
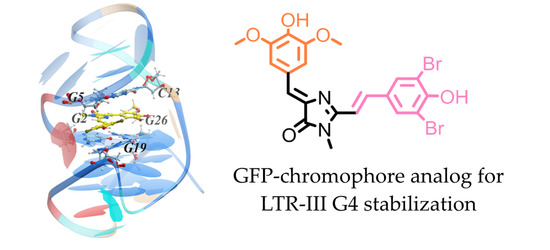
Probing GFP Chromophore Analogs as Anti-HIV Agents Targeting LTR-III G-Quadruplex

 ,
,  ,
,  ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Fret Melting Assay
2.2. NMR Experiments
2.3. Molecular Modeling
2.4. Biological Experiments
2.4.1. Cells and Viruses
2.4.2. Cytotoxicity Test
2.4.3. Cytopathic Effect Inhibition Test
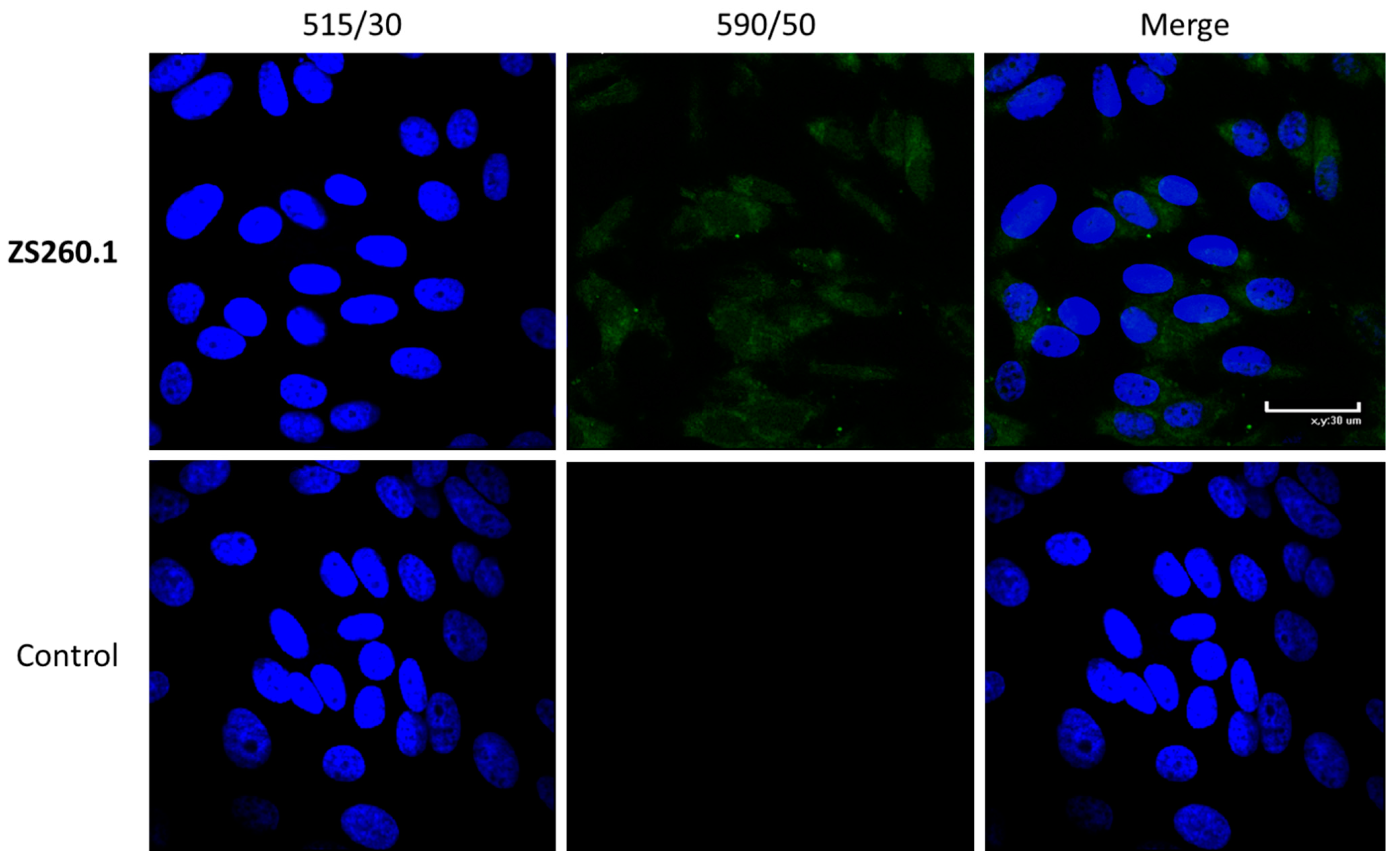
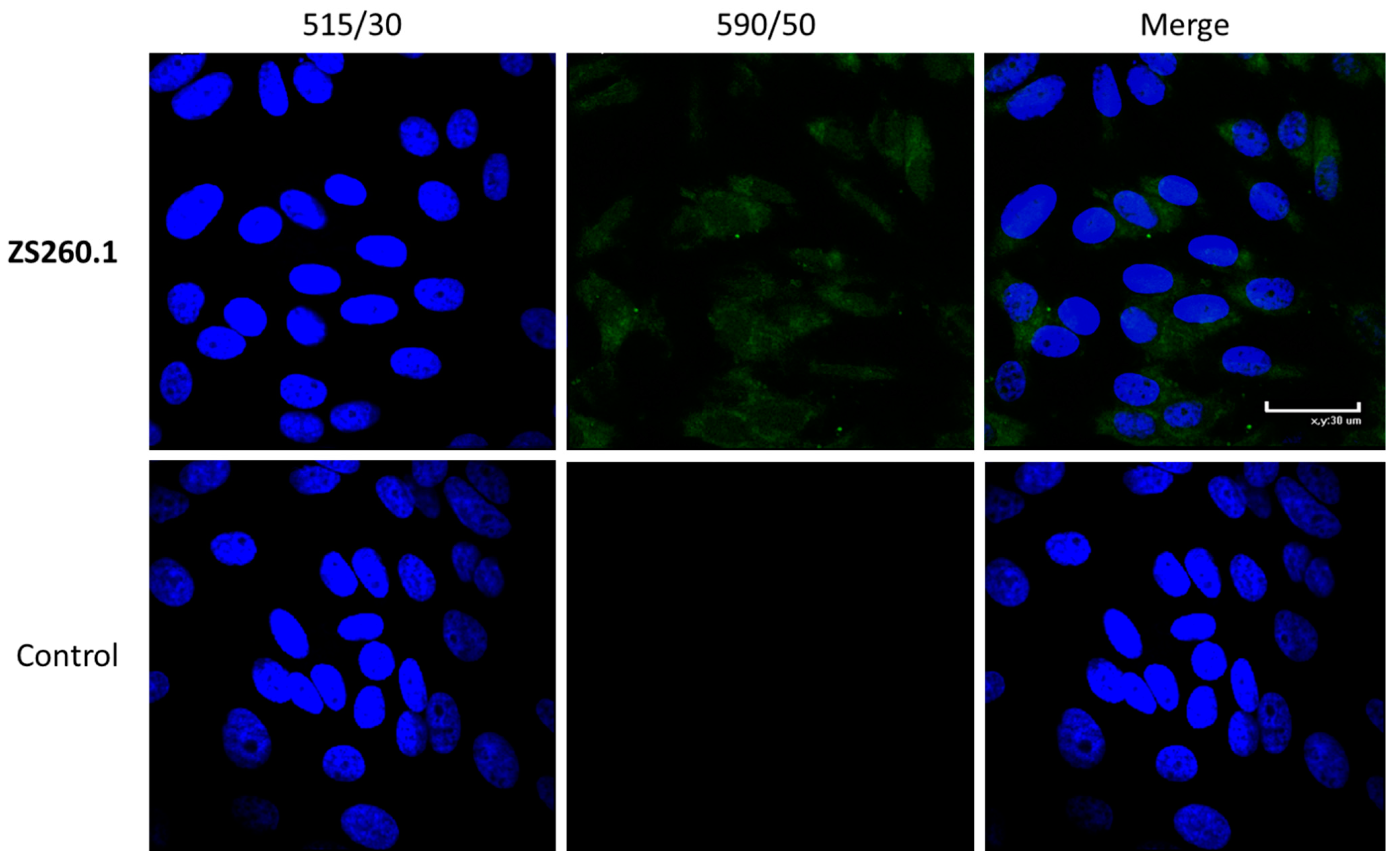
2.4.4. Cell Staining for Confocal Microscopy
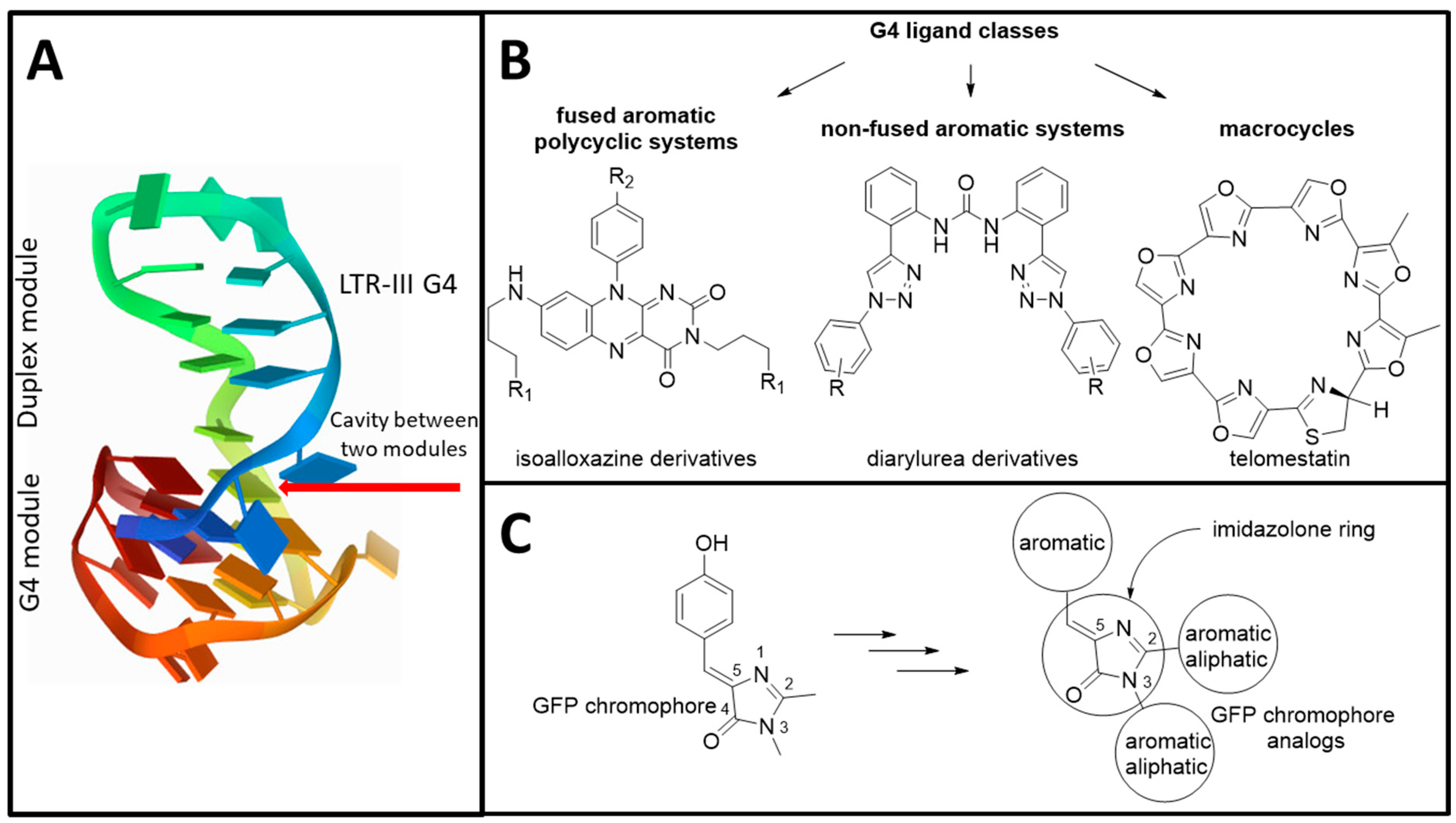
3. Results and Discussion
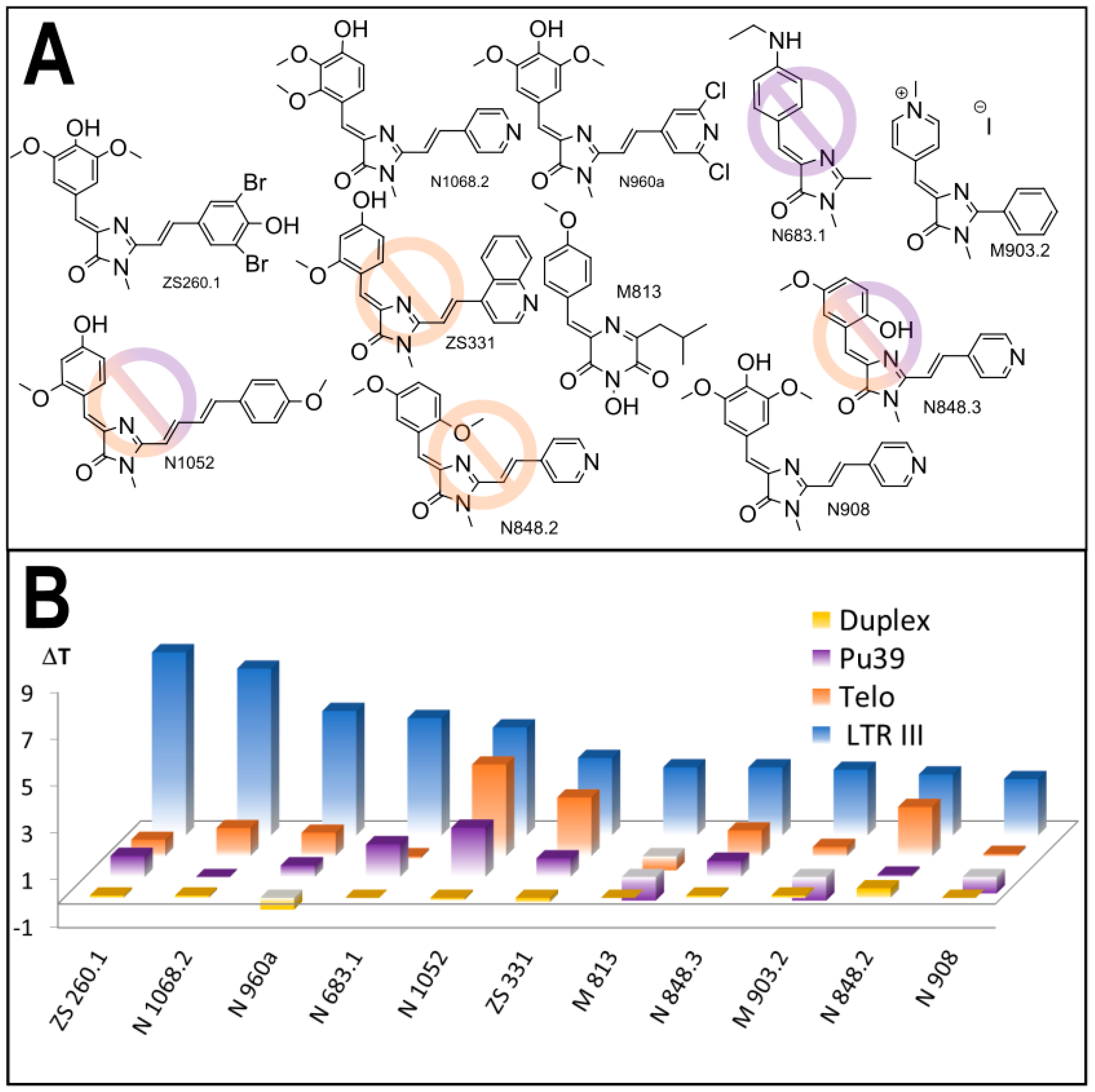
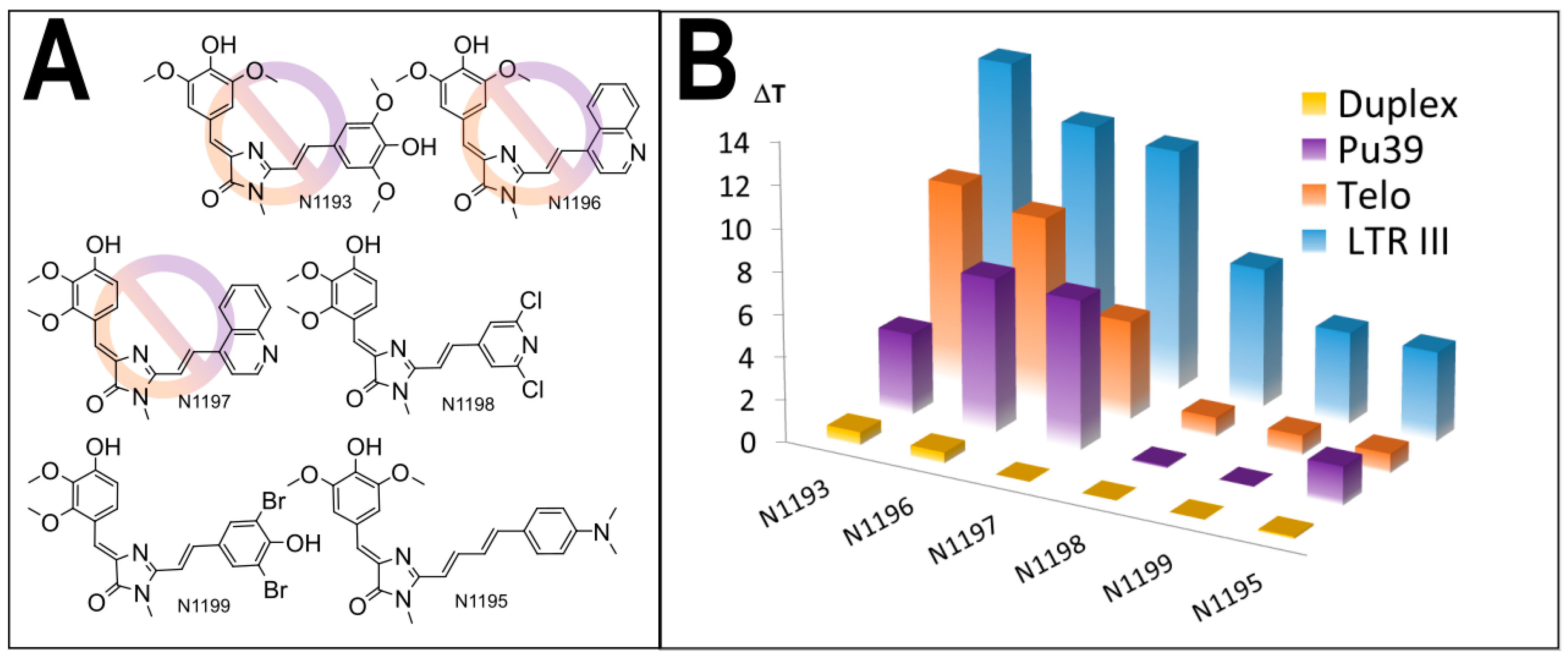
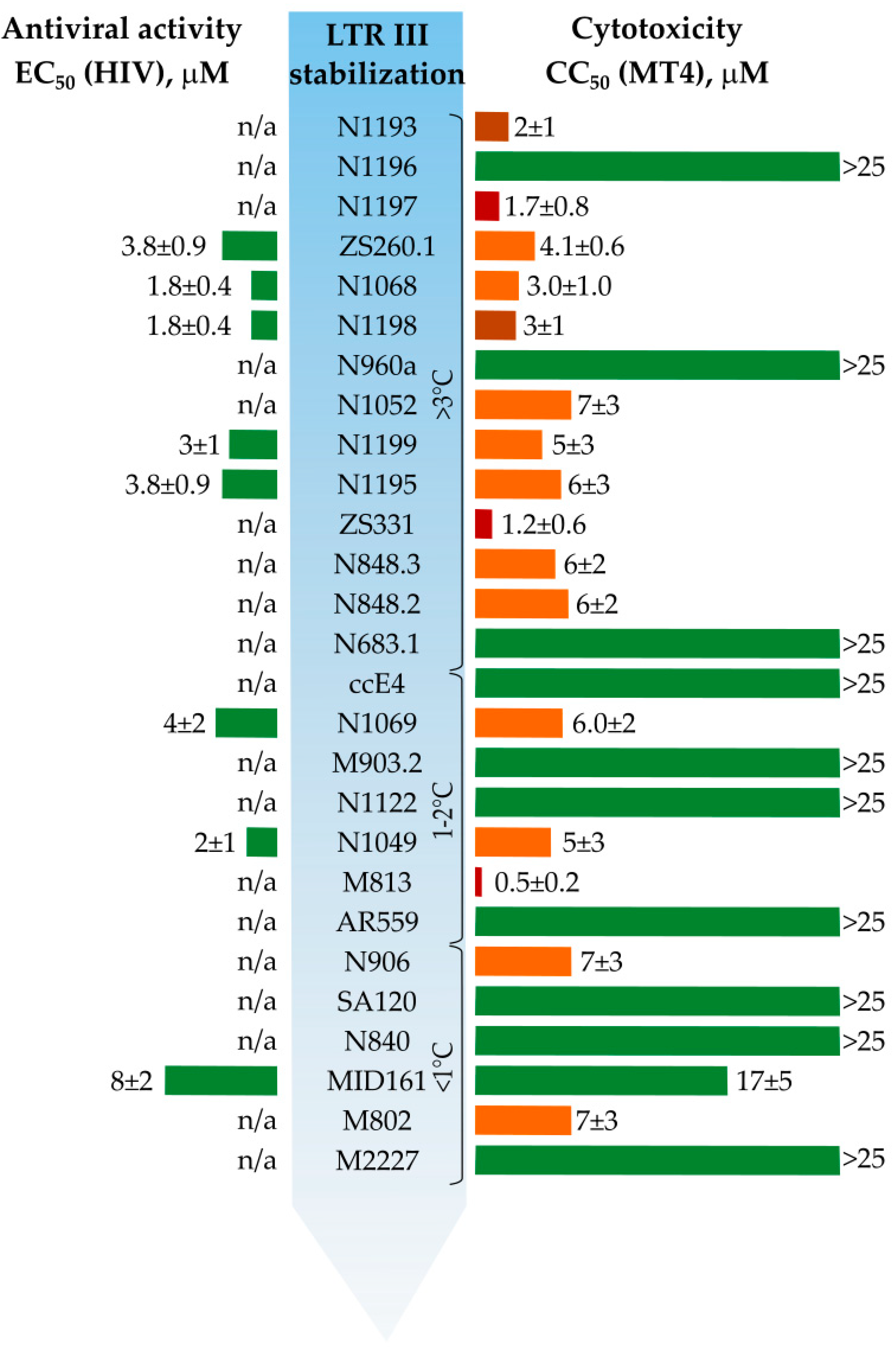
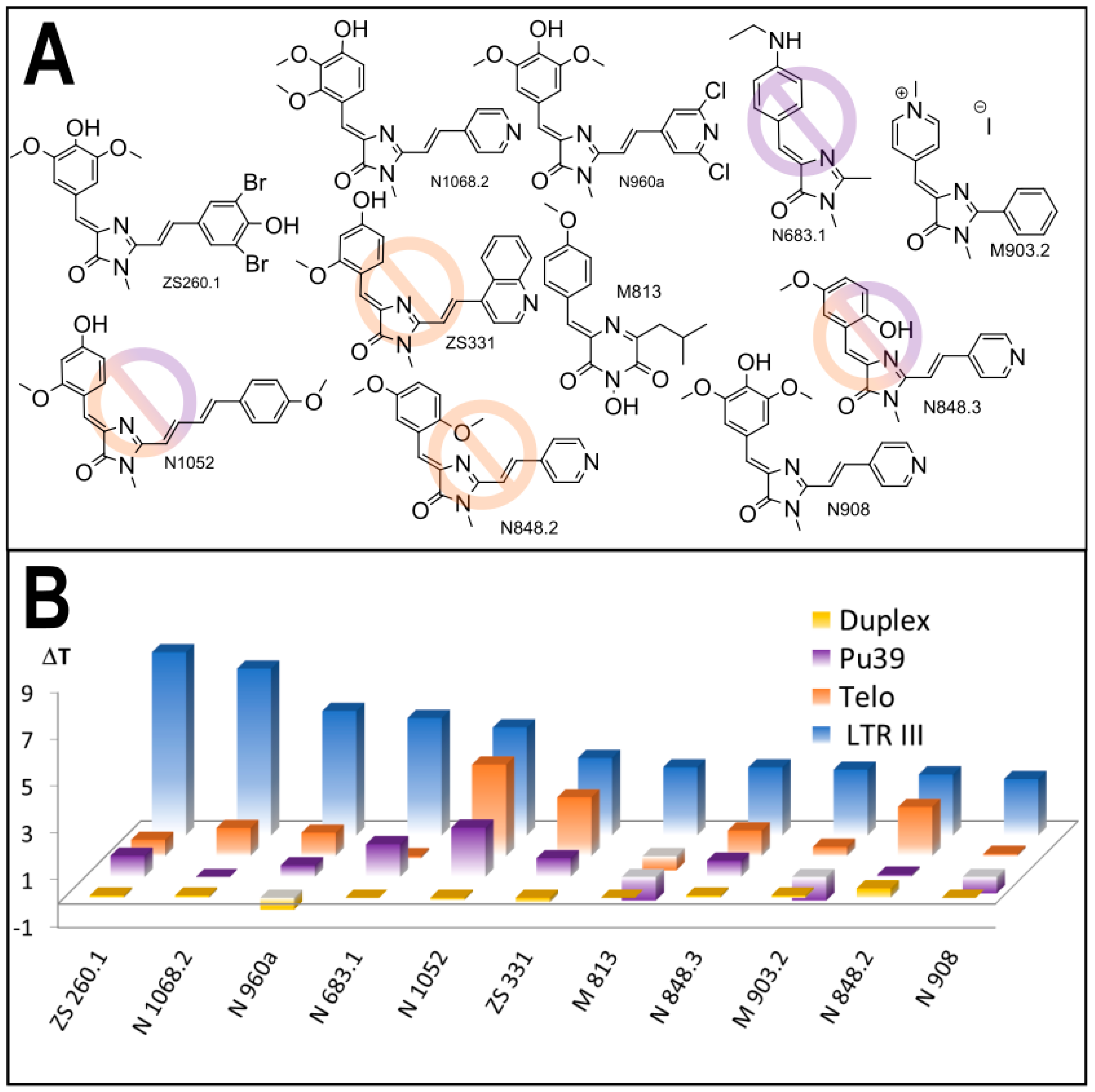
3.1. Primary Screeening of GFP Chromophore Analogs
3.2. Synthesis of the Lead Compounds Congeners
3.3. Mechanistic Studies
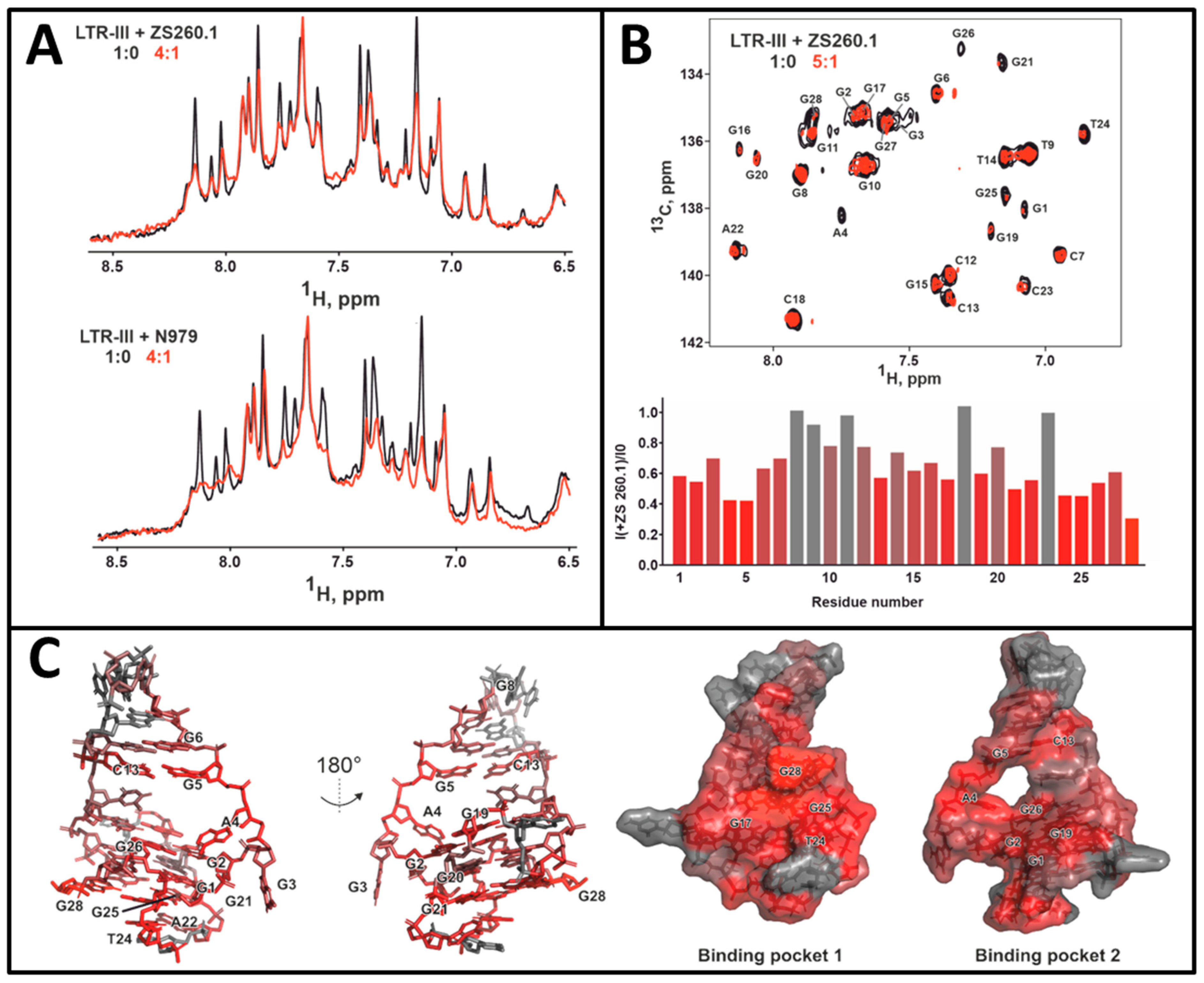
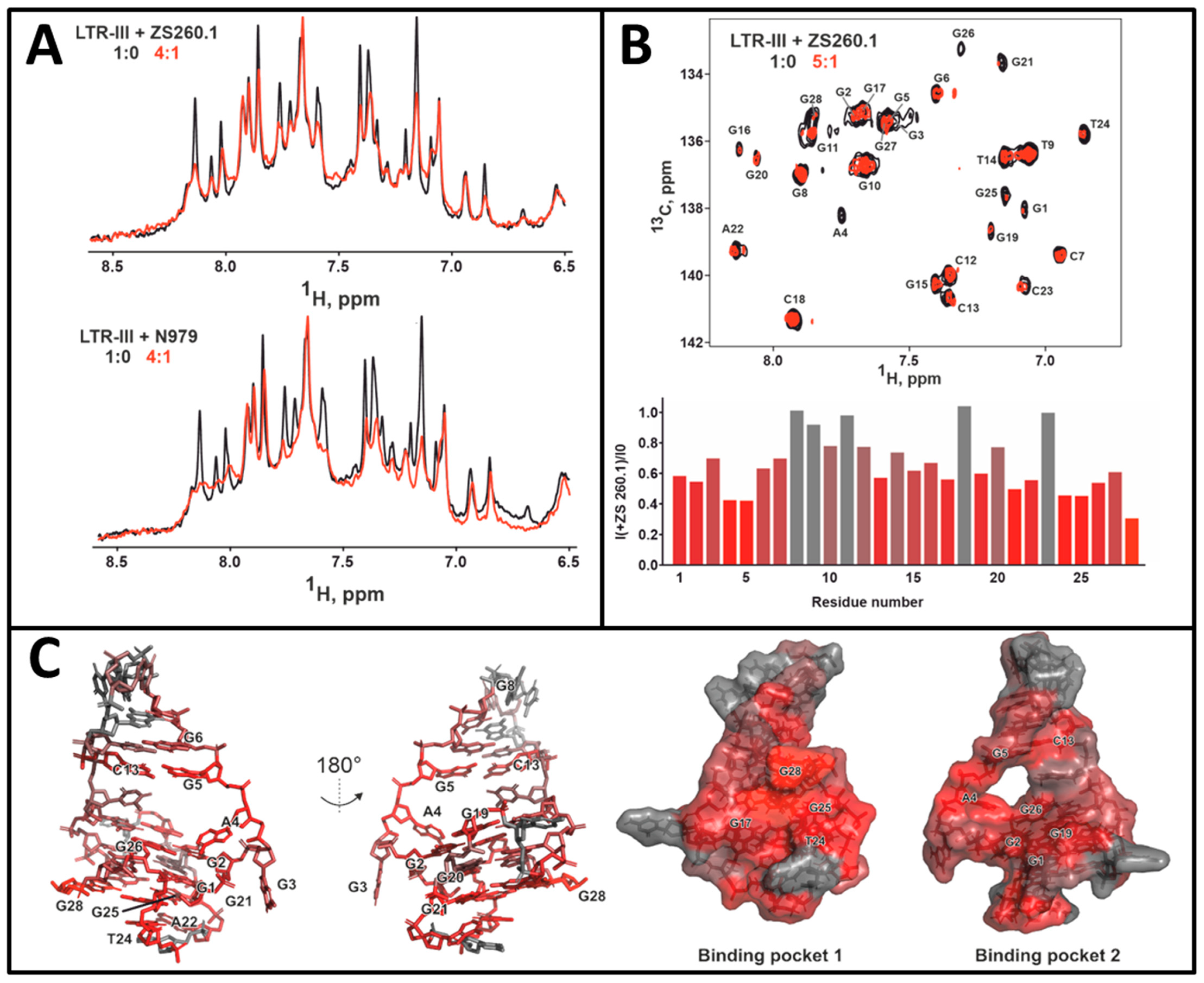
3.3.1. NMR Studies of the HIV-1 LTR-III-G4 Complex with ZS260.1
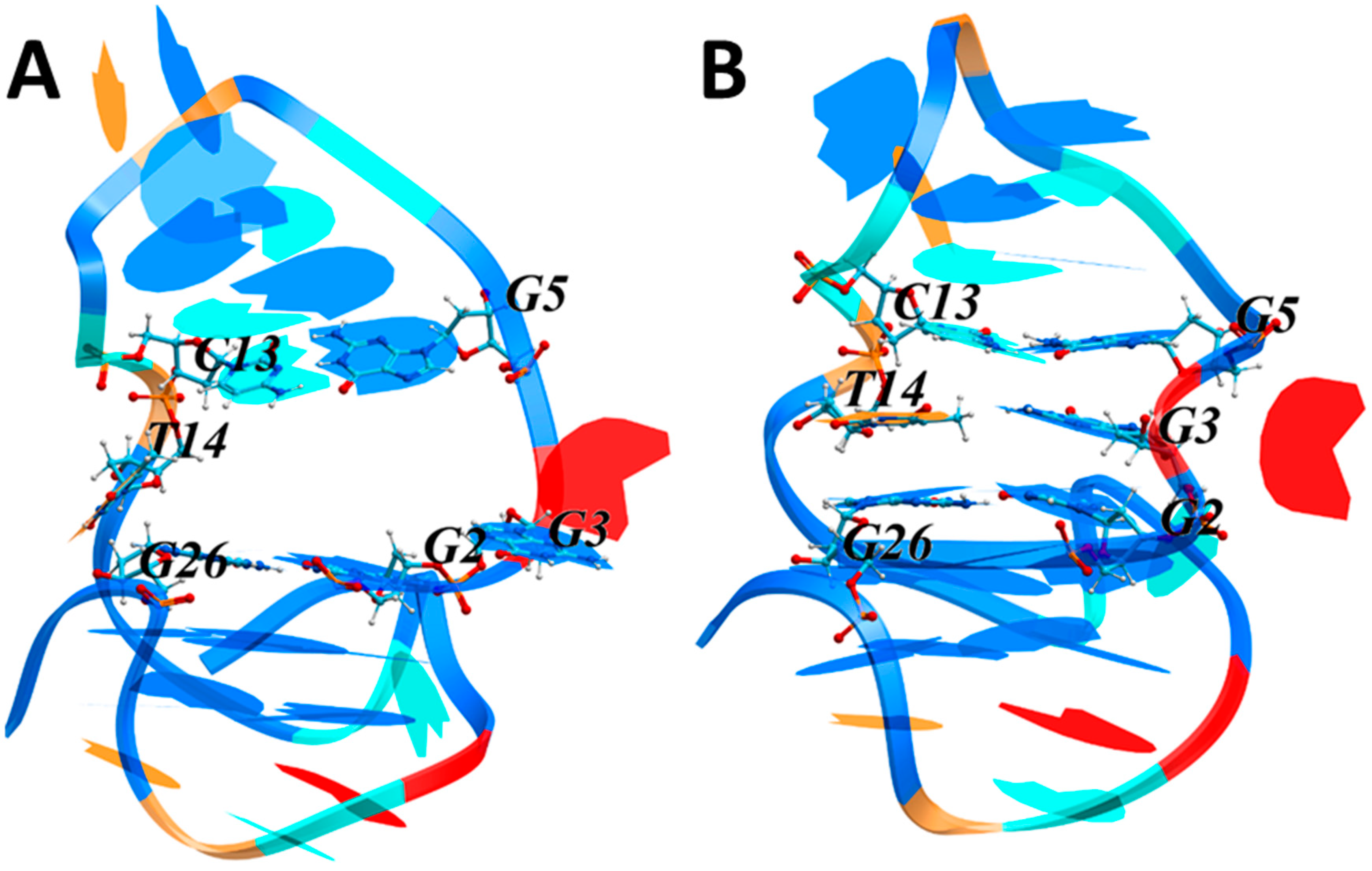
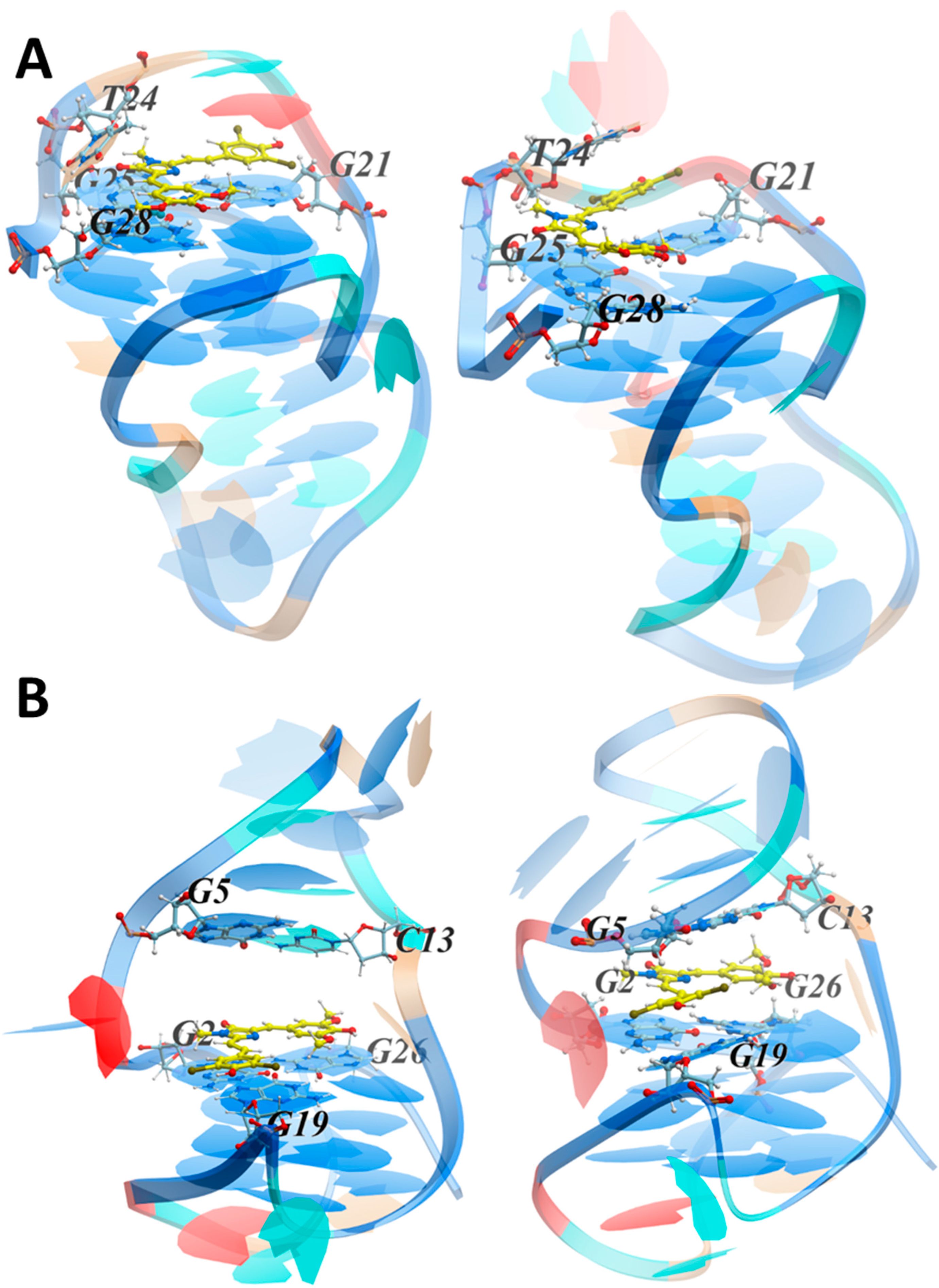
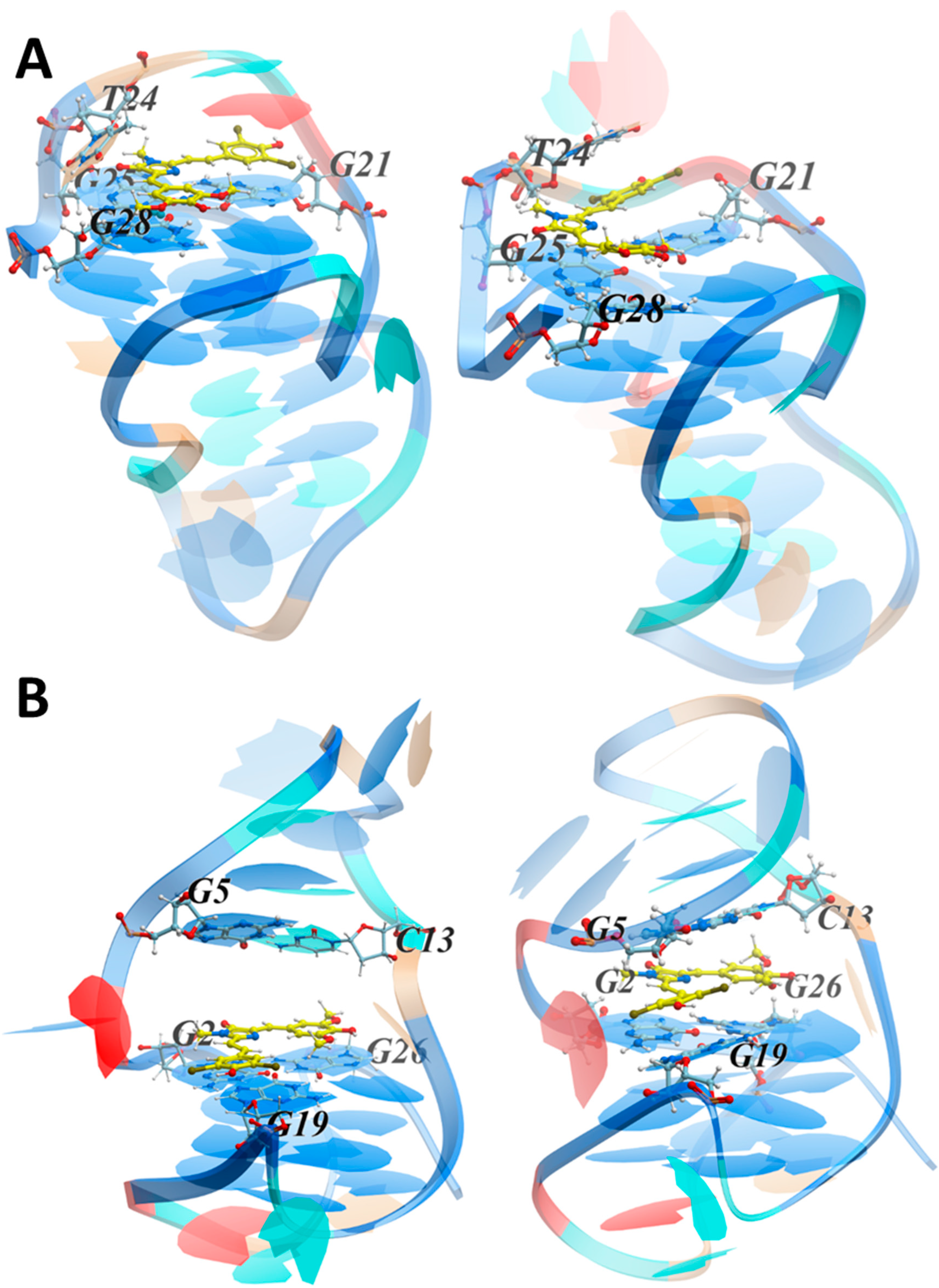
3.3.2. MD Simulations of the LTR-III Complex with ZS260.1
3.4. In Vitro Antiviral Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andreeva, D.V.; Tikhomirov, A.S.; Shchekotikhin, A.E. Ligands of G-Quadruplex Nucleic Acids. Russ. Chem. Rev. 2021, 90, 1–38. [Google Scholar] [CrossRef]
