Design, Synthesis and In Vitro Investigation of Novel Basic Celastrol Carboxamides as Bio-Inspired Leishmanicidal Agents Endowed with Inhibitory Activity against Leishmania Hsp90

 ,
,  , , , ,
, , , ,  and
and 
Abstract
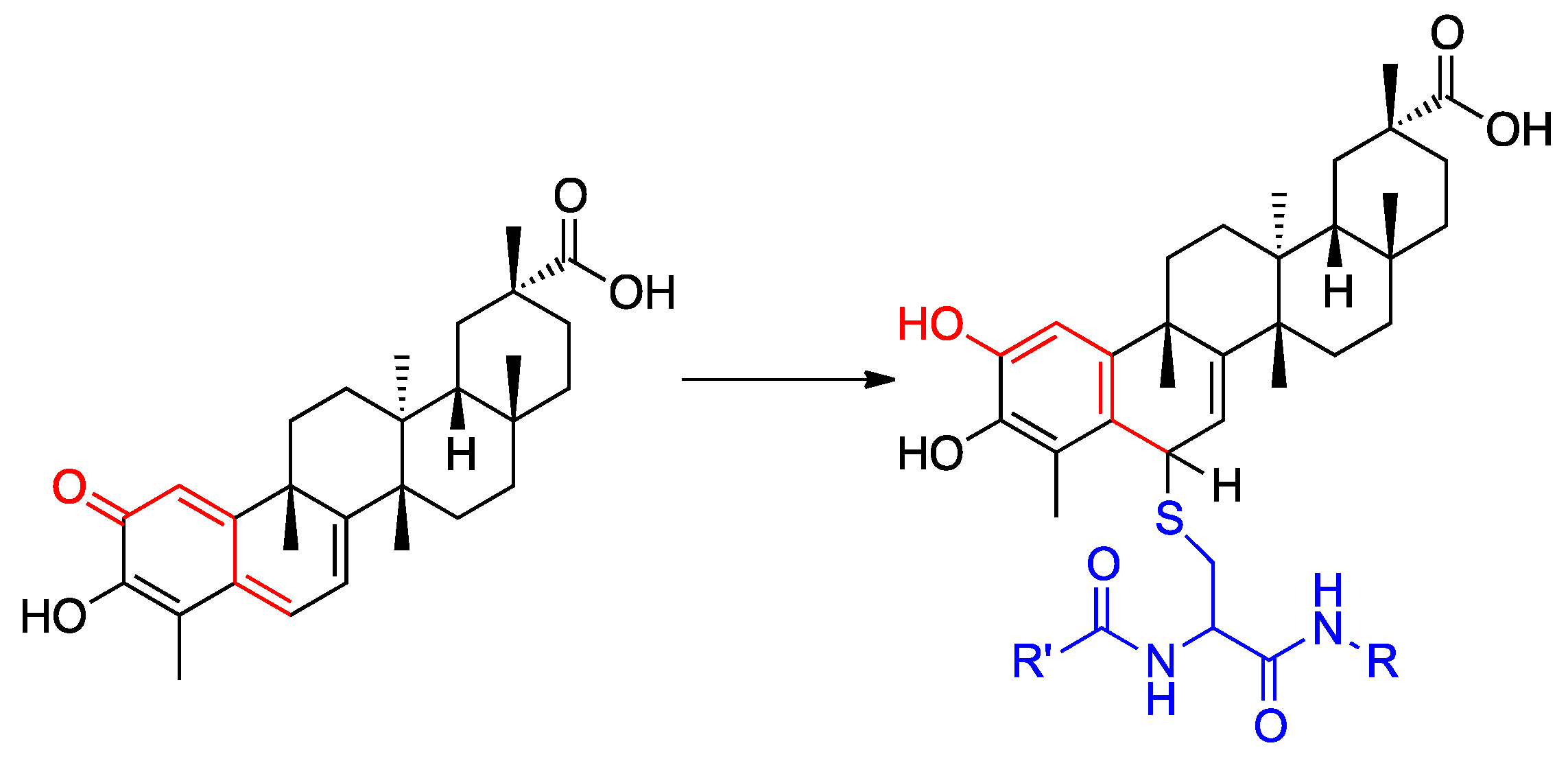
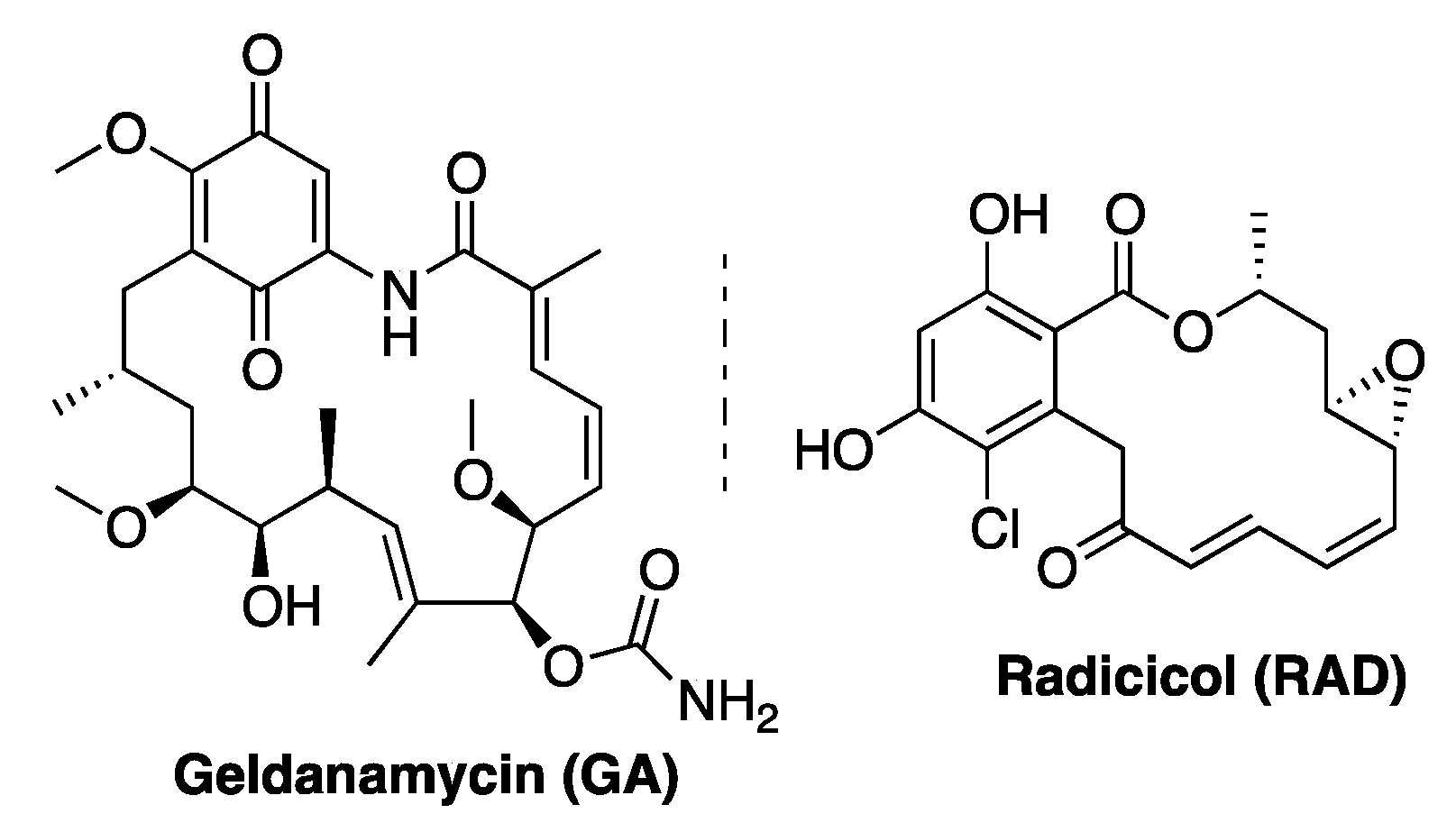
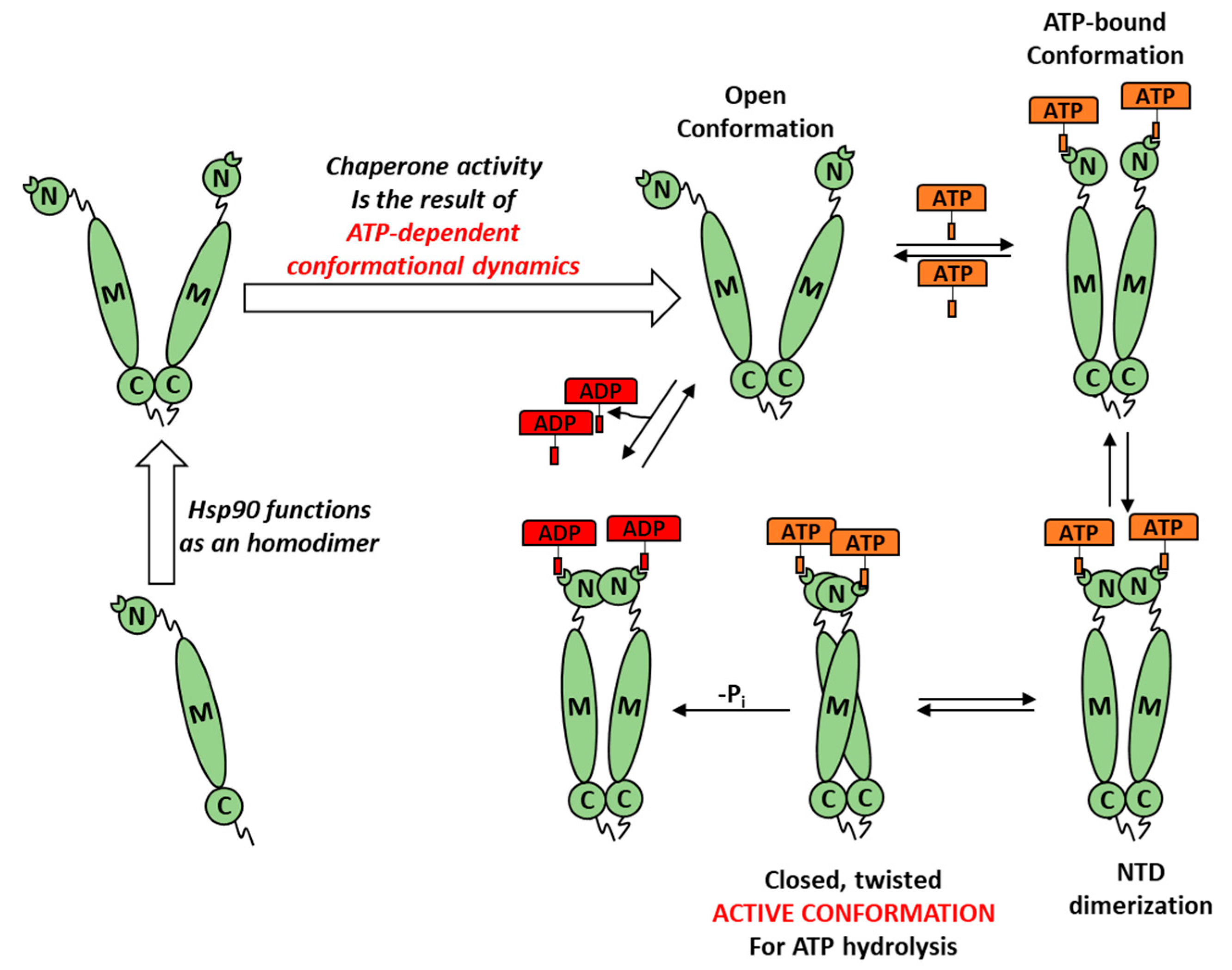
1. Introduction
2. Materials and Method
