
Mevalonate Kinase Deficiency and Squalene Synthase Inhibitor (TAK-475): The Balance to Extinguish the Inflammation


 ,
,  ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
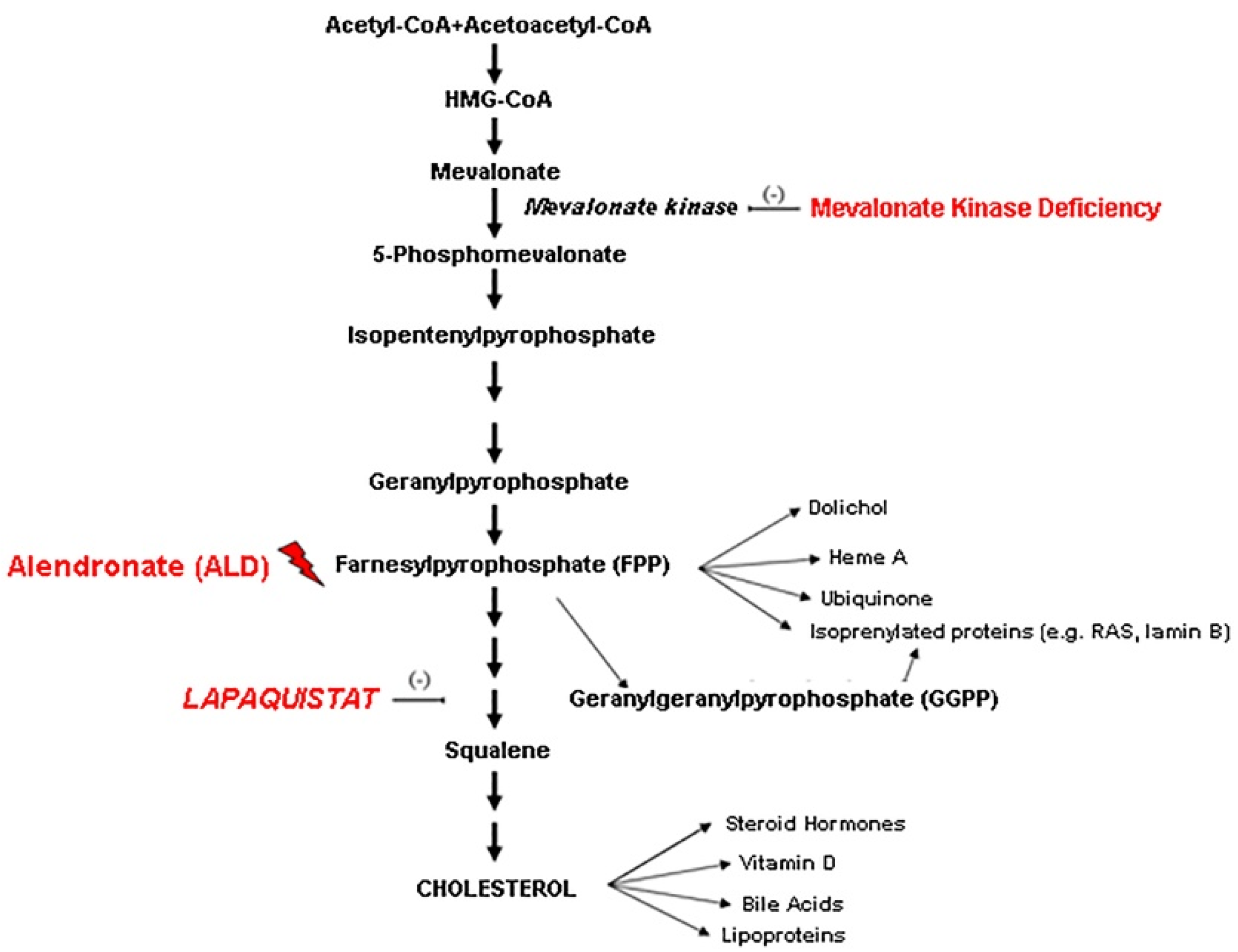
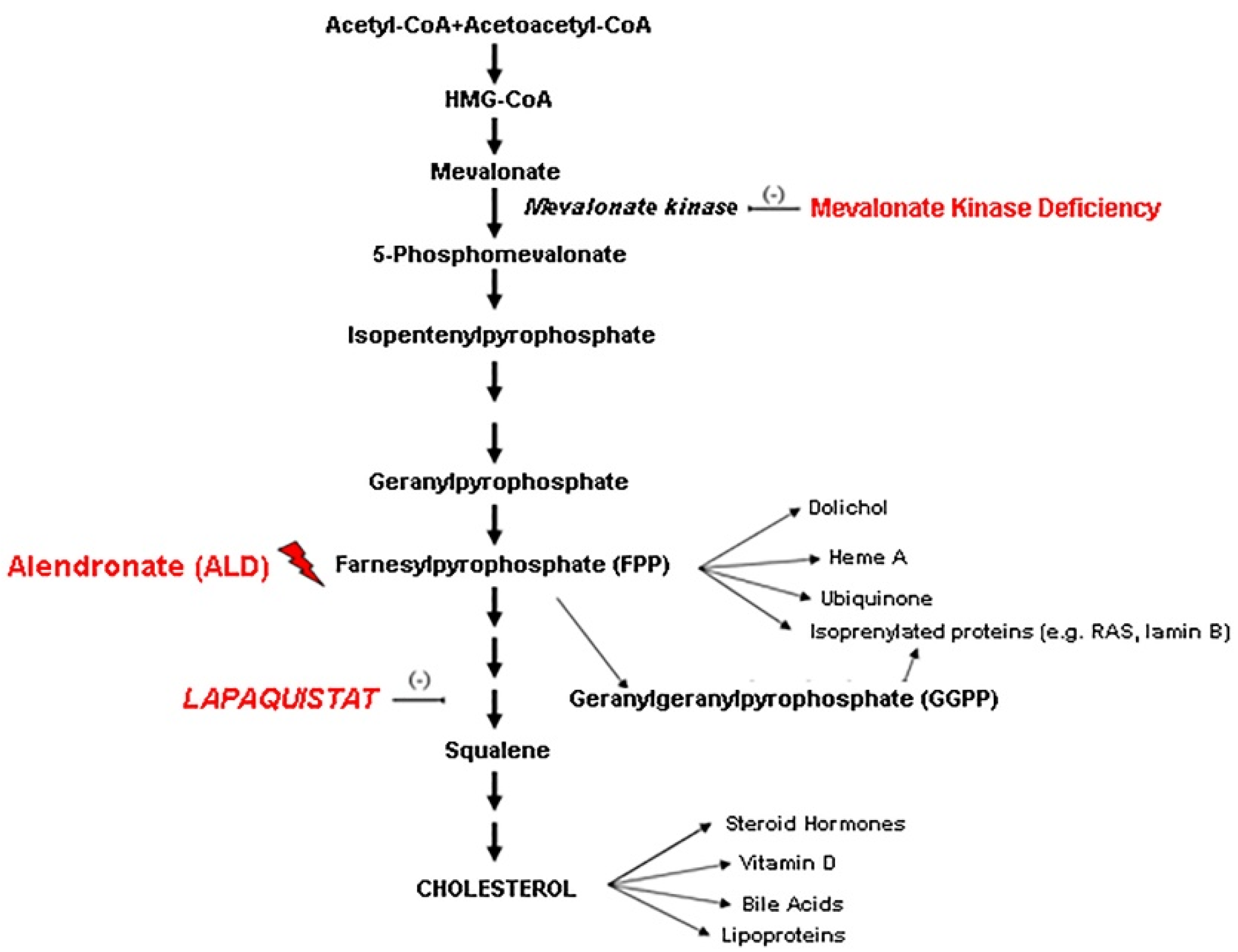
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Flow Cytometric Assessment of Apoptosis
2.4. The xCELLigence System and Impedance Measurement
2.5. Measurement of Cytokines in Cell Culture Supernatants
2.6. Transmission Electron Microscopy
2.7. Western Blot Analyses
2.8. Data Analysis
3. Results
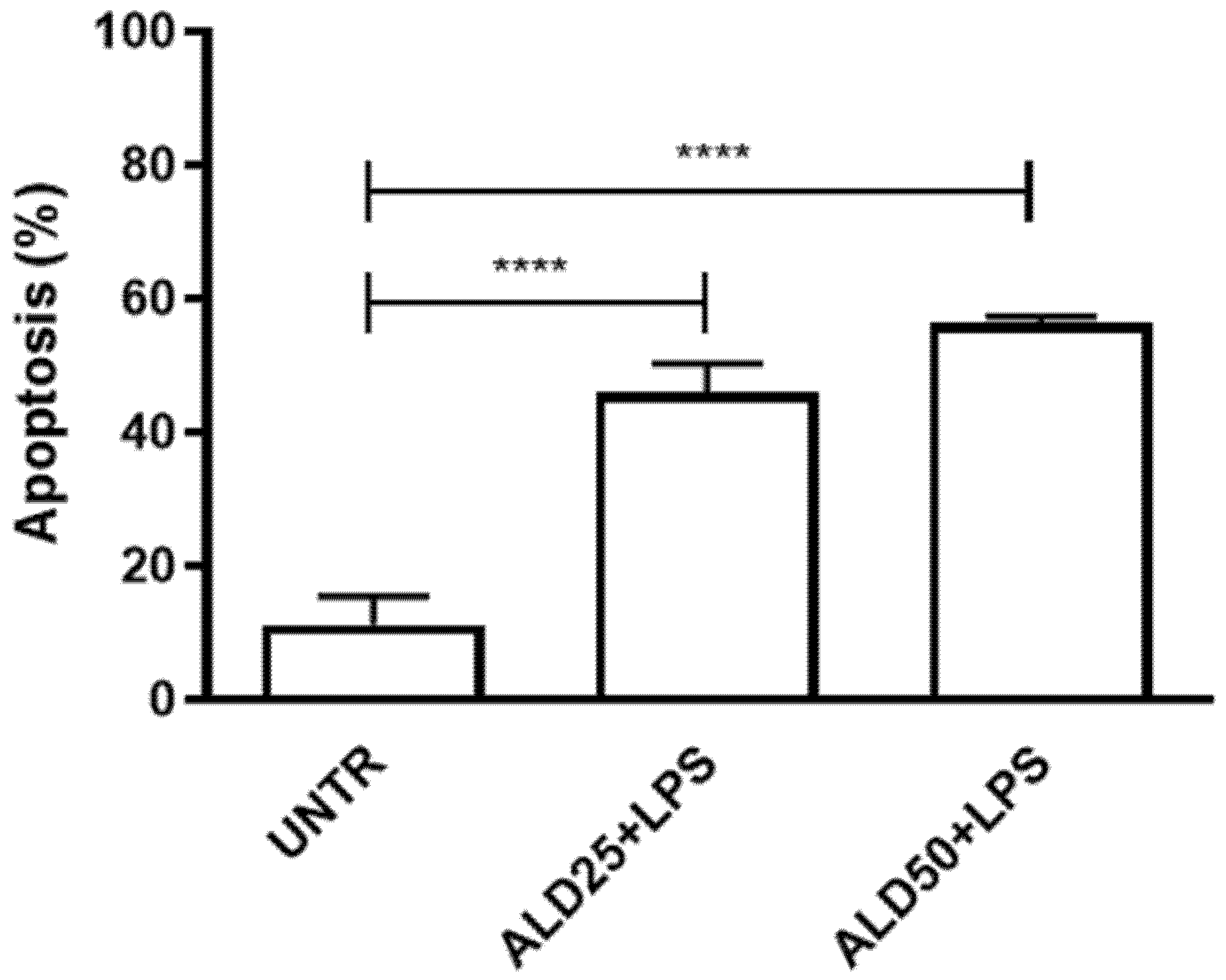
3.1. Alendronate Plus LPS Induced Apoptosis on RAW 264.7 Cells
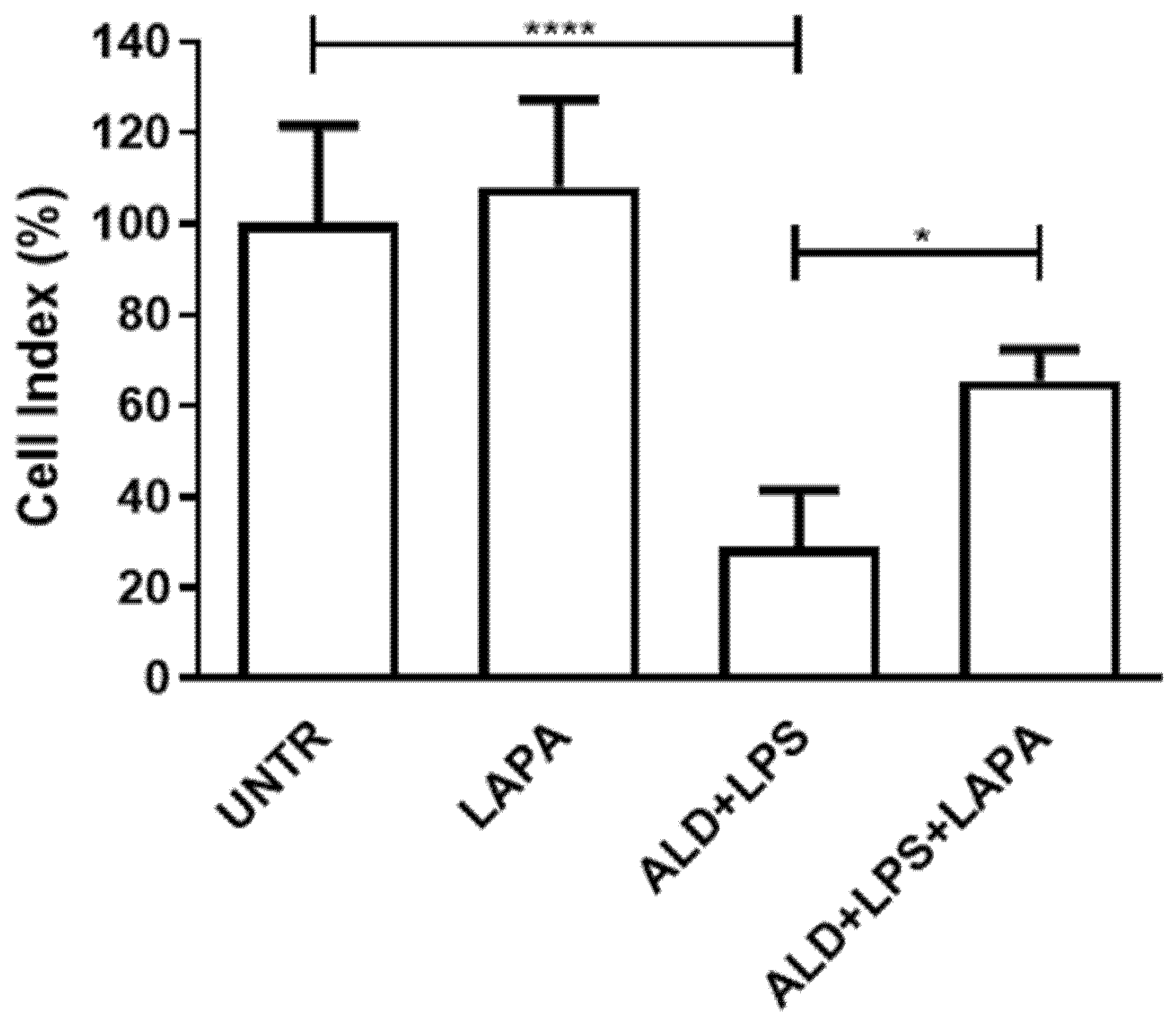
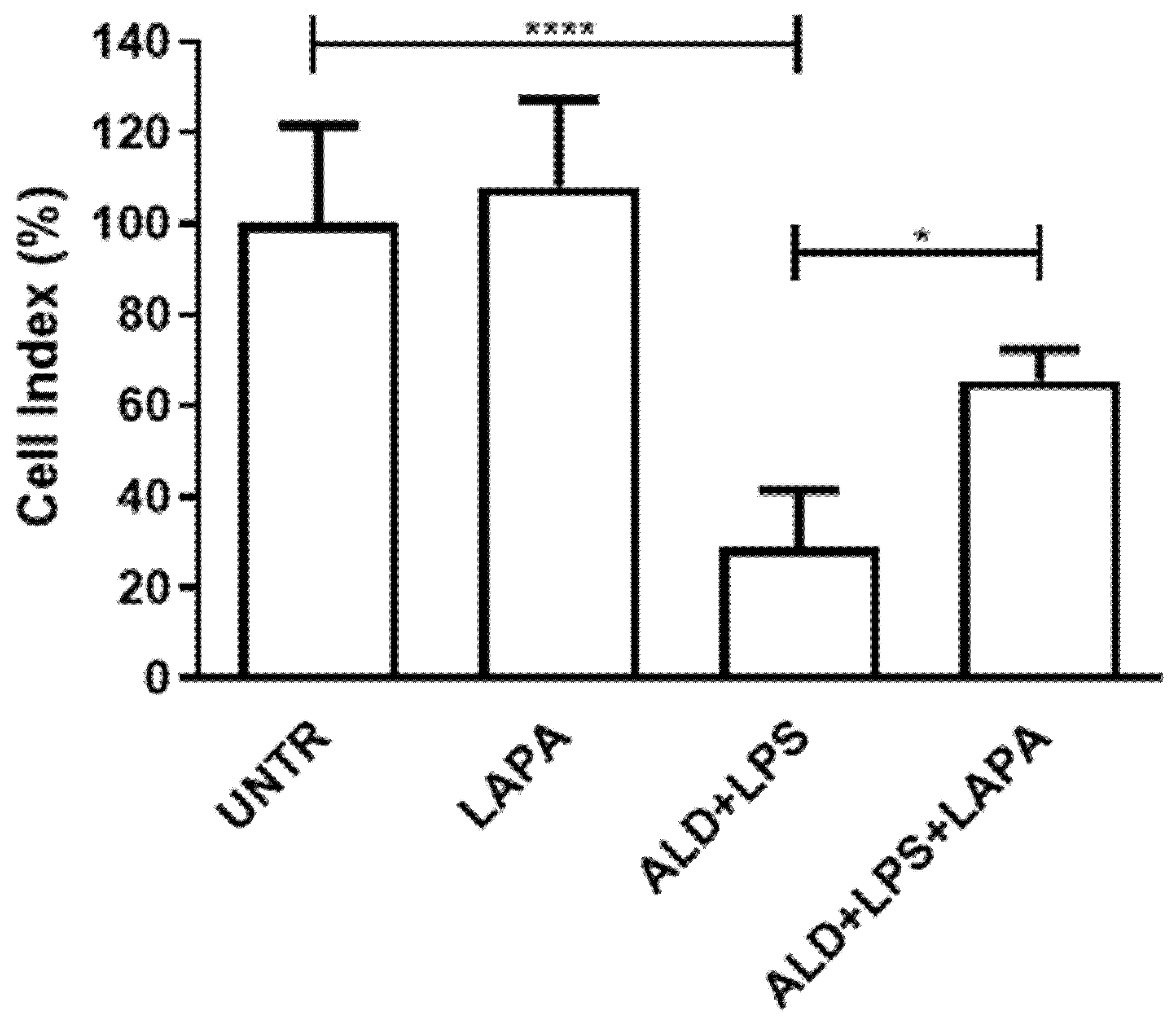
3.2. Lapaquistat Counteracted the Cytotoxic Effect of Alendronate Plus LPS
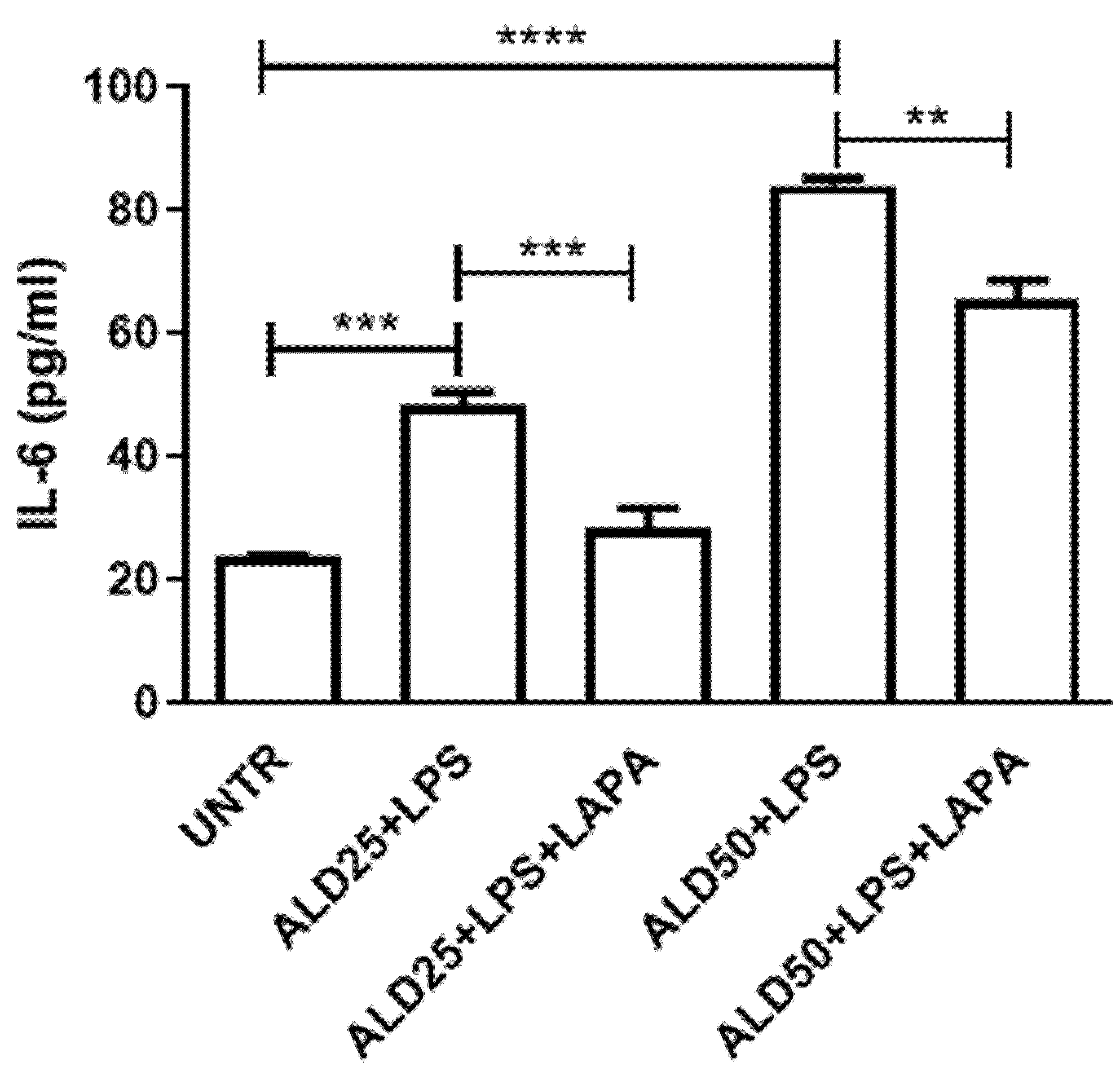
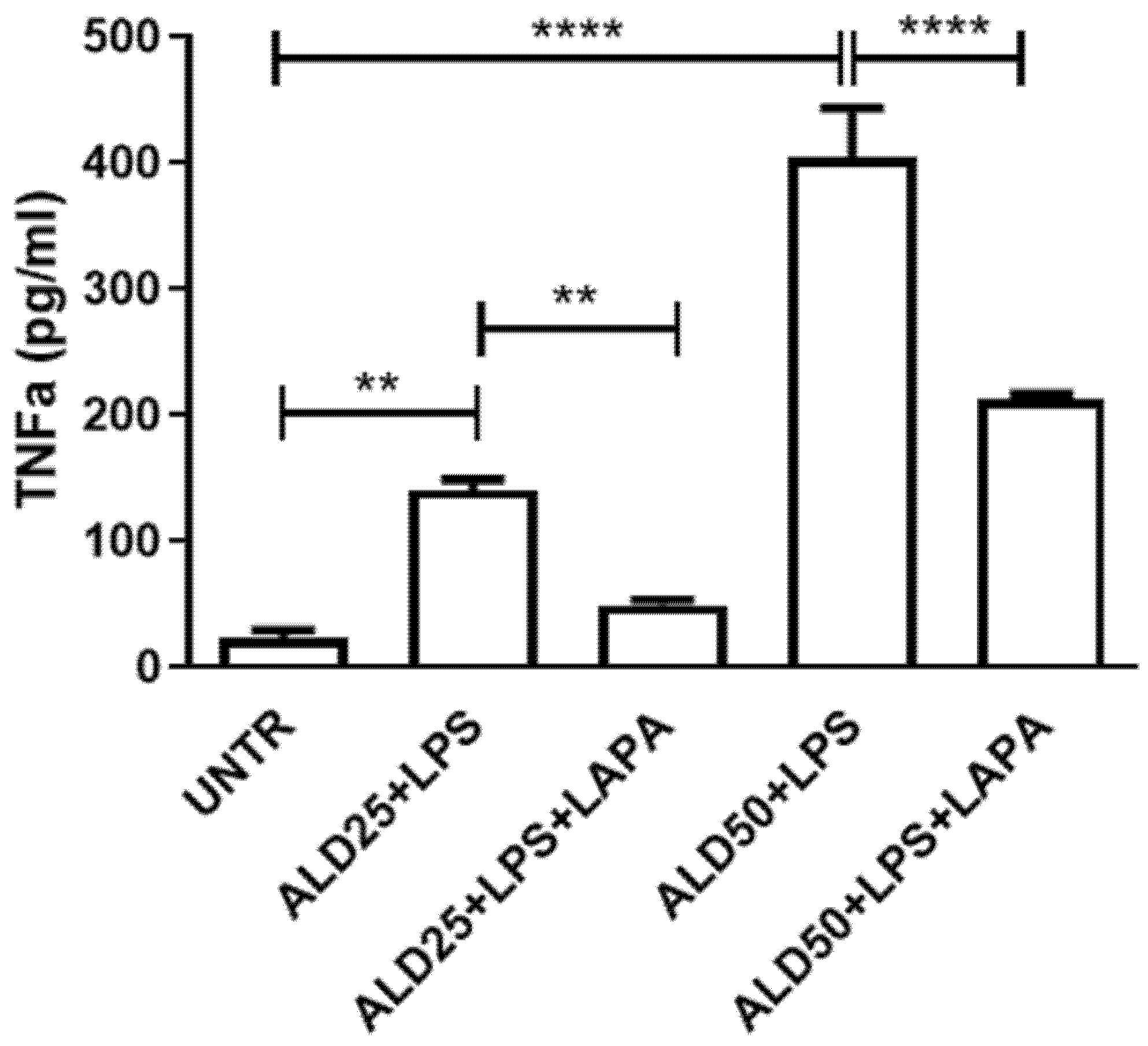
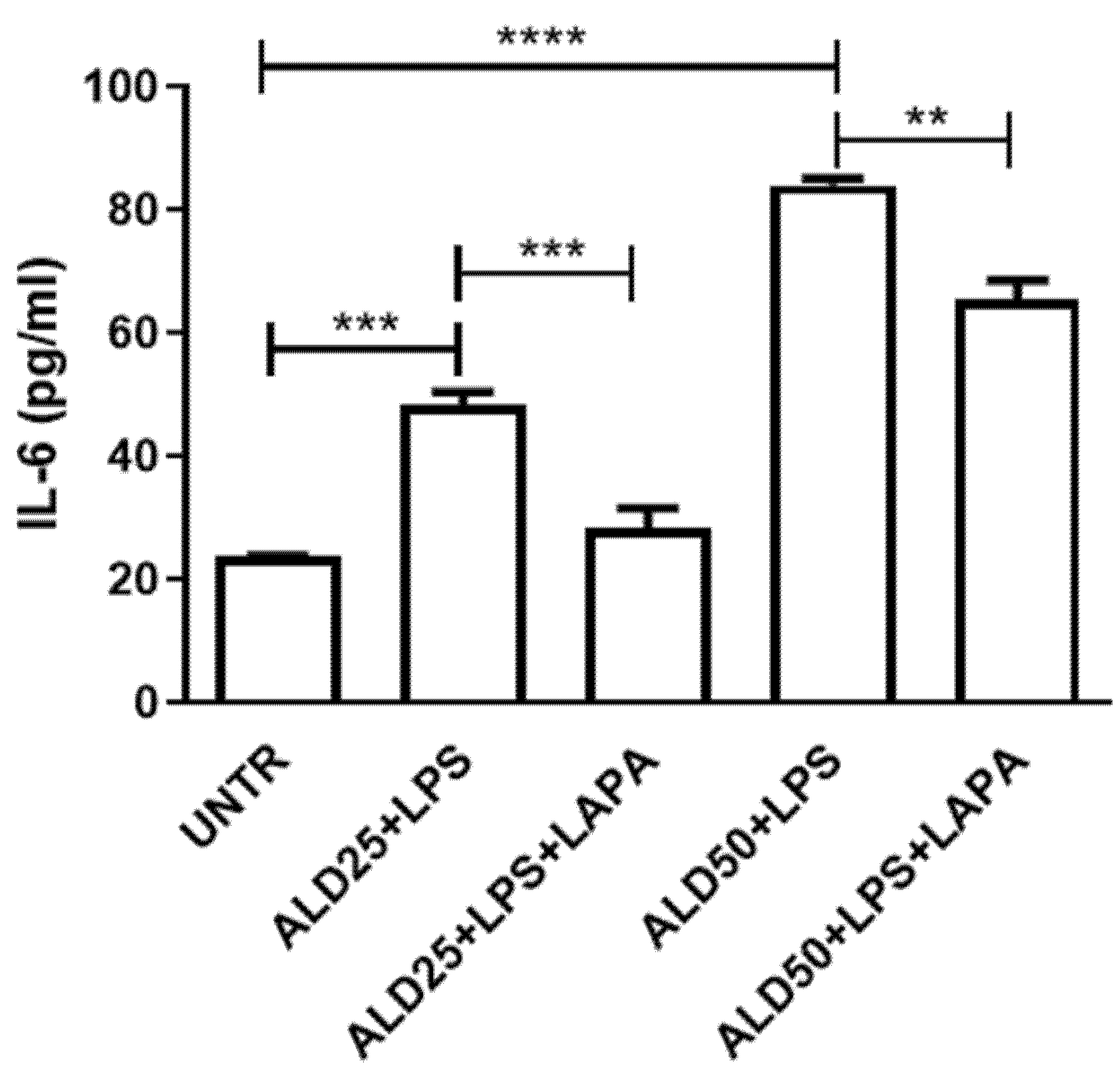
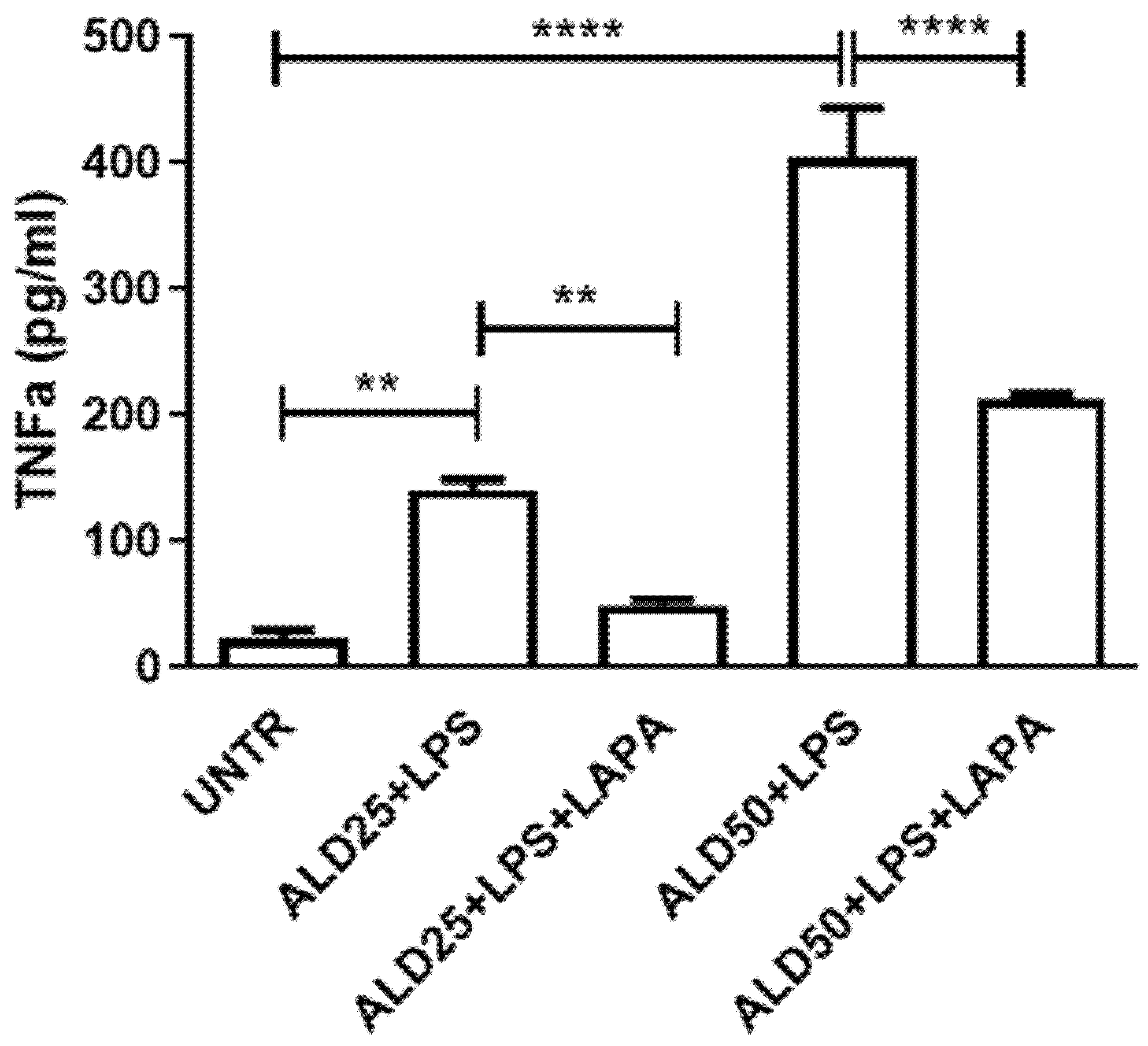
3.3. Anti-Inflammatory Effect of Lapaquistat in MKD Disease-Mimicking Conditions
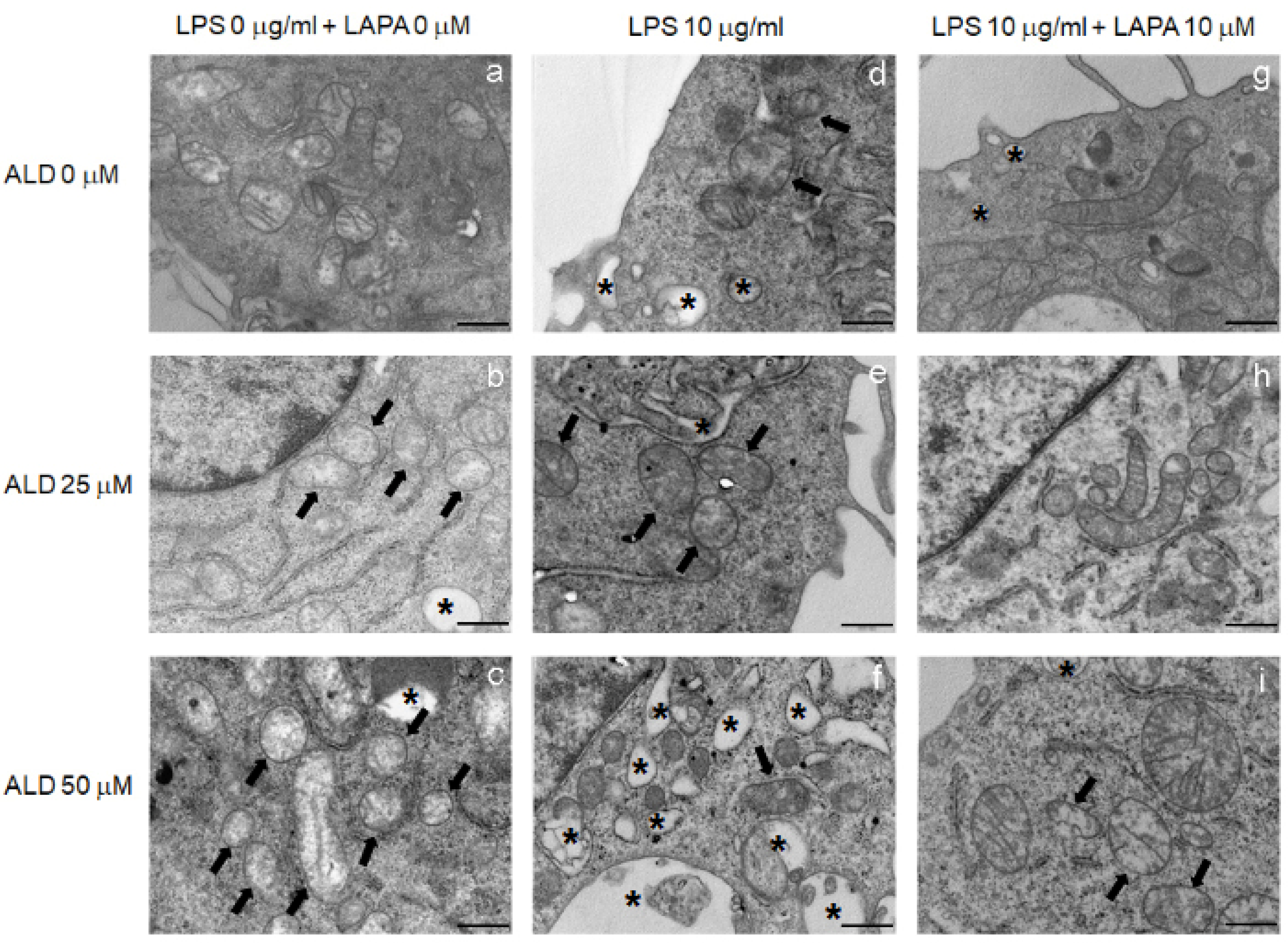
3.4. Lapaquistat Counteracted the Mitochondrial Damage Induced by LPS and Alendronate
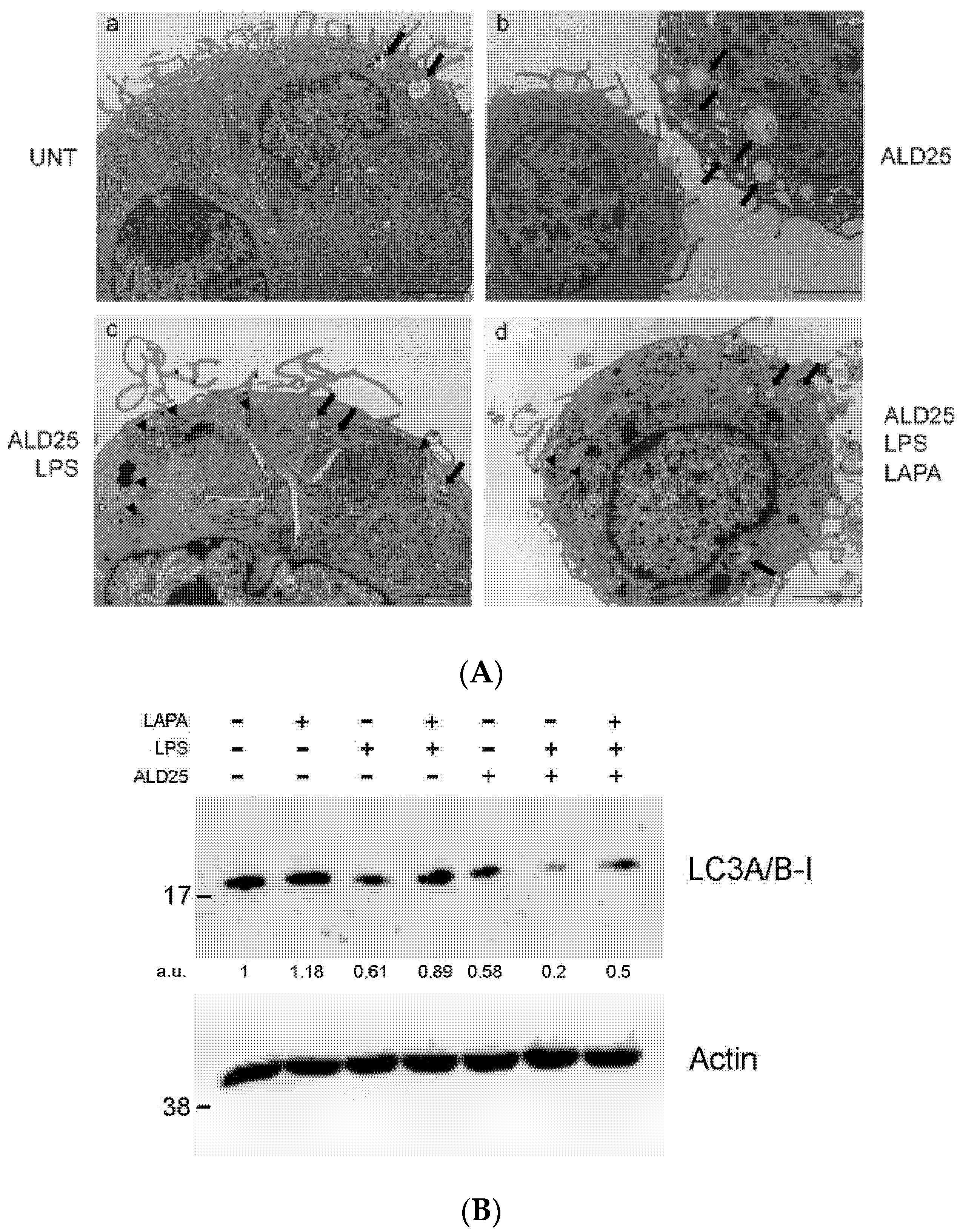