- Carvalho, J.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-Quadruplex, Friend or Foe: The Role of the G-Quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, E.; Richter, S.N. G-Quadruplexes and G-Quadruplex Ligands: Targets and Tools in Antiviral Therapy. Nucleic Acids Res. 2018, 46, 3270–3283. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Kim, N.; Kumari, M.; Verma, S.; Sharma, T.K.; Yadav, V.; Kumar, A. G-Quadruplex Structures in Bacteria-Biological Relevance and Potential as Antimicrobial Target. J. Bacteriol. 2021, 203, e0057720. [Google Scholar] [CrossRef] [PubMed]
- Métifiot, M.; Amrane, S.; Litvak, S.; Andreola, M.-L. G-Quadruplexes in Viruses: Function and Potential Therapeutic Applications. Nucleic Acids Res. 2014, 42, 12352–12366. [Google Scholar] [CrossRef] [Green Version]
- Zahin, M.; Dean, W.L.; Ghim, S.-J.; Joh, J.; Gray, R.D.; Khanal, S.; Bossart, G.D.; Mignucci-Giannoni, A.A.; Rouchka, E.C.; Jenson, A.B.; et al. Identification of G-Quadruplex Forming Sequences in Three Manatee Papillomaviruses. PLoS ONE 2018, 13, e0195625. [Google Scholar] [CrossRef] [Green Version]
- Perrone, R.; Nadai, M.; Frasson, I.; Poe, J.A.; Butovskaya, E.; Smithgall, T.E.; Palumbo, M.; Palù, G.; Richter, S.N. A Dynamic G-Quadruplex Region Regulates the HIV-1 Long Terminal Repeat Promoter. J. Med. Chem. 2013, 56, 6521–6530. [Google Scholar] [CrossRef] [Green Version]
- Perrone, R.; Nadai, M.; Poe, J.A.; Frasson, I.; Palumbo, M.; Palù, G.; Smithgall, T.E.; Richter, S.N. Formation of a Unique Cluster of G-Quadruplex Structures in the HIV-1 Nef Coding Region: Implications for Antiviral Activity. PLoS ONE 2013, 8, e73121. [Google Scholar] [CrossRef] [Green Version]
