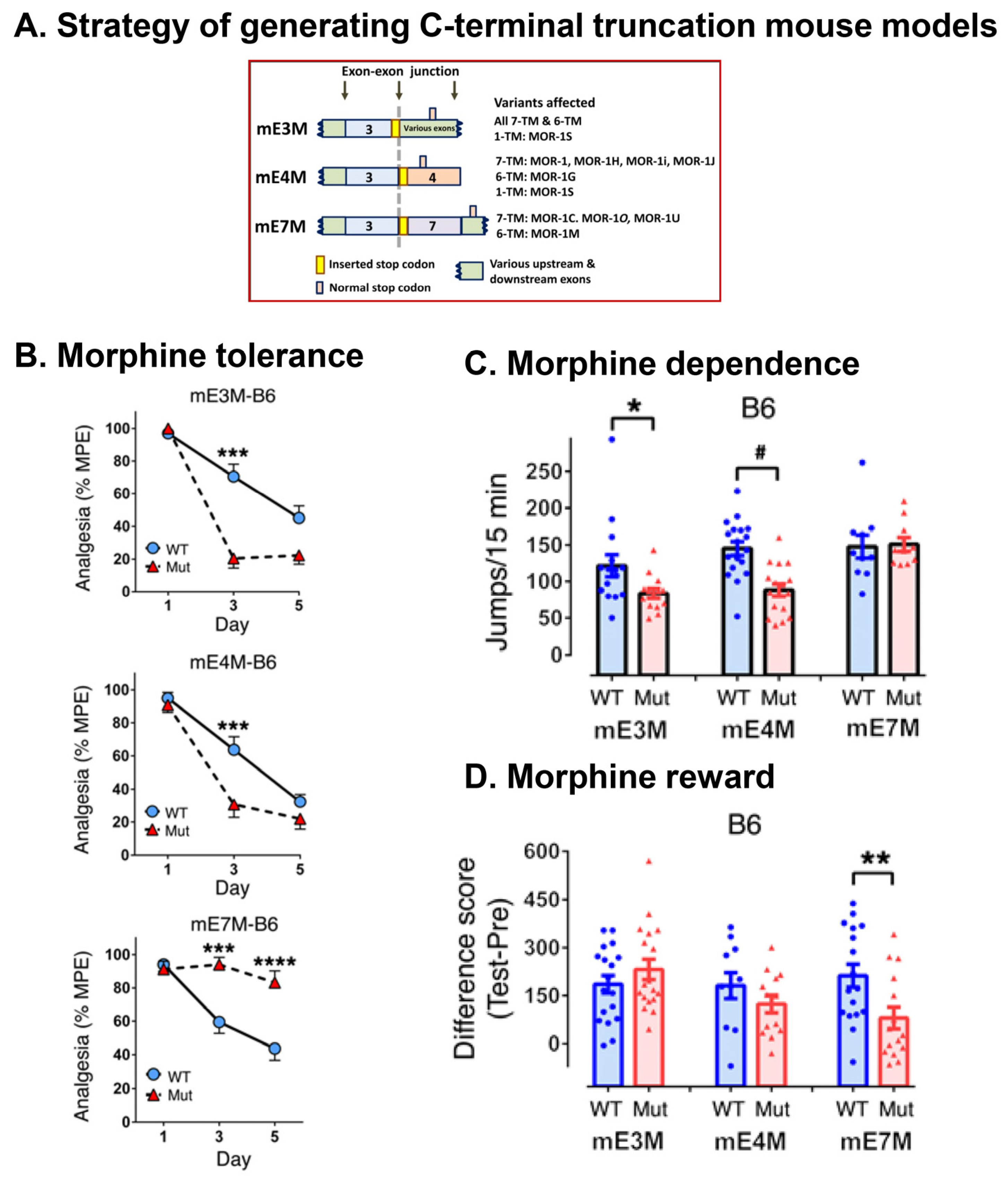
5.2.4. In Vivo Function of 7TM C-Terminal Variants
Morphine-Induced Itch (Pruritus)
Morphine Tolerance, Dependence, and Reward
5.3. The Function of Truncated 6TM Variants
5.3.1. Involvement of 6TM Variants in Heroin and M6G Analgesia
5.3.2. Essential Role of 6TM Variants in a Novel Type of Opioid Analgesic Drugs
5.3.3. Function of 6TM Variants in Delta/Kappa Opioids and Non-Opioids
5.3.4. A Chaperon-Like Function of 6TM Variants in Enhancing Expression of the 7TM MOR-1 at Protein Level
5.3.5. Role of 6TM Variants in Morphine Hyperalgesia
5.4. The Function of Single TM Variants
6. Molecular Mechanisms Underlying OPRM1 Alternative Splicing
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martin, W.R. Opioid antagonists. Pharmacol. Rev. 1967, 19, 463–521. [Google Scholar] [PubMed]
- Pert, C.B.; Pasternak, G.W.; Snyder, S.H. Opiate agonists and antagonists discriminated by receptor binding in brain. Science 1973, 182, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Terenius, L. Stereospecific interaction between narcotic analgesics and a synaptic plasma membrane fraction of rat cerebral cortex. Acta Pharmacol. Toxicol. 1973, 32, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.J.; Hiller, J.M.; Edelman, I. Stereospecific binding of the potent narcotic analgesice [3H]Etorphine to rat-brain homogenate. Proc. Natl. Acad. Sci. USA 1973, 70, 1947–1949. [Google Scholar] [CrossRef]
- Chen, Y.; Mestek, A.; Liu, J.; Hurley, J.A.; Yu, L. Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol. Pharmacol. 1993, 44, 8–12. [Google Scholar] [CrossRef]
- Thompson, R.C.; Mansour, A.; Akil, H.; Watson, S.J. Cloning and pharmacological characterization of a rat μ opioid receptor. Neuron 1993, 11, 903–913. [Google Scholar] [CrossRef]
- Wang, J.B.; Imai, Y.; Eppler, C.M.; Gregor, P.; Spivak, C.E.; Uhl, G.R. μ opiate receptor: cDNA cloning and expression. Proc. Natl. Acad. Sci. USA 1993, 90, 10230–10234. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Childers, S.R.; Snyder, S.H. Opiate analgesia: Evidence for mediation by a subpopulation of opiate receptors. Science 1980, 208, 514–516. [Google Scholar] [CrossRef]
- Wolozin, B.L.; Pasternak, G.W. Classification of multiple morphine and enkephalin binding sites in the central nervous system. Proc. Natl. Acad. Sci. USA 1981, 78, 6181–6185. [Google Scholar] [CrossRef]
- Pasternak, G.W. The pharmacology of mu analgesics: From patients to genes. Neuroscientist 2001, 7, 220–231. [Google Scholar] [CrossRef]
- Pan, Y.X. Diversity and complexity of the mu opioid receptor gene: Alternative pre-mRNA splicing and promoters. DNA Cell Biol. 2005, 24, 736–750. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Black, D.L. Activation of c-src neuron-specific splicing by an unusual RNA element in vivo and in vitro. Cell 1992, 69, 795–807. [Google Scholar] [CrossRef]
- Chan, R.C.; Black, D.L. The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol. Cell. Biol. 1997, 17, 4667–4676. [Google Scholar] [CrossRef]
- Chan, R.C.; Black, D.L. Conserved intron elements repress splicing of a neuron-specific c-src exon in vitro. Mol. Cell. Biol. 1997, 17, 2970. [Google Scholar] [CrossRef]
- Xu, R.; Teng, J.; Cooper, T.A. The cardiac troponin T alternative exon contains a novel purine-rich positive splicing element. Mol. Cell. Biol. 1993, 13, 3660–3674. [Google Scholar] [CrossRef]
- Ryan, K.J.; Cooper, T.A. Muscle-specific splicing enhancers regulate inclusion of the cardiac troponin T alternative exon in embryonic skeletal muscle. Mol. Cell. Biol. 1996, 16, 4014–4023. [Google Scholar] [CrossRef]
- Cooper, T.A. Muscle-specific splicing of a heterologous exon mediated by a single muscle-specific splicing enhancer from the cardiac troponin T gene. Mol. Cell. Biol. 1998, 18, 4519–4525. [Google Scholar] [CrossRef]
- Carstens, R.P.; Wagner, E.J.; Garcia-Blanco, M.A. An intronic splicing silencer causes skipping of the IIIb exon of fibroblast growth factor receptor 2 through involvement of polypyrimidine tract binding protein. Mol. Cell. Biol. 2000, 20, 7388–7400. [Google Scholar] [CrossRef]
- Carstens, R.P.; McKeehan, W.L.; Garcia-Blanco, M.A. An intronic sequence element mediates both activation and repression of rat fibroblast growth factor receptor 2 pre-mRNA splicing. Mol. Cell. Biol. 1998, 18, 2205–2217. [Google Scholar] [CrossRef]
- Carstens, R.P.; Eaton, J.V.; Krigman, H.R.; Walther, P.J.; Garcia-Blanco, M.A. Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene 1997, 15, 3059–3065. [Google Scholar] [CrossRef]
- Fu, X.D. The superfamily of arginine/serine-rich splicing factors. RNA 1995, 1, 663–680. [Google Scholar]
- Graveley, B.R. Sorting out the complexity of SR protein functions. RNA 2000, 6, 1197–1211. [Google Scholar] [CrossRef]
- Hastings, M.L.; Krainer, A.R. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell Biol. 2001, 13, 302–309. [Google Scholar] [CrossRef]
- Caceres, J.F.; Stamm, S.; Helfman, D.M.; Krainer, A.R. Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science 1994, 265, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Mayeda, A.; Krainer, A.R. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell 1992, 68, 365–375. [Google Scholar] [CrossRef]
- Yang, X.; Bani, M.R.; Lu, S.J.; Rowan, S.; Ben-David, Y.; Chabot, B. The A1 and A1B proteins of heterogeneous nuclear ribonucleoparticles modulate 5’ splice site selection in vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 6924–6928. [Google Scholar] [CrossRef] [PubMed]
- Fiset, S.; Chabot, B. hnRNP A1 may interact simultaneously with telomeric DNA and the human telomerase RNA in vitro. Nucleic Acids Res. 2001, 29, 2268–2275. [Google Scholar] [CrossRef] [PubMed]
- Ladd, A.N.; Charlet, N.; Cooper, T.A. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol. 2001, 21, 1285–1296. [Google Scholar] [CrossRef]
- Ladd, A.N.; Nguyen, N.H.; Malhotra, K.; Cooper, T.A. CELF6, a member of the CELF family of RNA-binding proteins, regulates muscle-specific splicing enhancer-dependent alternative splicing. J. Biol. Chem. 2004, 279, 17756–17764. [Google Scholar] [CrossRef]
- Ladd, A.N.; Taffet, G.; Hartley, C.; Kearney, D.L.; Cooper, T.A. Cardiac tissue-specific repression of CELF activity disrupts alternative splicing and causes cardiomyopathy. Mol. Cell. Biol. 2005, 25, 6267–6278. [Google Scholar] [CrossRef]
- Jensen, K.B.; Dredge, B.K.; Stefani, G.; Zhong, R.; Buckanovich, R.J.; Okano, H.J.; Yang, Y.Y.; Darnell, R.B. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 2000, 25, 359–371. [Google Scholar] [CrossRef]