3.5. Lapaquistat Inhibits Autophagy in RAW 264.7 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.P.; Cuisset, L.; Grateau, G.; Vasseur, C.; van de Velde-Visser, S.D.; de Jong, J.G.; Beckmann, J.S.; van der Meer, J.W.; Delpech, M. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat. Genet. 1999, 22, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; van Woerden, C.S.; Wijburg, F.A.; Wanders, R.J.; Waterham, H.R. Carrier frequency of the V377I (1129G>A) MVK mutation, associated with Hyper-IgD and periodic fever syndrome, in the Netherlands. Eur. J. Hum. Genet. 2003, 11, 196–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, G.; Gibson, K.M.; Brandt, I.K.; Bader, P.I.; Wappner, R.S.; Sweetman, L. Mevalonic aciduria—An inborn error of cholesterol and nonsterol isoprene biosynthesis. N. Engl. J. Med. 1986, 314, 1610–1614. [Google Scholar] [CrossRef] [PubMed]
- Akula, M.K.; Shi, M.; Jiang, Z.; Foster, C.E.; Miao, D.; Li, A.S.; Zhang, X.; Gavin, R.M.; Forde, S.D.; Germain, G.; et al. Control of the innate immune response by the mevalonate pathway. Nat. Immunol. 2016, 17, 922–929. [Google Scholar] [CrossRef]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef]