- Amrane, S.; Kerkour, A.; Bedrat, A.; Vialet, B.; Andreola, M.-L.; Mergny, J.-L. Topology of a DNA G-Quadruplex Structure Formed in the HIV-1 Promoter: A Potential Target for Anti-HIV Drug Development. J. Am. Chem. Soc. 2014, 136, 5249–5252. [Google Scholar] [CrossRef]
- Piekna-Przybylska, D.; Sullivan, M.A.; Sharma, G.; Bambara, R.A. U3 Region in the HIV-1 Genome Adopts a G-Quadruplex Structure in Its RNA and DNA Sequence. Biochemistry 2014, 53, 2581–2593. [Google Scholar] [CrossRef]
- Perrone, R.; Doria, F.; Butovskaya, E.; Frasson, I.; Botti, S.; Scalabrin, M.; Lago, S.; Grande, V.; Nadai, M.; Freccero, M.; et al. Synthesis, Binding and Antiviral Properties of Potent Core-Extended Naphthalene Diimides Targeting the HIV-1 Long Terminal Repeat Promoter G-Quadruplexes. J. Med. Chem. 2015, 58, 9639–9652. [Google Scholar] [CrossRef] [PubMed]
- Butovskaya, E.; Heddi, B.; Bakalar, B.; Richter, S.N.; Phan, A.T. Major G-Quadruplex Form of HIV-1 LTR Reveals a (3 + 1) Folding Topology Containing a Stem-Loop. J. Am. Chem. Soc. 2018, 140, 13654–13662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Chen, Y.-Q.; Wang, S.-R.; Zhou, X. G-Quadruplex: A Regulator of Gene Expression and Its Chemical Targeting. Chem 2018, 4, 1314–1344. [Google Scholar] [CrossRef] [Green Version]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef] [PubMed]