- Ule, J.; Stefani, G.; Mele, A.; Ruggiu, M.; Wang, X.; Taneri, B.; Gaasterland, T.; Blencowe, B.J.; Darnell, R.B. An RNA map predicting Nova-dependent splicing regulation. Nature 2006, 444, 580–586. [Google Scholar] [CrossRef]
- Ule, J.; Ule, A.; Spencer, J.; Williams, A.; Hu, J.S.; Cline, M.; Wang, H.; Clark, T.; Fraser, C.; Ruggiu, M.; et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005, 37, 844–852. [Google Scholar] [CrossRef]
- Cooper, T.A.; Mattox, W. The regulation of splice-site selection, and its role in human disease. Am. J. Hum. Genet. 1997, 61, 259–266. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef]
- Lin, C.L.; Bristol, L.A.; Jin, L.; Dykes-Hoberg, M.; Crawford, T.; Clawson, L.; Rothstein, J.D. Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 1998, 20, 589–602. [Google Scholar] [CrossRef]
- Meyer, T.; Fromm, A.; Munch, C.; Schwalenstocker, B.; Fray, A.E.; Ince, P.G.; Stamm, S.; Gron, G.; Ludolph, A.C.; Shaw, P.J. The RNA of the glutamate transporter EAAT2 is variably spliced in amyotrophic lateral sclerosis and normal individuals. J. Neurol. Sci. 1999, 170, 45–50. [Google Scholar] [CrossRef]
- Jaeckle, K.A.; Graus, F.; Houghton, A.; Cardon-Cardo, C.; Nielsen, S.L.; Posner, J.B. Autoimmune response of patients with paraneoplastic cerebellar degeneration to a Purkinje cell cytoplasmic protein antigen. Ann. Neurol. 1985, 18, 592–600. [Google Scholar] [CrossRef]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Lorson, C.L.; Rindt, H.; Shababi, M. Spinal muscular atrophy: Mechanisms and therapeutic strategies. Hum. Mol. Genet. 2010, 19, R111–R118. [Google Scholar] [CrossRef]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef]
- Resnick, D.K.; Resnick, N.M.; Welch, W.C.; Cooper, D.L. Differential expressions of CD44 variants in tumors affecting the central nervous system. Mol. Diagn. 1999, 4, 219–232. [Google Scholar] [CrossRef]
- Ge, K.; DuHadaway, J.; Du, W.; Herlyn, M.; Rodeck, U.; Prendergast, G.C. Mechanism for elimination of a tumor suppressor: Aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc. Natl. Acad. Sci. USA 1999, 96, 9689–9694. [Google Scholar] [CrossRef]
- Yamaguchi, F.; Saya, H.; Bruner, J.M.; Morrison, R.S. Differential expression of two fibroblast growth factor-receptor genes is associated with malignant progression in human astrocytomas. Proc. Natl. Acad. Sci. USA 1994, 91, 484–488. [Google Scholar] [CrossRef]
- Spraggon, L.; Cartegni, L. Antisense modulation of RNA processing as a therapeutic approach in cancer therapy. Drug Discov. Today Ther. Strat. 2013, 10, e139–e148. [Google Scholar] [CrossRef]
- Insel, P.A.; Sriram, K.; Gorr, M.W.; Wiley, S.Z.; Michkov, A.; Salmeron, C.; Chinn, A.M. GPCRomics: An approach to discover GPCR drug targets. Trends Pharm. Sci. 2019, 40, 378–387. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR drug targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef]
- Markovic, D.; Challiss, R.A. Alternative splicing of G protein-coupled receptors: Physiology and pathophysiology. Cell Mol. Life Sci. 2009, 66, 3337–3352. [Google Scholar] [CrossRef]
- Xu, J.; Lu, Z.; Narayan, A.; Le Rouzic, V.P.; Xu, M.; Hunkele, A.; Brown, T.G.; Hoefer, W.F.; Rossi, G.C.; Rice, R.C.; et al. Alternatively spliced mu opioid receptor C termini impact the diverse actions of morphine. J. Clin. Investig. 2017, 127, 1561–1573. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, J.; Xu, M.; Rossi, G.C.; Majumdar, S.; Pasternak, G.W.; Pan, Y.X. Truncated mu-opioid receptors with 6 transmembrane domains are essential for opioid analgesia. Anesth. Analg. 2018, 126, 1050–1057. [Google Scholar] [CrossRef]
- Xu, J.; Xu, M.; Brown, T.; Rossi, G.C.; Hurd, Y.L.; Inturrisi, C.E.; Pasternak, G.W.; Pan, Y.X. Stabilization of the mu-opioid receptor by truncated single transmembrane splice variants through a chaperone-like action. J. Biol. Chem. 2013, 288, 21211–21227. [Google Scholar] [CrossRef]
- Hawrylyshyn, K.A.; Michelotti, G.A.; Coge, F.; Guenin, S.P.; Schwinn, D.A. Update on human alpha1-adrenoceptor subtype signaling and genomic organization. Trends Pharm. Sci. 2004, 25, 449–455. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Price, D.T.; Schwinn, D.A. Alpha 1-adrenergic receptor regulation: Basic science and clinical implications. Pharm. Ther. 2000, 88, 281–309. [Google Scholar] [CrossRef]
- Chang, D.J.; Chang, T.K.; Yamanishi, S.S.; Salazar, F.H.; Kosaka, A.H.; Khare, R.; Bhakta, S.; Jasper, J.R.; Shieh, I.S.; Lesnick, J.D.; et al. Molecular cloning, genomic characterization and expression of novel human alpha1A-adrenoceptor isoforms. FEBS Lett. 1998, 422, 279–283. [Google Scholar] [CrossRef]
- Price, R.R.; Morris, D.P.; Biswas, G.; Smith, M.P.; Schwinn, D.A. Acute agonist-mediated desensitization of the human alpha 1a-adrenergic receptor is primarily independent of carboxyl terminus regulation: Implications for regulation of alpha 1aAR splice variants. J. Biol. Chem. 2002, 277, 9570–9579. [Google Scholar] [CrossRef] [PubMed]
- Coge, F.; Guenin, S.P.; Renouard-Try, A.; Rique, H.; Ouvry, C.; Fabry, N.; Beauverger, P.; Nicolas, J.P.; Galizzi, J.P.; Boutin, J.A.; et al. Truncated isoforms inhibit [3H]prazosin binding and cellular trafficking of native human alpha1A-adrenoceptors. Biochem. J. 1999, 343 Pt 1, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Tseng-Crank, J.; Kost, T.; Goetz, A.; Hazum, S.; Roberson, K.M.; Haizlip, J.; Godinot, N.; Robertson, C.N.; Saussy, D. The alpha 1C-adrenoceptor in human prostate: Cloning, functional expression, and localization to specific prostatic cell types. Br. J. Pharm. 1995, 115, 1475–1485. [Google Scholar] [CrossRef]
- Galiegue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carriere, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef]
- Ryberg, E.; Vu, H.K.; Larsson, N.; Groblewski, T.; Hjorth, S.; Elebring, T.; Sjogren, S.; Greasley, P.J. Identification and characterisation of a novel splice variant of the human CB1 receptor. FEBS Lett. 2005, 579, 259–264. [Google Scholar] [CrossRef]
- Shire, D.; Carillon, C.; Kaghad, M.; Calandra, B.; Rinaldi-Carmona, M.; Le Fur, G.; Caput, D.; Ferrara, P. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J. Biol. Chem. 1995, 270, 3726–3731. [Google Scholar] [CrossRef]
- Liu, Q.R.; Pan, C.H.; Hishimoto, A.; Li, C.Y.; Xi, Z.X.; Llorente-Berzal, A.; Viveros, M.P.; Ishiguro, H.; Arinami, T.; Onaivi, E.S.; et al. Species differences in cannabinoid receptor 2 (CNR2 gene): Identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009, 8, 519–530. [Google Scholar] [CrossRef]