- Ibrahim, J.N.; Jéru, I.; Lecron, J.C.; Medlej-Hashim, M. Cytokine signatures in hereditary fever syndromes (HFS). Cytokine Growth Factor Rev. 2017, 33, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Zanin, V.; Kleiner, G.; Monasta, L.; Crovella, S. Mouse model of mevalonate kinase deficiency: Comparison of cytokine and chemokine profile with that of human patients. Pediatr. Res. 2013, 74, 266–271. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, A.; Piscianz, E.; Vecchi Brumatti, L.; Tommasini, A. Mevalonate kinase deficiency: Therapeutic targets, treatments, and outcomes. Expert Opin. Orphan Drugs 2017, 5, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, A.; Loganes, C.; Valencic, E.; Piscianz, E.; Monasta, L.; Bilel, S.; Bortul, R.; Celeghini, C.; Zweyer, M.; Tommasini, A. Neuronal Dysfunction Associated with Cholesterol Deregulation. Int. J. Mol. Sci. 2018, 19, 1523. [Google Scholar] [CrossRef] [Green Version]
- De Benedetti, F.; Gattorno, M.; Anton, J.; Ben-Chetrit, E.; Frenkel, J.; Hoffman, H.M.; Koné-Paut, I.; Lachmann, H.J.; Ozen, S.; Simon, A.; et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N. Engl. J. Med. 2018, 378, 1908–1919. [Google Scholar] [CrossRef] [Green Version]