2.1. Chemistry
2.1.1. General Information
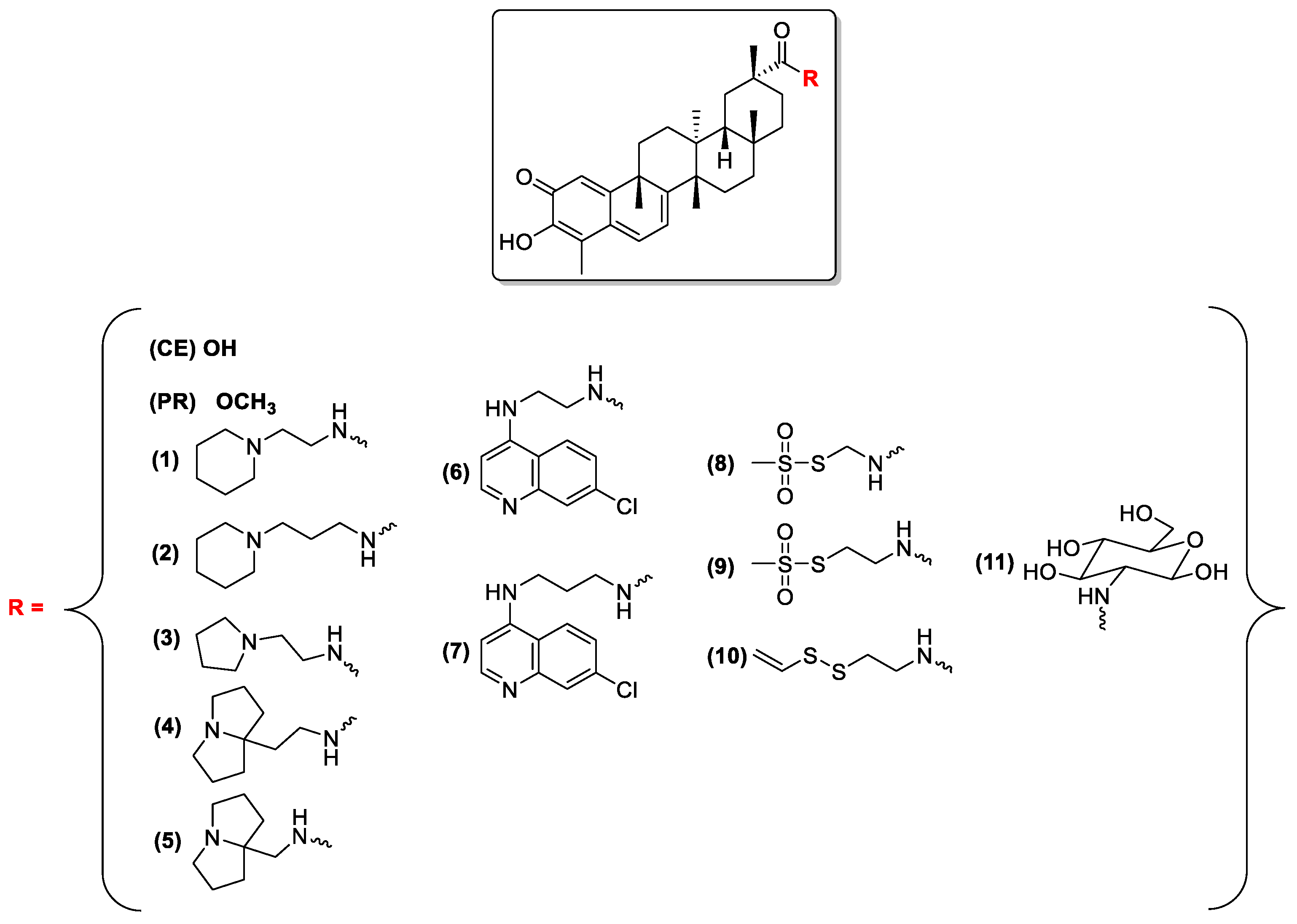
2.1.2. Preparation of Compounds 1–11
2.2. Biological Assays
2.2.1. Recombinant Expression of Full-Length Leishmania braziliensis Hsp90 (LbHsp90)
2.2.2. Determination of LbHsp90 ATPase Cycle Modulation In Vitro
- (1)
- ATP (25 mM) incubated at 27 °C for 60 min
- (2)