- Hillhouse, E.W.; Grammatopoulos, D.K. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: Implications for physiology and pathophysiology. Endocr. Rev. 2006, 27, 260–286. [Google Scholar] [CrossRef]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30. [Google Scholar]
- Munoz, M.; Covenas, R. Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 2014, 46, 1727–1750. [Google Scholar] [CrossRef]
- Piltonen, M.; Krokhotin, A.; Parisien, M.; Berube, P.; Djambazian, H.; Sladek, R.; Dokholyan, N.V.; Shabalina, S.A.; Diatchenko, L. Alternative splicing of opioid receptor genes shows a conserved pattern for 6tm receptor variants. Cell. Mol. Neurobiol. 2021, 41, 1039–1055. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Childers, S.R.; Pan, Y.X. Emerging Insights into mu opioid pharmacology. Handb. Exp. Pharmacol. 2020, 258, 89–125. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef]
- Hannon, J.; Hoyer, D. Molecular biology of 5-HT receptors. Behav. Brain Res. 2008, 195, 198–213. [Google Scholar] [CrossRef]
- Guest, P.C.; Salim, K.; Skynner, H.A.; George, S.E.; Bresnick, J.N.; McAllister, G. Identification and characterization of a truncated variant of the 5-hydroxytryptamine(2A) receptor produced by alternative splicing. Brain Res. 2000, 876, 238–244. [Google Scholar] [CrossRef]
- Canton, H.; Emeson, R.B.; Barker, E.L.; Backstrom, J.R.; Lu, J.T.; Chang, M.S.; Sanders-Bush, E. Identification, molecular cloning, and distribution of a short variant of the 5-hydroxytryptamine2C receptor produced by alternative splicing. Mol. Pharmacol. 1996, 50, 799–807. [Google Scholar]
- Wang, Q.; O’Brien, P.J.; Chen, C.X.; Cho, D.S.; Murray, J.M.; Nishikura, K. Altered G protein-coupling functions of RNA editing isoform and splicing variant serotonin2C receptors. J. Neurochem. 2000, 74, 1290–1300. [Google Scholar] [CrossRef]
- Medhurst, A.D.; Lezoualc’h, F.; Fischmeister, R.; Middlemiss, D.N.; Sanger, G.J. Quantitative mRNA analysis of five C-terminal splice variants of the human 5-HT4 receptor in the central nervous system by TaqMan real time RT-PCR. Brain Res. Mol. Brain Res. 2001, 90, 125–134. [Google Scholar] [CrossRef]
- Coupar, I.M.; Desmond, P.V.; Irving, H.R. Human 5-HT(4) and 5-HT(7) receptor splice variants: Are they important? Curr. Neuropharmacol. 2007, 5, 224–231. [Google Scholar] [CrossRef]
- Olsen, M.A.; Nawoschik, S.P.; Schurman, B.R.; Schmitt, H.L.; Burno, M.; Smith, D.L.; Schechter, L.E. Identification of a human 5-HT6 receptor variant produced by alternative splicing. Brain Res. Mol. Brain Res. 1999, 64, 255–263. [Google Scholar] [CrossRef]
- Mahe, C.; Bernhard, M.; Bobirnac, I.; Keser, C.; Loetscher, E.; Feuerbach, D.; Dev, K.K.; Schoeffter, P. Functional expression of the serotonin 5-HT7 receptor in human glioblastoma cell lines. Br. J. Pharm. 2004, 143, 404–410. [Google Scholar] [CrossRef]
- Krobert, K.A.; Bach, T.; Syversveen, T.; Kvingedal, A.M.; Levy, F.O. The cloned human 5-HT7 receptor splice variants: A comparative characterization of their pharmacology, function and distribution. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 363, 620–632. [Google Scholar] [CrossRef]
- Mahe, C.; Loetscher, E.; Dev, K.K.; Bobirnac, I.; Otten, U.; Schoeffter, P. Serotonin 5-HT7 receptors coupled to induction of interleukin-6 in human microglial MC-3 cells. Neuropharmacology 2005, 49, 40–47. [Google Scholar] [CrossRef]
- Jasper, J.R.; Kosaka, A.; To, Z.P.; Chang, D.J.; Eglen, R.M. Cloning, expression and pharmacology of a truncated splice variant of the human 5-HT7 receptor (h5-HT7b). Br. J. Pharmacol. 1997, 122, 126–132. [Google Scholar] [CrossRef]
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–579. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Goodman, R.; Snyder, S.H. An endogenous morphine like factor in mammalian brain. Life Sci. 1975, 16, 1765–1769. [Google Scholar] [CrossRef]
- Terenius, L.; Wahlstrom, A. Search for an endogenous ligand for the opiate receptor. Acta Physiol. Scand. 1975, 94, 74–81. [Google Scholar] [CrossRef]
- Goldstein, A. Opioid peptides (endorphins) in pituitary and brain. Science 1976, 193, 1081–1086. [Google Scholar] [CrossRef]
- Birdsall, N.J.M.; Hulme, E.C. C fragment of lipotropin has a high affinity for brain opiate receptors. Nature 1976, 260, 793–795. [Google Scholar]
- Evans, C.J.; Keith, D.J.; Morrison, H.M.; Edwards, R.H. Cloning of a cDNA encoding delta opioid receptor characteristics. Soc. Neurosci. 1992, 18, 21–21. [Google Scholar]
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The δ-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mestek, A.; Liu, J.; Yu, L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the μ and δ opioid receptors. Biochem. J. 1993, 295, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Xie, G.-X.; Thompson, R.C.; Mansour, A.; Goldstein, A.; Watson, S.J.; Akil, H. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 9954–9958. [Google Scholar] [CrossRef]
- Bunzow, J.R.; Saez, C.; Mortrud, M.; Bouvier, C.; Williams, J.T.; Low, M.; Grandy, D.K. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a μ, δ or kappa opioid receptor type. FEBS Lett. 1994, 347, 284–288. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, Y.; Liu, J.; Mestek, A.; Tian, M.; Kozak, C.A.; Yu, L. Molecular cloning, tissue distribution and chromosomal localization of a novel member of the opioid receptor gene family. FEBS Lett. 1994, 347, 279–283. [Google Scholar] [CrossRef]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.C. ORL-1, a novel member of the opioid family: Cloning, functional expression and localization. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef]
- Pan, Y.X.; Cheng, J.; Xu, J.; Pasternak, G.W. Cloning, expression and classification of A Kappa(3)-related opioid receptor using antisense oligodeoxynucleotides. Regul. Pept. 1994, 54, 217–218. [Google Scholar] [CrossRef]
- Pan, Y.X.; Cheng, J.; Xu, J.; Rossi, G.; Jacobson, E.; RyanMoro, J.; Brooks, A.I.; Dean, G.E.; Standifer, K.M.; Pasternak, G.W. Cloning and functional-characterization through antisense mapping of A Kappa(3)-related opioid receptor. Mol. Pharmacol. 1995, 47, 1180–1188. [Google Scholar]
- Kest, B.; Hopkins, E.; Palmese, C.A.; Adler, M.; Mogil, J.S. Genetic variation in morphine analgesic tolerance: A survey of 11 inbred mouse strains. Pharm. Biochem. Behav. 2002, 73, 821–828. [Google Scholar] [CrossRef]
- Kest, B.; Palmese, C.A.; Hopkins, E.; Adler, M.; Juni, A.; Mogil, J.S. Naloxone-precipitated withdrawal jumping in 11 inbred mouse strains: Evidence for common genetic mechanisms in acute and chronic morphine physical dependence. Neuroscience 2002, 115, 463–469. [Google Scholar] [CrossRef]