- Malcova, H.; Strizova, Z.; Milota, T.; Striz, I.; Sediva, A.; Cebecauerova, D.; Horvath, R. IL-1 Inhibitors in the Treatment of Monogenic Periodic Fever Syndromes: From the Past to the Future Perspectives. Front. Immunol. 2021, 11, 619257. [Google Scholar] [CrossRef]
- Schneiders, M.S.; Houten, S.M.; Turkenburg, M.; Wanders, R.J.; Waterham, H.R. Manipulation of isoprenoid biosynthesis as a possible therapeutic option in mevalonate kinase deficiency. Arthritis Rheum. 2006, 54, 2306–2313. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T.; Amano, Y.; Tozawa, R.; Ishikawa, E.; Imura, Y.; Yukimasa, H.; Sugiyama, Y. Lipid-lowering properties of TAK-475, a squalene synthase inhibitor, in vivo and in vitro. Br. J. Pharmacol. 2003, 139, 911–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, Y.; Nishimoto, T.; Tozawa, R.; Ishikawa, E.; Imura, Y.; Sugiyama, Y. Lipid-lowering effects of TAK-475, a squalene synthase inhibitor, in animal models of familial hypercholesterolemia. Eur. J. Pharmacol. 2003, 466, 155–161. [Google Scholar] [CrossRef]
- Henneman, L.; van Cruchten, A.G.; Kulik, W.; Waterham, H.R. Inhibition of the isoprenoid biosynthesis pathway; detection of intermediates by UPLC-MS/MS. Biochim. Biophys. Acta 2011, 1811, 227–233. [Google Scholar] [CrossRef]