- Compounds (different concentrations) incubated with ATP (25 mM) at 27 °C for 60 min
- (3)
- LbHsp90 (7 μM) at 27 °C for 60 min
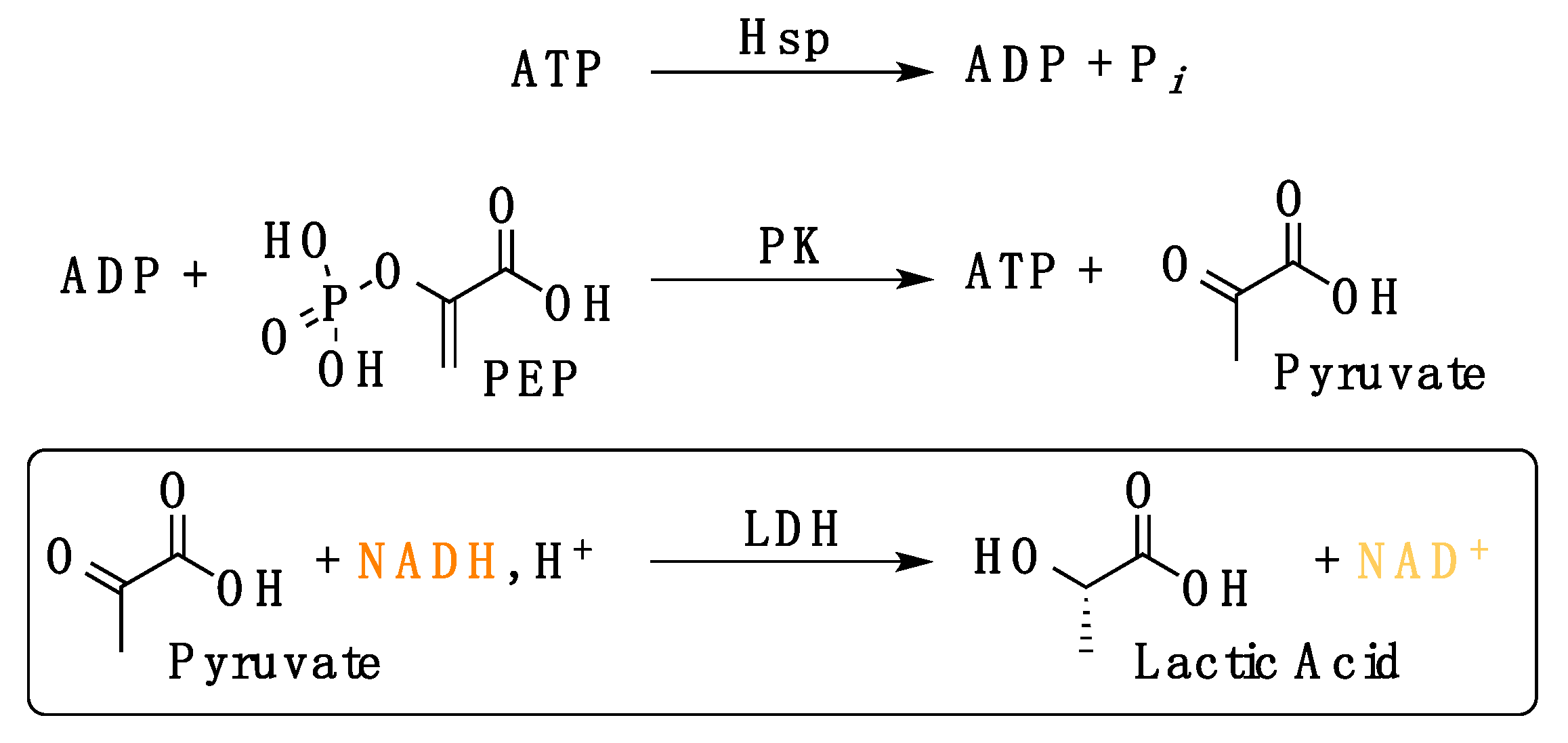
2.2.3. Evaluation of LbHsp90 ATPase Kinetics In Vitro
- (1)
- Efficiency of the enzymatic cascade by the used compounds: HEPES buffer (20 mM, pH 7.5) containing KCl (100 mM), MgCl2 (1 mM), ADP (1 mM), NADH (0.18 mM), L-lactate dehydrogenase (4 U mL−1), phosphoenolpyruvate (1 mM), pyruvate kinase (2.5 U mL−1);
- (2)
- Interference of the tested compound(s) with the enzymatic cascade: HEPES buffer (20 mM, pH 7.5) containing KCl (100 mM), MgCl2 (1 mM), ADP (1 mM), NADH (0.18 mM), L-lactate dehydrogenase (4 U mL−1), phosphoenolpyruvate (1 mM), pyruvate kinase (2.5 U mL−1) and 50 μM compound.