- McLuckie, K.I.E.; Waller, Z.A.E.; Sanders, D.A.; Alves, D.; Rodriguez, R.; Dash, J.; McKenzie, G.J.; Venkitaraman, A.R.; Balasubramanian, S. G-Quadruplex-Binding Benzo[a]Phenoxazines down-Regulate c-KIT Expression in Human Gastric Carcinoma Cells. J. Am. Chem. Soc. 2011, 133, 2658–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Casado, L.; Serrano-Chacón, I.; Montalvillo-Jiménez, L.; Corzana, F.; Bastida, A.; Santana, A.G.; González, C.; Asensio, J.L. De Novo Design of Selective Quadruplex–Duplex Junction Ligands and Structural Characterisation of Their Binding Mode: Targeting the G4 Hot-spot. Chem. Eur. J. 2021, 27, 6204–6212. [Google Scholar] [CrossRef]
- Ehrhardt, D. GFP Technology for Live Cell Imaging. Curr. Opin. Plant Biol. 2003, 6, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Burgess, K. Syntheses of Highly Fluorescent GFP-Chromophore Analogues. J. Am. Chem. Soc. 2008, 130, 4089–4096. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tachibana, S.R.; Baleeva, N.S.; Myasnyanko, I.N.; Bogdanov, A.M.; Gavrikov, A.S.; Mishin, A.S.; Malyshevskaya, K.K.; Baranov, M.S.; Fang, C. Developing Bright GFP-Like Fluorogens for Live-cell Imaging with Nonpolar Protein-chromophore Interactions. Chem. Eur. J. 2021, 27, 8946–8950. [Google Scholar] [CrossRef] [PubMed]