- Lutz, R.A.; Cruciani, R.A.; Costa, T.; Munson, P.J.; Rodbard, D. A very high affinity opioid binding site in rat brain: Demonstration by computer modeling. Biochem. Biophys. Res. Commun. 1984, 122, 265–269. [Google Scholar] [CrossRef]
- Munson, P.J.; Cruciani, R.A.; Lutz, R.A.; Rodbard, D. New methods for characterization of complex receptor systems involving 3 or more biniding sites: Application to brain opiate receptors. J. Recept. Res. 1984, 4, 339–355. [Google Scholar] [CrossRef]
- Moskowitz, A.S.; Goodman, R.R. Autoradiographic distribution of μ1 and μ2 opioid binding in the mouse central nervous system. Brain Res. 1985, 360, 117–129. [Google Scholar] [CrossRef]
- Moskowitz, A.S.; Goodman, R.R. Autoradiographic analysis of mu1, mu2, and delta opioid binding in the central nervous of C57BL/6BY and CXBK (opioid receptor-deficient) mice. Brain Res. 1985, 360, 108–116. [Google Scholar] [CrossRef]
- Goodman, R.R.; Pasternak, G.W. Visualization of mu1 opiate receptors in rat brain using a computerized autoradiographic subtraction technique. Proc. Natl. Acad. Sci. USA 1985, 82, 6667–6671. [Google Scholar] [CrossRef]
- Rossi, G.C.; Brown, G.P.; Leventhal, L.; Yang, K.; Pasternak, G.W. Novel receptor mechanisms for heroin and morphine-6β -glucuronide analgesia. Neurosci. Lett. 1996, 216, 1–4. [Google Scholar] [CrossRef]
- Brown, G.P.; Yang, K.; Ouerfelli, O.; Standifer, K.M.; Byrd, D.; Pasternak, G.W. 3H-morphine-6β-glucuronide binding in brain membranes and an MOR-1-transfected cell line. J. Pharmacol. Exp. Ther. 1997, 282, 1291–1297. [Google Scholar]
- Brown, G.P.; Yang, K.; King, M.A.; Rossi, G.C.; Leventhal, L.; Chang, A.; Pasternak, G.W. 3-Methoxynaltrexone, a selective heroin/morphine-6β-glucuronide antagonist. FEBS Lett. 1997, 412, 35–38. [Google Scholar] [CrossRef]
- Rossi, G.C.; Pan, Y.-X.; Brown, G.P.; Pasternak, G.W. Antisense mapping the MOR-1 opioid receptor: Evidence for alternative splicing and a novel morphine-6β-glucuronide receptor. FEBS Lett. 1995, 369, 192–196. [Google Scholar] [CrossRef]
- Schuller, A.G.; King, M.A.; Zhang, J.; Bolan, E.; Pan, Y.X.; Morgan, D.J.; Chang, A.; Czick, M.E.; Unterwald, E.M.; Pasternak, G.W.; et al. Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat. Neurosci. 1999, 2, 151–156. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Xu, M.; Rossi, G.C.; Matulonis, J.E.; Pasternak, G.W. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proc. Natl. Acad. Sci. USA 2009, 106, 4917–4922. [Google Scholar] [CrossRef]
- Ohno, S. Gene duplication and the uniqueness of vertebrate genomes circa 1970-1999. Semin. Cell Dev. Biol. 1999, 10, 517–522. [Google Scholar] [CrossRef]
- Escriva, H.; Manzon, L.; Youson, J.; Laudet, V. Analysis of lamprey and hagfish genes reveals a complex history of gene duplications during early vertebrate evolution. Mol. Biol. Evol. 2002, 19, 1440–1450. [Google Scholar] [CrossRef]
- Lundin, L.G.; Larhammar, D.; Hallbook, F. Numerous groups of chromosomal regional paralogies strongly indicate two genome doublings at the root of the vertebrates. J. Struct. Funct. Genom. 2003, 3, 53–63. [Google Scholar] [CrossRef]
- Dreborg, S.; Sundstrom, G.; Larsson, T.A.; Larhammar, D. Evolution of vertebrate opioid receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 15487–15492. [Google Scholar] [CrossRef]
- Larhammar, D.; Dreborg, S.; Larsson, T.A.; Sundstrom, G. Early duplications of opioid receptor and Peptide genes in vertebrate evolution. Ann. N. Y. Acad. Sci. 2009, 1163, 451–453. [Google Scholar] [CrossRef]
- Stevens, C.W. The evolution of vertebrate opioid receptors. Front. Biosci. 2009, 14, 1247–1269. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-X.; Pasternak, G.W. Molecular Biology of Mu Opioid Receptors. In Opiate, 2nd ed.; Pasternak, G.W., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 121–160. [Google Scholar]
- Herrero-Turrion, M.J.; Rodríguez, R.E. Bioinformatic analysis of the origin, sequence and diversification of [mu] opioid receptors in vertebrates. Mol. Phylogenet. Evol. 2008, 49, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Kozak, C.A.; Filie, J.; Adamson, M.C.; Chen, Y.; Yu, L. Murine chromosomal location of the μ and kappa opioid receptor genes. Genomics 1994, 21, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Giros, B.; Pohl, M.; Rochelle, J.M.; Seldin, M.F. Chromosomal localization of opioid peptide and receptor genes in the mouse. Life Sci. 1995, 56, PL369–PL375. [Google Scholar] [CrossRef]
- Kaufman, D.L.; Keith, D.E., Jr.; Anton, B.; Tian, J.; Magendzo, K.; Newman, D.; Tran, T.H.; Lee, D.S.; Wen, C.; Xia, Y.-R.; et al. Characterization of the murine μ opioid receptor gene. J. Biol. Chem. 1995, 270, 15877–15883. [Google Scholar] [CrossRef]
- Wang, J.B.; Johnson, P.S.; Persico, A.M.; Hawkins, A.L.; Griffin, C.A.; Uhl, G.R. Human [mu] opiate receptor: cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett. 1994, 338, 217–222. [Google Scholar] [CrossRef]
- Min, B.H.; Augustin, L.B.; Felsheim, R.F.; Fuchs, J.A.; Loh, H.H. Genomic structure and analysis of promoter sequence of a mouse μ opioid receptor gene. Proc. Natl. Acad. Sci. USA 1994, 91, 9081–9085. [Google Scholar] [CrossRef]
- Ko, J.L.; Minnerath, S.R.; Loh, H.H. Murine chromosomal location of the μ and kappa opioid receptor genes. Biochem. Biophys. Res. Commun. 1997, 234, 351–357. [Google Scholar] [CrossRef]
- Liang, Y.; Carr, L.G. Transcription of the mouse mu-opioid receptor gene is regulated by two promoters. Brain Res. 1997, 769, 372–374. [Google Scholar] [CrossRef]
- Xu, Y.; Carr, L.G. Transcriptional regulation of the human mu opioid receptor (hMOR) gene: Evidence of positive and negative cis-acting elements in the proximal promoter and presence of a distal promoter. DNA Cell Biol. 2001, 20, 391–402. [Google Scholar] [CrossRef]
- Xu, J.; Xu, M.; Pan, Y.X. Characterizing exons 11 and 1 promoters of the mu opioid receptor (Oprm) gene in transgenic mice. BMC Mol. Biol. 2006, 7, 41. [Google Scholar] [CrossRef]
- Bare, L.A.; Mansson, E.; Yang, D. Expression of two variants of the human μ opioid receptor mRNA in SK-N-SH cells and human brain. FEBS Lett. 1994, 354, 213–216. [Google Scholar] [CrossRef]