2.2.4. Promastigote Stage of Leishmania spp. Cultures and Antileishmanial Activity
2.2.5. Intramacrophage Amastigotes Stage of Leishmania spp. Cultures and Antileishmanial Activity
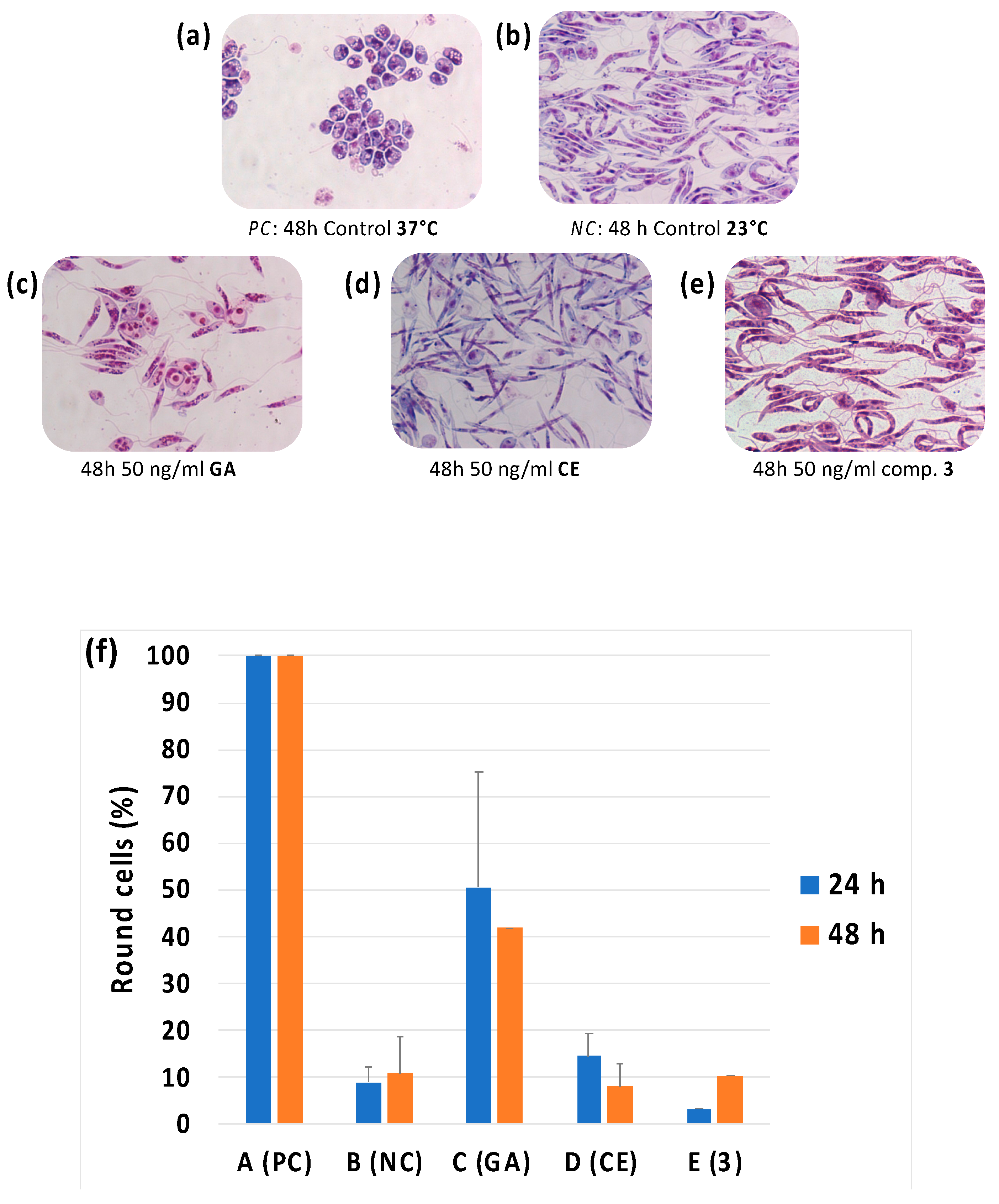
2.2.6. Evaluation of Morphological Differentiation of Leishmania Cells
2.2.7. Cytotoxicity Assay on Different Cell Lines (HMEC-1, BMDM, THP1)
3. Results
3.1. Leishmanicidal Activity on Promastigotes Cultures and Cytotoxicity Evalutation
3.2. Leishmanicidal Activity on Amastigotes Cultures and Selectivity of Action
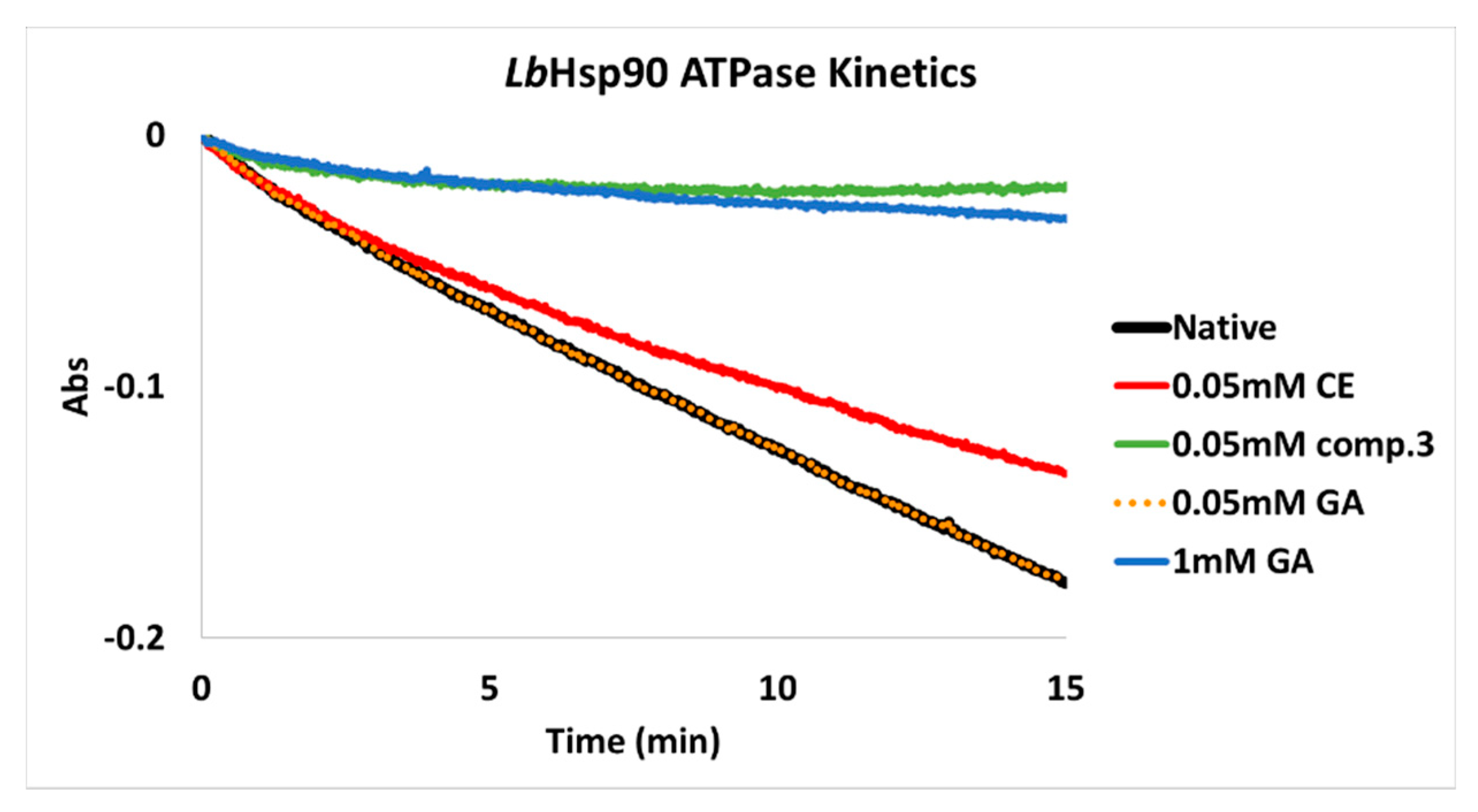
3.3. In Vitro Inhibition of LbHsp90
3.4. ATP-Competitve vs. Non-Competitive Hsp90 Modulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMF | dimethylformamide |
| DCM | dichloromethane |
| HOBt | hydroxybenzotriazole |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| TEA | triethylamine |
| s | singlet |
| bs | broad singlet |
| d | doublet |
| bd | broad doublet |
| t | triplet. |
Appendix A
- -
- Phase A: H2O/CH3CN/TFA 97:3:0.1
- -
- Phase B: H2O/CH3CN/TFA 30:70:0.1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | % A | % B | Flux (mL/min) |
|---|---|---|---|
| 0 | 100 | 0 | 0 |
| 1 | 100 | 0 | 1 |
| 18 | 0 | 100 | 1 |
| 22 | 0 | 100 | 1 |
| 24 | 0 | 100 | 1 |
| 30 | 100 | 0 | 1 |
| 31 | 100 | 0 | 0 |
Appendix B

k = ΔAbs/min = extrapolated apparent kinetic constant (Abs/min)
| Compounds (50 μM) | LbHsp90 Inhibition (%) 1 |
|---|---|