- Bozhanova, N.G.; Baranov, M.S.; Klementieva, N.V.; Sarkisyan, K.S.; Gavrikov, A.S.; Yampolsky, I.V.; Zagaynova, E.V.; Lukyanov, S.A.; Lukyanov, K.A.; Mishin, A.S. Protein Labeling for Live Cell Fluorescence Microscopy with a Highly Photostable Renewable Signal. Chem. Sci. 2017, 8, 7138–7142. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, V.B.; Turaev, A.V.; Petrunina, N.A.; Melnik, D.M.; Khodarovich, Y.M.; Pozmogova, G.E.; Zatsepin, T.S.; Varizhuk, A.M.; Aralov, A.V. Phenoxazine Pseudonucleotides in DNA I-Motifs Allow Precise Profiling of Small Molecule Binders by Fluorescence Monitoring. Analyst 2021, 146, 4436–4440. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An Approach to Computing Electrostatic Charges for Molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A New Method for Protein Modeling and Design: Applications to Docking and Structure Prediction from the Distorted Native Conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Izadi, S.; Onufriev, A.V. Accuracy Limit of Rigid 3-Point Water Models. J. Chem. Phys. 2016, 145, 074501. [Google Scholar] [CrossRef] [Green Version]
- Zgarbová, M.; Luque, F.J.; Šponer, J.; Cheatham, T.E.; Otyepka, M.; Jurečka, P. Toward Improved Description of DNA Backbone: Revisiting Epsilon and Zeta Torsion Force Field Parameters. J. Chem. Theory Comput. 2013, 9, 2339–2354. [Google Scholar] [CrossRef]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E.; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born Radii in the Generalized Born Approximation: The Importance of Being Perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Tassinari, M.; Lena, A.; Butovskaya, E.; Pirota, V.; Nadai, M.; Freccero, M.; Doria, F.; Richter, S. A