- Zimprich, A.; Simon, T.; Hollt, V. Cloning and expression of an isoform of the rat μ opioid receptor (rMOR 1 B) which differs in agonist induced desensitization from rMOR1. FEBS Lett. 1995, 359, 142–146. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Bolan, E.; Abbadie, C.; Chang, A.; Zuckerman, A.; Rossi, G.; Pasternak, G.W. Identification and characterization of three new alternatively spliced mu-opioid receptor isoforms. Mol. Pharmacol. 1999, 56, 396–403. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Bolan, E.; Chang, A.; Mahurter, L.; Rossi, G.; Pasternak, G.W. Isolation and expression of a novel alternatively spliced mu opioid receptor isoform, MOR-1F. FEBS Lett. 2000, 466, 337–340. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Mahurter, L.; Xu, M.; Gilbert, A.K.; Pasternak, G.W. Identification and characterization of two new human mu opioid receptor splice variants, hMOR-1O and hMOR-1X. Biochem. Biophys. Res. Commun. 2003, 301, 1057–1061. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Bolan, E.; Moskowitz, H.S.; Xu, M.; Pasternak, G.W. Identification of four novel exon 5 splice variants of the mouse mu-opioid receptor gene: Functional consequences of C-terminal splicing. Mol. Pharmacol. 2005, 68, 866–875. [Google Scholar] [CrossRef]
- Pan, L.; Xu, J.; Yu, R.; Xu, M.M.; Pan, Y.X.; Pasternak, G.W. Identification and characterization of six new alternatively spliced variants of the human mu opioid receptor gene, Oprm. Neuroscience 2005, 133, 209–220. [Google Scholar] [CrossRef]
- Pasternak, D.A.; Pan, L.; Xu, J.; Yu, R.; Xu, M.M.; Pasternak, G.W.; Pan, Y.X. Identification of three new alternatively spliced variants of the rat mu opioid receptor gene: Dissociation of affinity and efficacy. J. Neurochem. 2004, 91, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Doyle, G.A.; Sheng, X.R.; Lin, S.S.; Press, D.M.; Grice, D.E.; Buono, R.J.; Ferraro, T.N.; Berrettini, W.H. Identification of five mouse mu-opioid receptor (MOR) gene (Oprm1) splice variants containing a newly identified alternatively spliced exon. Gene 2007, 395, 98–107. [Google Scholar] [CrossRef]
- Doyle, G.A.; Sheng, X.R.; Lin, S.S.; Press, D.M.; Grice, D.E.; Buono, R.J.; Ferraro, T.N.; Berrettini, W.H. Identification of three mouse micro-opioid receptor (MOR) gene (Oprm1) splice variants containing a newly identified alternatively spliced exon. Gene 2007, 388, 135–147. [Google Scholar] [CrossRef]
- Kvam, T.M.; Baar, C.; Rakvag, T.T.; Kaasa, S.; Krokan, H.E.; Skorpen, F. Genetic analysis of the murine mu opioid receptor: Increased complexity of Oprm gene splicing. J. Mol. Med. 2004, 82, 250–255. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell 2017, 170, 457–469. [Google Scholar] [CrossRef]
- Xu, J.; Xu, M.; Bolan, E.; Gilbert, A.K.; Pasternak, G.W.; Pan, Y.X. Isolating and characterizing three alternatively spliced mu opioid receptor variants: mMOR-1A, mMOR-1O, and mMOR-1P. Synapse 2014, 68, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.X.; Xu, J.; Mahurter, L.; Bolan, E.; Xu, M.; Pasternak, G.W. Generation of the mu opioid receptor (MOR-1) protein by three new splice variants of the Oprm gene. Proc. Natl. Acad. Sci. USA 2001, 98, 14084–14089. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, M.; Hurd, Y.L.; Pasternak, G.W.; Pan, Y.X. Isolation and characterization of new exon 11-associated N-terminal splice variants of the human mu opioid receptor gene. J. Neurochem. 2009, 108, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, M.; Rossi, G.C.; Pasternak, G.W.; Pan, Y.X. Identification and characterization of seven new exon 11-associated splice variants of the rat mu opioid receptor gene, OPRM1. Mol. Pain 2011, 7, 9. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Zaykin, D.V.; Gris, P.; Ogurtsov, A.Y.; Gauthier, J.; Shibata, K.; Tchivileva, I.E.; Belfer, I.; Mishra, B.; Kiselycznyk, C.; et al. Expansion of the human {micro}-opioid receptor gene architecture: Novel functional variants. Hum. Mol. Genet. 2009, 18, 1037–1051. [Google Scholar] [CrossRef]
- Stefano, G.B.; Hartman, A.; Bilfinger, T.V.; Magazine, H.I.; Liu, Y.; Casares, F.; Goligorsky, M.S. Presence of the mu3 opiate receptor in endothelial cells. Coupling to nitric oxide production and vasodilation. J. Biol. Chem. 1995, 270, 30290–30293. [Google Scholar] [CrossRef]
- Du, Y.-L.; Elliot, K.; Pan, Y.-X.; Pasternak, G.W.; Inturrisi, C.E. A splice variant of the mu opioid receptor is present in human SHSY-5Y cells. Soc. Neurosci. 1997, 23, 1206. [Google Scholar]
- Choi, H.S.; Kim, C.S.; Hwang, C.K.; Song, K.Y.; Wang, W.; Qiu, Y.; Law, P.Y.; Wei, L.N.; Loh, H.H. The opioid ligand binding of human mu-opioid receptor is modulated by novel splice variants of the receptor. Biochem. Biophys. Res. Commun. 2006, 343, 1132–1140. [Google Scholar] [CrossRef]
- Raynor, K.; Kong, H.; Mestek, A.; Bye, L.S.; Tian, M.; Liu, J.; Yu, L.; Reisine, T. Characterization of the cloned human mu opioid receptor. J. Pharmacol. Exp. Ther. 1995, 272, 423–428. [Google Scholar]
- Xu, J.; Lu, Z.; Xu, M.; Rossi, G.C.; Kest, B.; Waxman, A.R.; Pasternak, G.W.; Pan, Y.X. Differential expressions of the alternatively spliced variant mRNAs of the micro opioid receptor gene, OPRM1, in brain regions of four inbred mouse strains. PLoS ONE 2014, 9, e111267. [Google Scholar] [CrossRef]
- Xu, J.; Faskowitz, A.J.; Rossi, G.C.; Xu, M.; Lu, Z.; Pan, Y.X.; Pasternak, G.W. Stabilization of morphine tolerance with long-term dosing: Association with selective upregulation of mu-opioid receptor splice variant mRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, 279–284. [Google Scholar] [CrossRef]
- Dever, S.M.; Xu, R.; Fitting, S.; Knapp, P.E.; Hauser, K.F. Differential expression and HIV-1 regulation of mu-opioid receptor splice variants across human central nervous system cell types. J. Neurovirol. 2012, 18, 181–190. [Google Scholar] [CrossRef]
- Dever, S.M.; Costin, B.N.; Xu, R.; El-Hage, N.; Balinang, J.; Samoshkin, A.; O’Brien, M.A.; McRae, M.; Diatchenko, L.; Knapp, P.E.; et al. Differential expression of the alternatively spliced OPRM1 isoform mu-opioid receptor-1K in HIV-infected individuals. AIDS 2014, 28, 19–30. [Google Scholar] [CrossRef]
- Brown, T.G.; Xu, J.; Hurd, Y.L.; Pan, Y.X. Dysregulated expression of the alternatively spliced variant mRNAs of the mu opioid receptor gene, OPRM1, in the medial prefrontal cortex of male human heroin abusers and heroin self-administering male rats. J. Neurosci. Res. 2020. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, H.; Qin, F.; Wang, Q.; Sun, Q.; Xie, S.; Wang, Q.; Tang, Z.; Lu, Z. Sex associated differential expressions of the alternatively spliced variants mRNA of OPRM1 in brain regions of C57BL/6 mouse. Cell Physiol. Biochem. 2018, 50, 1441–1459. [Google Scholar] [CrossRef]