- Shidoji, Y.; Tabata, Y. Unequivocal evidence for endogenous geranylgeranoic acid biosynthesized from mevalonate in mammalian cells. J. Lipid Res. 2019, 60, 579–593. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, A.; Piscianz, E.; Zweyer, M.; Bortul, R.; Loganes, C.; Girardelli, M.; Baj, G.; Monasta, L.; Celeghini, C. Geranylgeraniol and Neurological Impairment: Involvement of Apoptosis and Mitochondrial Morphology. Int. J. Mol. Sci. 2016, 17, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcuzzi, A.; Loganes, C.; Celeghini, C.; Kleiner, G. Repositioning Of Tak-475 in Mevalonate Kinase Disease: Translating Theory into Practice. Curr. Med. Chem. 2018, 25, 2783–2796. [Google Scholar] [CrossRef]
- Ebihara, T.; Teshima, K.; Kondo, T.; Tagawa, Y.; Moriwaki, T.; Asahi, S. Pharmacokinetics of TAK-475, a Squalene Synthase Inhibitor, in Rats and Dogs. Drug Res. 2016, 66, 287–292. [Google Scholar] [CrossRef]
- Seiki, S.; Frishman, W.H. Pharmacologic inhibition of squalene synthase and other downstream enzymes of the cholesterol synthesis pathway: A new therapeutic approach to treatment of hypercholesterolemia. Cardiol. Rev. 2009, 17, 70–76. [Google Scholar] [CrossRef]
- Liao, J.K. Squalene synthase inhibitor lapaquistat acetate: Could anything be better than statins? Circulation 2011, 123, 1925–1928. [Google Scholar] [CrossRef]