| CE | 28.9 ± 5.4 |
| 3 | 97.8 ± 1.2 |
| GA | <0.05 |
| GA Concentration (μM) | LbHsp90 Inhibition (%) 1 |
|---|---|
| 10 | 0.1 ± 0.02 |
| 15 | 0.2 ± 0.05 |
| 20 | 0.3 ± 0.08 |
| 50 | 0.7 ± 0.04 |
| 100 | 6.5 ± 0.9 |
| 200 | 30.7 ± 1.2 |
| 400 | 52.3 ± 0.8 |
| 600 | 76.9 ± 3.0 |
| 800 | 90.5 ± 2.5 |
| 1000 | 99 ± 0.9 |
| 1200 | >99 |
References
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Chan, Y.; Chellappan, D.K.; Madheswaran, T.; Zeeshan, F.; Chan, Y.L.; Collet, T.; Gupta, G.; Oliver, B.G.; Wark, P.; et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed. Pharmacother. 2019, 109, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.; Das, M.; Dua, K.; Malipeddi, H. Evaluation of in vitro and in vivo anti-urolithiatic activity of silver nanoparticles containing aqueous leaf extract of Tragia involucrata. Drug Deliv. Transl. Res. 2017, 7, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Dua, K.; Sheshala, R.; Ying Ling, T.; Hui Ling, S.; Gorajana, A. Anti-Inflammatory, Antibacterial and Analgesic Potential of Cocos Nucifera Linn.: A Review. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2013, 12, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Shanmugam, M.K.; Sethi, G. Molecular targets of celastrol derived from Thunder of God Vine: Potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011, 303, 9–20. [Google Scholar] [CrossRef]