- Abbadie, C.; Pan, Y.X.; Drake, C.T.; Pasternak, G.W. Comparative immunohistochemical distributions of carboxy terminus epitopes from the mu-opioid receptor splice variants MOR-1D, MOR-1 and MOR-1C in the mouse and rat CNS. Neuroscience 2000, 100, 141–153. [Google Scholar] [CrossRef]
- Abbadie, C.; Pan, Y.X.; Pasternak, G.W. Differential distribution in rat brain of mu opioid receptor carboxy terminal splice variants MOR-1C-like and MOR-1-like immunoreactivity: Evidence for region-specific processing. J. Comp. Neurol. 2000, 419, 244–256. [Google Scholar] [CrossRef]
- Abbadie, C.; Pan, Y.X.; Pasternak, G.W. Immunohistochemical study of the expression of exon11-containing mu opioid receptor variants in mouse brain. Neuroscience 2004, 127, 419–430. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Y.X.; Kolesnikov, Y.; Pasternak, G.W. Immunohistochemical labeling of the mu opioid receptor carboxy terminal splice variant mMOR-1B4 in the mouse central nervous system. Brain Res. 2006, 1099, 33–43. [Google Scholar] [CrossRef]
- Schulz, S.; Schreff, M.; Koch, T.; Zimprich, A.; Gramsch, C.; Elde, R.; Hollt, V. Immunolocalization of two mu-opioid receptor isoforms (MOR1 and MOR1B) in the rat central nervous system. Neuroscience 1998, 82, 613–622. [Google Scholar] [CrossRef]
- Abbadie, C.; Pasternak, G.W.; Aicher, S.A. Presynaptic localization of the carboxy-terminus epitopes of the [mu] opioid receptor splice variants MOR-1C and MOR-1D in the superficial laminae of the rat spinal cord. Neuroscience 2001, 106, 833–842. [Google Scholar] [CrossRef]
- Bolan, E.A.; Pan, Y.X.; Pasternak, G.W. Functional analysis of MOR-1 splice variants of the mouse mu opioid receptor gene Oprm. Synapse 2004, 51, 11–18. [Google Scholar] [CrossRef]
- Law, P.Y.; Wong, Y.H.; Loh, H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 389–430. [Google Scholar] [CrossRef] [PubMed]
- Koch, T.; Schulz, S.; Schroder, H.; Wolf, R.; Raulf, E.; Hollt, V. Carboxyl-terminal splicing of the rat mu opioid receptor modulates agonist-mediated internalization and receptor resensitization. J. Biol. Chem. 1998, 273, 13652–13657. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [PubMed]
- Von Zastrow, M.; Svingos, A.; Haberstock-Debic, H.; Evans, C. Regulated endocytosis of opioid receptors: Cellular mechanisms and proposed roles in physiological adaptation to opiate drugs. Curr. Opin. Neurobiol. 2003, 13, 348–353. [Google Scholar] [CrossRef]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of mu-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef]
- Lau, E.K.; Trester-Zedlitz, M.; Trinidad, J.C.; Kotowski, S.J.; Krutchinsky, A.N.; Burlingame, A.L.; von Zastrow, M. Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci. Signal. 2011, 4, ra52. [Google Scholar] [CrossRef]
- Mann, A.; Illing, S.; Miess, E.; Schulz, S. Different mechanisms of homologous and heterologous mu-opioid receptor phosphorylation. Br. J. Pharmacol. 2015, 172, 311–316. [Google Scholar] [CrossRef]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Koch, T.; Schulz, S.; Pfeiffer, M.; Klutzny, M.; Schroder, H.; Kahl, E.; Hollt, V. C-terminal splice variants of the mouse mu-opioid receptor differ in morphine-induced internalization and receptor resensitization. J. Biol. Chem. 2001, 276, 31408–31414. [Google Scholar] [CrossRef]
- Deng, H.B.; Yu, Y.; Pak, Y.; O’Dowd, B.F.; George, S.R.; Surratt, C.K.; Uhl, G.R.; Wang, J.B. Role for the C-terminus in agonist-induced mu opioid receptor phosphorylation and desensitization. Biochemistry 2000, 39, 5492–5499. [Google Scholar] [CrossRef]
- Wang, X.F.; Barbier, E.; Chiu, Y.T.; He, Y.; Zhan, J.; Bi, G.H.; Zhang, H.Y.; Feng, B.; Liu-Chen, L.Y.; Wang, J.B.; et al. T394A Mutation at the mu Opioid receptor blocks opioid tolerance and increases vulnerability to heroin self-administration in mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 10392–10403. [Google Scholar] [CrossRef]
- Abbadie, C.; Gultekin, S.H.; Pasternak, G.W. Immunohistochemical localization of the carboxy terminus of the novel mu opioid receptor splice variant MOR-1C within the human spinal cord. Neuroreport 2000, 11, 1953–1957. [Google Scholar] [CrossRef]
- Tanowitz, M.; von Zastrow, M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J. Biol. Chem. 2003, 278, 45978–45986. [Google Scholar] [CrossRef]
- Tanowitz, M.; Hislop, J.N.; von Zastrow, M. Alternative splicing determines the post-endocytic sorting fate of G-protein-coupled receptors. J. Biol. Chem. 2008, 283, 35614–35621. [Google Scholar] [CrossRef]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorganic Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef]
- Grim, T.W.; Acevedo-Canabal, A.; Bohn, L.M. Toward directing opioid receptor signaling to refine opioid therapeutics. Biol. Psychiatry 2020, 87, 15–21. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased opioid ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Varadi, A.; Marrone, G.F.; Palmer, T.C.; Narayan, A.; Szabo, M.R.; Le Rouzic, V.; Grinnell, S.G.; Subrath, J.J.; Warner, E.; Kalra, S.; et al. Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit beta-arrestin-2. J. Med. Chem. 2016, 59, 8381–8397. [Google Scholar] [CrossRef]
- Uprety, R.; Che, T.; Zaidi, S.A.; Grinnell, S.G.; Varga, B.R.; Faouzi, A.; Slocum, S.T.; Allaoa, A.; Varadi, A.; Nelson, M.; et al. Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site. Elife 2021, 10, e56519. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.; Hunkele, A.; Xu, J.; Bassoni, D.L.; Pasternak, G.W.; Pan, Y.X. Mu opioids induce biased signaling at the full-length seven transmembrane c-terminal splice variants of the mu opioid receptor gene, Oprm1. Cell. Mol. Neurobiol. 2020, 41, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, J.C.; Loach, A.B.; Carr, D.B. Itching after epidural and spinal opiates. Pain 1988, 33, 149–160. [Google Scholar] [CrossRef]
- Chaney, M.A. Side effects of intrathecal and epidural opioids. Can. J. Anaesth. 1995, 42, 891–903. [Google Scholar] [CrossRef]
- Hales, P. Pruritus after epidural morphine. Lancet 1980, 2, 204. [Google Scholar] [CrossRef]
- Liu, X.Y.; Liu, Z.C.; Sun, Y.G.; Ross, M.; Kim, S.; Tsai, F.F.; Li, Q.F.; Jeffry, J.; Kim, J.Y.; Loh, H.H.; et al. Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell 2011, 147, 447–458. [Google Scholar] [CrossRef]
- Liu, X.Y.; Ginosar, Y.; Yazdi, J.; Hincker, A.; Chen, Z.F. Cross-talk between human spinal cord mu-opioid receptor 1Y isoform and gastrin-releasing peptide receptor mediates opioid-induced scratching behavior. Anesthesiology 2019, 131, 381–391. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 10494–10500. [Google Scholar] [CrossRef]