- Eskelinen, E.L. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008, 4, 257–260. [Google Scholar] [CrossRef]
- Tolkovsky, A.M. Mitophagy. Biochim. Biophys. Acta 2009, 1793, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorana, D.; Bonatti, F.; Mozzoni, P.; Vaglio, A.; Percesepe, A. Monogenic Autoinflammatory Diseases with Mendelian Inheritance: Genes, Mutations, and Genotype/Phenotype Correlations. Front. Immunol. 2017, 8, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaratnam, J.; Frenkel, J. Management of Mevalonate Kinase Deficiency: A Pediatric Perspective. Front Immunol. 2020, 11, 1150. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Bao, M.; Yi, Z.; Fu, Y. Activation of TLR7 Inhibition of Mycobacterium Tuberculosis Survival by Autophagy in RAW 264.7 Macrophages. J. Cell. Biochem. 2017, 118, 4222–4229. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.; Drewe, E.; van der Meer, J.W.; Powell, R.J.; Kelley, R.I.; Stalenhoef, A.F.; Drenth, J.P. Simvastatin treatment for inflammatory attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Clin. Pharmacol. Ther. 2004, 75, 476–483. [Google Scholar] [CrossRef]
- Hoffmann, G.F.; Charpentier, C.; Mayatepek, E.; Mancini, J.; Leichsenring, M.; Gibson, K.M.; Divry, P.; Hrebicek, M.; Lehnert, W.; Sartor, K.; et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics 1993, 91, 915–921. [Google Scholar]
- Hübner, C.; Hoffmann, G.F.; Charpentier, C.; Gibson, K.M.; Finckh, B.; Puhl, H.; Lehr, H.A.; Kohlschütter, A. Decreased plasma ubiquinone-10 concentration in patients with mevalonate kinase deficiency. Pediatr. Res. 1993, 34, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, B.N.; Schlesinger, P.H.; Ory, D.S.; Baker, N.A. 25-Hydroxycholesterol increases the availability of cholesterol in phospholipid membranes. Biophys. J. 2011, 100, 948–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J.; Frank, M. Repurposed drugs in metabolic disorders. Curr. Top. Med. Chem. 2013, 13, 2386–2394. [Google Scholar] [CrossRef]
- Lotfi Shahreza, M.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J.R. A review of network-based approaches to drug repositioning. Brief. Bioinform. 2018, 19, 878–892. [Google Scholar] [CrossRef]
- Suzuki, N.; Ito, T.; Matsui, H.; Takizawa, M. Anti-inflammatory and cytoprotective effects of a squalene synthase inhibitor, TAK-475 active metabolite-I, in immune cells simulating mevalonate