Fragment-Based Approach for the Development of G-Quadruplex Ligands: Role of the Amidoxime Moiety. Molecules 2018, 23, 1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A Highly Conserved Repetitive DNA Sequence, (TTAGGG)n, Present at the Telomeres of Human Chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Carver, M.; Punchihewa, C.; Jones, R.A.; Yang, D. Structure of the Hybrid-2 Type Intramolecular Human Telomeric G-Quadruplex in K+ Solution: Insights into Structure Polymorphism of the Human Telomeric Sequence. Nucleic Acids Res. 2007, 35, 4927–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Tang, Q.; Li, Y.; Zhang, Y.; Zhao, C.; Yan, J.; You, H. Folding/Unfolding Kinetics of G-Quadruplexes Upstream of the P1 Promoter of the Human BCL-2 Oncogene. J. Biol. Chem. 2019, 294, 5890–5895. [Google Scholar] [CrossRef]
- Dexheimer, T.S.; Sun, D.; Hurley, L.H. Deconvoluting the Structural and Drug-Recognition Complexity of the G-Quadruplex-Forming Region Upstream of the Bcl-2 P1 Promoter. J. Am. Chem. Soc. 2006, 128, 5404–5415. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, V.B.; Varizhuk, A.M.; Lizunova, S.A.; Nikolenko, T.A.; Ivanov, I.A.; Severov, V.V.; Belyaev, E.S.; Shitikov, E.A.; Pozmogova, G.E.; Aralov, A.V. Phenoxazine-Based Scaffold for Designing G4-Interacting Agents. Org. Biomol. Chem. 2020, 18, 6147–6154. [Google Scholar] [CrossRef]