- Raehal, K.M.; Bohn, L.M. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 2011, 60, 58–65. [Google Scholar] [CrossRef]
- Raehal, K.M.; Walker, J.K.; Bohn, L.M. Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Sotnikova, T.D.; Medvedev, I.O.; Lefkowitz, R.J.; Dykstra, L.A.; Caron, M.G. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J. Neurosci. 2003, 23, 10265–10273. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Majumdar, S.; Grinnell, S.; Le Rouzic, V.; Burgman, M.; Polikar, L.; Ansonoff, M.; Pintar, J.; Pan, Y.X.; Pasternak, G.W. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc. Natl. Acad. Sci. USA 2011, 108, 19778–19783. [Google Scholar] [CrossRef]
- Majumdar, S.; Subrath, J.; Le Rouzic, V.; Polikar, L.; Burgman, M.; Nagakura, K.; Ocampo, J.; Haselton, N.; Pasternak, A.R.; Grinnell, S.; et al. Synthesis and Evaluation of aryl-naloxamide opiate analgesics targeting truncated exon 11-associated mu opioid receptor (MOR-1) splice variants. J. Med. Chem. 2012, 55, 6352–6362. [Google Scholar] [CrossRef]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, J.; Rossi, G.C.; Majumdar, S.; Pasternak, G.W.; Pan, Y.X. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J. Clin. Investig. 2015, 125, 2626–2630. [Google Scholar] [CrossRef]
- Grinnell, S.G.; Uprety, R.; Varadi, A.; Subrath, J.; Hunkele, A.; Pan, Y.X.; Pasternak, G.W.; Majumdar, S. Synthesis and characterization of azido aryl analogs of IBNtxA for Radio-photoaffinity labeling opioid receptors in cell lines and in mouse brain. Cell Mol. Neurobiol. 2021, 41, 977–993. [Google Scholar] [CrossRef]
- Pickett, J.E.; Varadi, A.; Palmer, T.C.; Grinnell, S.G.; Schrock, J.M.; Pasternak, G.W.; Karimov, R.R.; Majumdar, S. Mild, Pd-catalyzed stannylation of radioiodination targets. Bioorg. Med. Chem. Lett. 2015, 25, 1761–1764. [Google Scholar] [CrossRef]
- Wieskopf, J.S.; Pan, Y.X.; Marcovitz, J.; Tuttle, A.H.; Majumdar, S.; Pidakala, J.; Pasternak, G.W.; Mogil, J.S. Broad-spectrum analgesic efficacy of IBNtxA is mediated by exon 11-associated splice variants of the mu-opioid receptor gene. Pain 2014, 155, 2063–2070. [Google Scholar] [CrossRef]
- Marrone, G.F.; Grinnell, S.G.; Lu, Z.; Rossi, G.C.; Le Rouzic, V.; Xu, J.; Majumdar, S.; Pan, Y.X.; Pasternak, G.W. Truncated mu opioid GPCR variant involvement in opioid-dependent and opioid-independent pain modulatory systems within the CNS. Proc. Natl. Acad. Sci. USA 2016, 113, 3663–3668. [Google Scholar] [CrossRef] [PubMed]
- Samoshkin, A.; Convertino, M.; Viet, C.T.; Wieskopf, J.S.; Kambur, O.; Marcovitz, J.; Patel, P.; Stone, L.S.; Kalso, E.; Mogil, J.S.; et al. Structural and functional interactions between six-transmembrane mu-opioid receptors and beta2-adrenoreceptors modulate opioid signaling. Sci. Rep. 2015, 5, 18198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, J.; Pan, Y.X. A truncated six transmembrane splice variant MOR-1G enhances expression of the full-length seven transmembrane mu-opioid receptor through heterodimerization. Mol. Pharmacol. 2020, 98, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.F.; Le Rouzic, V.; Varadi, A.; Xu, J.; Rajadhyaksha, A.M.; Majumdar, S.; Pan, Y.X.; Pasternak, G.W. Genetic dissociation of morphine analgesia from hyperalgesia in mice. Psychopharmacology 2017, 234, 1891–1900. [Google Scholar] [CrossRef]
- Juni, A.; Klein, G.; Pintar, J.E.; Kest, B. Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 2007, 147, 439–444. [Google Scholar] [CrossRef]
- Oladosu, F.A.; Conrad, M.S.; O’Buckley, S.C.; Rashid, N.U.; Slade, G.D.; Nackley, A.G. Mu opioid splice variant MOR-1K contributes to the development of opioid-induced hyperalgesia. PLoS ONE 2015, 10, e0135711. [Google Scholar] [CrossRef]
- He, S.Q.; Zhang, Z.N.; Guan, J.S.; Liu, H.R.; Zhao, B.; Wang, H.B.; Li, Q.; Yang, H.; Luo, J.; Li, Z.Y.; et al. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron 2011, 69, 120–131. [Google Scholar] [CrossRef]
- Xu, J.; Lu, Z.; Xu, M.; Pan, L.; Deng, Y.; Xie, X.; Liu, H.; Ding, S.; Hurd, Y.L.; Pasternak, G.W.; et al. A heroin addiction severity-associated intronic single nucleotide polymorphism modulates alternative pre-mRNA splicing of the mu opioid receptor gene OPRM1 via hnRNPH interactions. J. Neurosci. 2014, 34, 11048–11066. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPCR Receptors | Gene Symbol | Receptor Name (Number of Variants) | Agonist | Antagonist | Tissue Distribution | Reference |
|---|---|---|---|---|---|---|
| Adrenergic Receptors | ADRA1A | α1A-AR C-term variant (4) 6-TM variant (11) | Dabuzalgron | Amitriptyline Promethazine Nortriptyline | Mainly liver, heart, and brain | [59,60,61,62,63] |
| ADRA1B | α1B-AR: 6-TM variant (1) | Phenylephrine | Conopeptide p-TIA | Brain | [64] | |
| Cannabinoid Receptors | CNR1 | CB1 N-term variant (2) | THC 2-AG ACEA | TM38837 VD60 | Mainly brain, DRG, and kidney | [65,66,67] |
| CNR2 | CB2 N-term variant (2) | THC 2-AG Nabilone | SR144528 AM-630 | Mainly testis and spleen | [68] | |
| Corticotropin-Releasing Hormone Receptor | CRHR1 | CRH-R1 IL variant (1) C-term variant (1) N-term variant (2) | Stressin I Ovine CRH | Antalarmin | Mainly brain | [69] |
| CRHR2 | CRH-R2 N-term variant (3) | Urocortin II Urocortin III | K41,498 | Mainly brain | [69] | |
| Tachykinin Receptor | TACR1 | NK-1R C-term variant (2) | GR73632 | CP96345 | Mainly brain, GI tract, and lung | [70,71] |
| Opioid Receptors | OPRD1 | DOR-1 6TM variant (4) 1TM variant (3) | DPDPE Enkephalin Deltorphin SNC-80 | Naltrindole | Mainly brain | [72] |
| OPRK1 | KOR-1 N-term variant (5) 6TM variant (2) 1TM variant (1) | U50,488H U69,593 Nalfurafine Dynorphins | nor-BNI DIPPA JDTic | Mainly brain and prostate | [72] | |
| OPRL1 | ORL-1/KOR-3 N-term variant (18) 6TM variant (3) 1TM variant (1) | SR-8993 Ro65-6570 N/OFQ | J-113,397 JTC-801 | Mainly brain and blood | [72] | |
| OPRM1 | MOR-1 C-term variant (12) N-term variant (1) 6TM variant (4) 1TM variant (4) | DAMGO Morphine Fentanyl Methadone Β-Endorphin | β-FNA Naloxone Naltrexone Cyprodime CTAP | Mainly brain | [11,12,73] | |
| Prostaglandin E Receptors | PTGER3 | EP3 C-term variant (6) | SC-46275 ONO-AE-248 | DG-041 | Mainly kidney, uterus, and stomach | [74] |
| Serotonin Receptors | HTR2A | 5-HT2A 6-TM variant (1) | TCB-2 | Volinanserin | Mainly brain | [75,76] |
| HTR2C | 5-HT2C C-term variant (1) 6-TM variant (1) | Loreaserin | Agomelatine | Mainly brain | [77,78] | |