kinase deficiency (MKD)-like condition. Springerplus 2016, 5, 1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rimondi, E.; Valencic, E.; Tommasini, A.; Secchiero, P.; Melloni, E.; Marcuzzi, A. Mevalonate Kinase Deficiency and Squalene Synthase Inhibitor (TAK-475): The Balance to Extinguish the Inflammation. Biomolecules 2021, 11, 1438. https://doi.org/10.3390/biom11101438
Rimondi E, Valencic E, Tommasini A, Secchiero P, Melloni E, Marcuzzi A. Mevalonate Kinase Deficiency and Squalene Synthase Inhibitor (TAK-475): The Balance to Extinguish the Inflammation. Biomolecules. 2021; 11(10):1438. https://doi.org/10.3390/biom11101438
Chicago/Turabian StyleRimondi, Erika, Erica Valencic, Alberto Tommasini, Paola Secchiero, Elisabetta Melloni, and Annalisa Marcuzzi. 2021. "Mevalonate Kinase Deficiency and Squalene Synthase Inhibitor (TAK-475): The Balance to Extinguish the Inflammation" Biomolecules 11, no. 10: 1438. https://doi.org/10.3390/biom11101438
APA StyleRimondi, E., Valencic, E., Tommasini, A., Secchiero, P., Melloni, E., & Marcuzzi, A. (2021). Mevalonate Kinase Deficiency and Squalene Synthase Inhibitor (TAK-475): The Balance to Extinguish the Inflammation. Biomolecules, 11(10), 1438. https://doi.org/10.3390/biom11101438