- Salminen, A.; Lehtonen, M.; Paimela, T.; Kaarniranta, K. Celastrol: Molecular targets of Thunder God Vine. Biochem. Biophys. Res. Commun. 2010, 394, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Brinker, A.M.; Ma, J.; Lipsky, P.E.; Raskin, I. Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae). Phytochemistry 2007, 68, 732–766. [Google Scholar] [CrossRef]
- Cascão, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and Function BT—Molecular Chaperones. In Molecular Chaperones; Springer: Berlin/Heidelberg, Germany, 2012; Volume 328, pp. 155–240. ISBN 9783642345517. [Google Scholar]
- Dos Santos, V.A.F.F.M.; Leite, K.M.; Da Costa Siqueira, M.; Regasini, L.O.; Martinez, I.; Nogueira, C.T.; Galuppo, M.K.; Stolf, B.S.; Pereira, A.M.S.; Cicarelli, R.M.B.; et al. Antiprotozoal activity of quinonemethide triterpenes from Maytenus ilicifolia (Celastraceae). Molecules 2013, 18, 1053–1062. [Google Scholar] [CrossRef]
- Tallorin, L.; Durrant, J.D.; Nguyen, Q.G.; McCammon, J.A.; Burkart, M.D. Celastrol inhibits Plasmodium falciparum enoyl-acyl carrier protein reductase. Bioorg. Med. Chem. 2014, 22, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Malaria Report and WHO|Leishmaniasis. Available online: https://www.who.int/leishmaniasis/en/ (accessed on 15 February 2020).
- Sundar, S.; Chakravarty, J. Leishmaniasis: An update of current pharmacotherapy. Expert Opin. Pharmacother. 2013, 14, 53–63. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 13 February 2019).