- Ilyinsky, N.S.; Shchyolkina, A.K.; Borisova, O.F.; Mamaeva, O.K.; Zvereva, M.I.; Azhibek, D.M.; Livshits, M.A.; Mitkevich, V.A.; Balzarini, J.; Sinkevich, Y.B.; et al. Novel Multi-Targeting Anthra [2,3-b]Thiophene-5,10-Diones with Guanidine-Containing Side Chains: Interaction with Telomeric G-Quadruplex, Inhibition of Telomerase and Topoisomerase I and Cytotoxic Properties. Eur. J. Med. Chem. 2014, 85, 605–614. [Google Scholar] [CrossRef]
- Lim, K.W.; Jenjaroenpun, P.; Low, Z.J.; Khong, Z.J.; Ng, Y.S.; Kuznetsov, V.A.; Phan, A.T. Duplex Stem-Loop-Containing Quadruplex Motifs in the Human Genome: A Combined Genomic and Structural Study. Nucleic Acids Res. 2015, 43, 5630–5646. [Google Scholar] [CrossRef] [Green Version]
- Povarova, N.V.; Bozhanova, N.G.; Sarkisyan, K.S.; Gritcenko, R.; Baranov, M.S.; Yampolsky, I.V.; Lukyanov, K.A.; Mishin, A.S. Docking-Guided Identification of Protein Hosts for GFP Chromophore-like Ligands. J. Mater. Chem. C 2016, 4, 3036–3040. [Google Scholar] [CrossRef] [Green Version]
- Mineev, K.S.; Goncharuk, S.A.; Goncharuk, M.V.; Povarova, N.V.; Sokolov, A.I.; Baleeva, N.S.; Smirnov, A.Y.; Myasnyanko, I.N.; Ruchkin, D.A.; Bukhdruker, S.; et al. NanoFAST: Structure-Based Design of a Small Fluorogen-Activating Protein with Only 98 Amino Acids. Chem. Sci. 2021, 12, 6719–6725. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Sequence (5′→3′) |
|---|---|
| LTR-III | FAM-GGGAGGCGTGGCCTGGGCGGGACTGGGG-TAMRA |
| Telo | FAM-AGGGTTAGGGTTAGGGTTAGGG-TAMRA |
| dsDNA | FAM-CTATAGCGCGCTATAG-TAMRA |