| HTR4 | 5-HT4 C-term variant (9) EL variant (1) | Mosapride Prucalopride Velusetrag | SDZ 205–557 SB-207266 GR113808 | Mainly intestine and brain | [79,80] | |
| HTR6 | 5-HT6 6-TM variant (1) | WAY-181187 EMD-386088 E-6801 | Ro 04-6790 SB271046 | Mainly brain | [81] | |
| HTR7 | 5-HT7 C-term variant (3) | AS-19 LP-12 E-55888 | SB-269970 SB-258719 JNJ-18038683 | Mainly brain, heart, and GI tract | [82,83,84,85] |
| Species | Variant | Exons | Amino Acid Sequence Downstream of Exon3 |
|---|---|---|---|
| Mouse | mMOR-1 | 4 | LENLEAETAPLP |
| mMOR-1A | 3b | VCAF | |
| mMOR-1B1 | 5a | KIDLF | |
| mMOR-1B2 | 5ab | KLLMWRAMPTFKRHLAIMLSLDN | |
| mMOR-1B3 | 5abc | TSLTLQ | |
| mMOR-1B4 | 5abcd | AHQKPQECLKCRCLSLTILVICLHFQHQQFFIMIKKNVS | |
| mMOR-1B5 | 5abcde | CV | |
| mMOR-1C | 7/8/9 | PTLAVSVAQIFTGYPSPTHVEKPCKSCMDRGMRNLLPDDGPRQESGEGQLGR | |
| mMOR-1D | 8/9 | RNEEPSS | |
| mMOR-1E/Eii/Eiiii/Eiv | 6/7/8/9 | KKKLDSQRGCVQHPV | |
| mMOR-1F | 10/6/7/8/9 | APCACVPGANRGQTKASDLLDLELETVGSHQADAETNPGPYEGSKCAEPLAISLVPLY | |
| mMOR-1H/-1i/-1J | 4 | LENLEAETAPLP | |
| mMOR-1O | 7ab | PTLAVSVAQIFTGYPSPTHVEKPCKSCMDR | |
| mMOR-1P | 15 | IMKFEAIYPKLSFKSWALKYFTFIREKKRNTKAGALPPLPTCHAGSPSQAHRGVAAWLLPLRHMGPSYPS | |
| mMOR-1V/-1Vii | 18/6/7/8/9 | KQEKTKTKSAWEIWEQKEHTLLLGETHLTIQHLS | |
| mMOR-1U | 7/19/8/9 | PTLAVSVAQIFTGYPSPTHVEKPCKSCMDSVDCYNRKQQTGSLRKNKKKKKRRKNKQNILEAGISRGMRNLLPDDGPRQESGEGQLGR | |
| mMOR-1W(D2) | 18/6/7/8/9 | LAFGCCNEHHDQR | |
| mMOR-1T(D2) | 4 | LENLEAETAPLP | |
| Human | hMOR-1 | 4 | LENLEAETAPLP |
| hMOR-1A | 3b | VRSL | |
| hMOR-1B1 | 5a | KIDLFQKSSLLNCE | |
| hMOR-1B2 | 5ab | RERRQKSDW | |
| hMOR-1B3 | 5abc | GPPAKFVADQLAG | |
| hMOR-1B4 | 5abcd | S | |
| hMOR-1B5 | 5abcde | VELNLDCHCENAKPWPLSYNAG | |
| hMOR-1O | O | PPLAVSMAQIFTRYSPPTHREKTCNDYMKR | |
| hMOR-1X | X | CLPIPSLSCWALEHGCLVVYPGPLQGPLVRYDLPAILHSSCLRGNTAPSPSGGAFLLS | |
| hMOR-1Y | Y/5abc | IRDPISNLPRVSVF | |
| hMOR-1i | 4 | LENLEAETAPLP | |
| Rat | rMOR-1 | 4 | LENLEAETAPLP |
| rMOR-1A | 3b | VCAF | |
| rMOR-1B1 | 5a | KIDLF | |
| rMOR-1B2 | 5ab | EPQSVET | |
| rMOR-1C1 | 5abc | PALAVSVAQIFTGYPSPTHGEKPCKSYRDRPRPCGRTWSLKSRAESNVENHFHCGAALIYNNVNFI | |
| rMOR-1C2 | 5abcd | PALAVSVAQIFTGYPSPTHGEKPCKSYRDRPRPCGRTWSLKSRAESNVENHFHCGAALIYNNELKIGPVSWLQMPAHVLVRPW | |
| rMOR-1D | 5abcde | T | |
| rMOR-1H1/H2/i1/i2/i3 | 4 | LENLEAETAPLP | |
| rMOR-1P | 15 | GAEL |
| Strain | Brain Region | All 7TM Variants (mE1-2) | All 6TM Variants | All 1TM Variants | |||
|---|---|---|---|---|---|---|---|
| Expression (E-ΔC(t)) | % of all 7TM Variants | Expression (E-ΔC(t)) | % of All 7TM Variants | Expression (E-ΔC(t)) | % of All 7TM Variants | ||
| 129P3 | Pfc | 0.012789 | 100 | 0.000284 | 3.0 | 0.000406 | 4.2 |
| Str | 0.027871 | 100 | 0.001179 | 2.2 | 0.002181 | 4.0 | |
| Tha | 0.026575 | 100 | 0.001264 | 2.4 | 0.001814 | 3.4 | |
| Hyp | 0.036192 | 100 | 0.001170 | 2.6 | 0.002670 | 6.0 | |
| Hip | 0.021809 | 100 | 0.000873 | 3.2 | 0.001654 | 6.1 | |
| PAG | 0.020924 | 100 | 0.001090 | 2.4 | 0.001510 | 3.3 | |
| BS | 0.016948 | 100 | 0.001291 | 4.1 | 0.001646 | 5.2 | |
| Cb | 0.002093 | 100 | 0.000057 | 5.0 | 0.000175 | 15.5 | |
| Spc | 0.009637 | 100 | 0.001971 | 11.9 | 0.001239 | 7.4 | |
| WB | 0.008726 | 100 | 0.000647 | 13.5 | 0.001634 | 34.1 | |
| C57Bl/6J | Pfc | 0.003384 | 100 | 0.000215 | 7.6 | 0.000206 | 7.3 |
| Str | 0.008820 | 100 | 0.000327 | 3.2 | 0.000603 | 5.9 | |
| Tha | 0.029864 | 100 | 0.003211 | 8.0 | 0.002239 | 5.5 | |
| Hyp | 0.011862 | 100 | 0.000978 | 4.6 | 0.001090 | 5.1 | |
| Hip | 0.011638 | 100 | 0.000491 | 3.7 | 0.000720 | 5.4 | |
| PAG | 0.029910 | 100 | 0.013060 | 20.9 | 0.003317 | 5.3 | |
| BS | 0.012993 | 100 | 0.000670 | 3.9 | 0.001145 | 6.7 | |
| Cb | 0.003403 | 100 | 0.000078 | 6.2 | 0.000187 | 14.8 | |
| Spc | 0.009516 | 100 | 0.001087 | 7.5 | 0.001087 | 7.5 | |
| WB | 0.008982 | 100 | 0.000477 | 9.0 | 0.000416 | 7.8 | |
| SJL/J | Pfc | 0.006287 | 100 | 0.000141 | 4.9 | 0.000375 | 13.0 |
| Str | 0.011500 | 100 | 0.000568 | 6.7 | 0.000518 | 6.1 | |
| Tha | 0.022085 | 100 | 0.000645 | 2.4 | 0.001472 | 5.4 | |
| Hyp | 0.032938 | 100 | 0.002588 | 6.2 | 0.002166 | 5.2 | |
| Hip | 0.016593 | 100 | 0.000701 | 5.6 | 0.001084 | 8.7 | |
| PAG | 0.017955 | 100 | 0.000748 | 3.0 | 0.000869 | 3.5 | |
| BS | 0.029092 | 100 | 0.001523 | 4.4 | 0.001917 | 5.5 | |
| Cb | 0.003475 | 100 | 0.000244 | 17.7 | 0.000253 | 18.3 | |
| Spc | 0.030211 | 100 | 0.002908 | 5.9 | 0.002011 | 4.1 | |
| WB | 0.010318 | 100 | 0.000371 | 2.7 | 0.001296 | 9.3 | |
| SWR/J | Pfc | 0.042361 | 100 | 0.000371 | 4.1 | 0.000681 | 7.5 |
| Str | 0.012889 | 100 | 0.000384 | 2.2 | 0.001083 | 6.3 | |
| Tha | 0.010931 | 100 | 0.000280 | 1.5 | 0.000962 | 5.0 | |
| Hyp | 0.030392 | 100 | 0.001608 | 2.2 | 0.003108 | 4.2 | |
| Hip | 0.010829 | 100 | 0.000232 | 2.2 | 0.000661 | 6.3 | |
| PAG | 0.026677 | 100 | 0.000858 | 1.5 | 0.001792 | 3.2 | |
| BS | 0.020672 | 100 | 0.001450 | 4.0 | 0.001437 | 4.0 | |
| Cb | 0.014594 | 100 | 0.000598 | 9.8 | 0.000583 | 9.6 | |
| Spc | 0.031986 | 100 | 0.002458 | 2.8 | 0.003501 | 4.0 | |
| WB | 0.018681 | 100 | 0.000599 | 2.4 | 0.001033 | 4.2 | |
| KO Mouse Model | Variant | ||
|---|---|---|---|
| 7TM | 6TM | 1TM | |
| E1 KO | Lost | Retained | Lost |
| E11 KO | Retained | Lost | Retained |
| E1/E11 KO | Lost | Lost | Lost |
| Triple KO (E1+DOR-1+KOR-1) | Lost | Retained | Lost |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Kang, W.-J.; Abrimian, A.; Xu, J.; Cartegni, L.; Majumdar, S.; Hesketh, P.; Bekker, A.; Pan, Y.-X. Alternative Pre-mRNA Splicing of the Mu Opioid Receptor Gene, OPRM1: Insight into Complex Mu Opioid Actions. Biomolecules 2021, 11, 1525. https://doi.org/10.3390/biom11101525
Liu S, Kang W-J, Abrimian A, Xu J, Cartegni L, Majumdar S, Hesketh P, Bekker A, Pan Y-X. Alternative Pre-mRNA Splicing of the Mu Opioid Receptor Gene, OPRM1: Insight into Complex Mu Opioid Actions. Biomolecules. 2021; 11(10):1525. https://doi.org/10.3390/biom11101525
Chicago/Turabian StyleLiu, Shan, Wen-Jia Kang, Anna Abrimian, Jin Xu, Luca Cartegni, Susruta Majumdar, Patrick Hesketh, Alex Bekker, and Ying-Xian Pan. 2021. "Alternative Pre-mRNA Splicing of the Mu Opioid Receptor Gene, OPRM1: Insight into Complex Mu Opioid Actions" Biomolecules 11, no. 10: 1525. https://doi.org/10.3390/biom11101525
APA StyleLiu, S., Kang, W.-J., Abrimian, A., Xu, J., Cartegni, L., Majumdar, S., Hesketh, P., Bekker, A., & Pan, Y.-X. (2021). Alternative Pre-mRNA Splicing of the Mu Opioid Receptor Gene, OPRM1: Insight into Complex Mu Opioid Actions. Biomolecules, 11(10), 1525. https://doi.org/10.3390/biom11101525