| Pu39 | FAM-AGGGGCGGGCGCGGGAGGAAGGGGGCGGGAGCGGGGCTG-TAMRA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryazantsev, D.Y.; Myshkin, M.Y.; Alferova, V.A.; Tsvetkov, V.B.; Shustova, E.Y.; Kamzeeva, P.N.; Kovalets, P.V.; Zaitseva, E.R.; Baleeva, N.S.; Zatsepin, T.S.; et al. Probing GFP Chromophore Analogs as Anti-HIV Agents Targeting LTR-III G-Quadruplex. Biomolecules 2021, 11, 1409. https://doi.org/10.3390/biom11101409
Ryazantsev DY, Myshkin MY, Alferova VA, Tsvetkov VB, Shustova EY, Kamzeeva PN, Kovalets PV, Zaitseva ER, Baleeva NS, Zatsepin TS, et al. Probing GFP Chromophore Analogs as Anti-HIV Agents Targeting LTR-III G-Quadruplex. Biomolecules. 2021; 11(10):1409. https://doi.org/10.3390/biom11101409
Chicago/Turabian StyleRyazantsev, Dmitriy Y., Mikhail Yu. Myshkin, Vera A. Alferova, Vladimir B. Tsvetkov, Elena Y. Shustova, Polina N. Kamzeeva, Polina V. Kovalets, Elvira R. Zaitseva, Nadezhda S. Baleeva, Timofei S. Zatsepin, and et al. 2021. "Probing GFP Chromophore Analogs as Anti-HIV Agents Targeting LTR-III G-Quadruplex" Biomolecules 11, no. 10: 1409. https://doi.org/10.3390/biom11101409
APA StyleRyazantsev, D. Y., Myshkin, M. Y., Alferova, V. A., Tsvetkov, V. B., Shustova, E. Y., Kamzeeva, P. N., Kovalets, P. V., Zaitseva, E. R., Baleeva, N. S., Zatsepin, T. S., Shenkarev, Z. O., Baranov, M. S., Kozlovskaya, L. I., & Aralov, A. V. (2021). Probing GFP Chromophore Analogs as Anti-HIV Agents Targeting LTR-III G-Quadruplex. Biomolecules, 11(10), 1409. https://doi.org/10.3390/biom11101409