- Wiesgigl, M.; Clos, J. Heat Shock Protein 90 Homeostasis Controls Stage Differentiation in Leishmania donovani. Mol. Biol. Cell 2001, 12, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Clos, J. No stress—Hsp90 and signal transduction in Leishmania. Parasitology 2014, 141, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Wiest, R.; Garcia-Cardena, G.; Cadelina, G.; Groszmann, R.J.; Sessa, W.C. Hsp90 regulation of endothelial nitric oxide synthase contributes to vascular control in portal hypertension. Am. J. Physiol. 1999, 277, 463–468. [Google Scholar] [CrossRef]
- Neckers, L. Development of small molecule Hsp90 inhibitors: Utilizing both forward and reverse chemical genomics for drug identification. Curr. Med. Chem. 2003, 10, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; Trump, D.L.; Eiseman, J.L.; Belani, C.P.; Agarwala, S.S.; Zuhowski, E.G.; Lan, J.; Potter, D.M.; Ivy, S.P.; Ramalingam, S.; et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin. Cancer Res. 2005, 11, 3385–3391. [Google Scholar] [CrossRef]
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24. [Google Scholar] [CrossRef]
- Jarosz, D.F.; Lindquist, S. Hsp90 and Environmental Stress Transform the Adaptive Value of Natural Genetic Variation. Science 2010, 330, 1820–1824. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.; Xu, J.; Shi, J.; Jiang, L.; Yao, N.; Ye, W. Discovery and development of natural heat shock protein 90 inhibitors in cancer treatment. Acta Pharm. Sin. B 2012, 2, 238–245. [Google Scholar] [CrossRef]
- Petersen, A.L.D.O.A.; Guedes, C.E.S.; Versoza, C.L.; Lima, J.G.B.; de Freitas, L.A.R.; Borges, V.M.; Veras, P.S.T. 17-AAG Kills Intracellular Leishmania amazonensis while Reducing Inflammatory Responses in Infected Macrophages. PLoS ONE 2012, 7, e49496. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.M.; Petersen, A.L.O.A.; Celes, F.S.; Borges, V.M.; Veras, P.S.T.; de Oliveira, C.I. Chemotherapeutic Potential of 17-AAG against Cutaneous Leishmaniasis Caused by Leishmania (Viannia) braziliensis. PLoS Negl. Trop. Dis. 2014, 8, e3275. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.L.D.O.A.; Campos, T.A.; Dantas, D.A.D.S.; Rebouças, J.D.S.; da Silva, J.C.; de Menezes, J.P.B.; Formiga, F.R.; de Melo, J.V.; Machado, G.; Veras, P.S.T. Encapsulation of the HSP-90 Chaperone Inhibitor 17-AAG in Stable Liposome Allow Increasing the Therapeutic Index as Assessed, in vitro, on Leishmania (L) amazonensis Amastigotes-Hosted in Mouse CBA Macrophages. Front. Cell. Infect. Microbiol. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Wiesgigl, M.; Clos, J. The heat shock protein 90 of Leishmania donovani. Med. Microbiol. Immunol. 2001, 190, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.A.H.; Ramos, S.L.; Tassone, G.; Leitão, A.; Montanari, C.A.; Botta, M.; Mori, M.; Borges, J.C. Discovery of small molecule inhibitors of Leishmania braziliensis Hsp90 chaperone. J. Enzym. Inhib. Med. Chem. 2020, 35, 639–649. [Google Scholar] [CrossRef]
- Mohammadi-Ostad-Kalayeh, S.; Stahl, F.; Scheper, T.; Kock, K.; Herrmann, C.; Heleno Batista, F.A.; Borges, J.C.; Sasse, F.; Eichner, S.; Ongouta, J.; et al. Heat Shock Proteins Revisited: Using a Mutasynthetically Generated Reblastatin Library to Compare the Inhibition of Human and Leishmania Hsp90s. ChemBioChem 2018, 19, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Seraphim, T.V.; Ramos, C.H.I.; Borges, J.C. The Interaction Networks of Hsp70 and Hsp90 in the Plasmodium and Leishmania Parasites. In The Molecular Chaperones Interaction Networks in Protein Folding and Degradation; Houry, W.A., Ed.; Springer: Berlin, Germany, 2014; pp. 445–485. ISBN 9781493911301. [Google Scholar]
- Bassanini, I.; D’Annessa, I.; Costa, M.; Monti, D.; Colombo, G.; Riva, S. Chemo-enzymatic synthesis of (E)-2,3-diaryl-5-styryl- trans-2,3-dihydrobenzofuran-based scaffolds and their in vitro and in silico evaluation as a novel sub-family of potential allosteric modulators of the 90 kDa heat shock protein (Hsp90). Org. Biomol. Chem. 2018, 16, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Sattin, S.; Tao, J.; Vettoretti, G.; Moroni, E.; Pennati, M.; Lopergolo, A.; Morelli, L.; Bugatti, A.; Zuehlke, A.; Moses, M.; et al. Activation of Hsp90 Enzymatic Activity and Conformational Dynamics through Rationally Designed Allosteric Ligands. Chem. Eur. J. 2015, 21, 13598–13608. [Google Scholar] [CrossRef]
- Vettoretti, G.; Moroni, E.; Sattin, S.; Tao, J.; Agard, D.A.; Bernardi, A.; Colombo, G. Molecular Dynamics Simulations Reveal the Mechanisms of Allosteric Activation of Hsp90 by Designed Ligands. Sci. Rep. 2016, 6, 23830. [Google Scholar] [CrossRef]
- Rehn, A.; Moroni, E.; Zierer, B.K.; Tippel, F.; Morra, G.; John, C.; Richter, K.; Colombo, G.; Buchner, J. Allosteric Regulation Points Control the Conformational Dynamics of the Molecular Chaperone Hsp90. J. Mol. Biol. 2016, 428, 4559–4571. [Google Scholar] [CrossRef]
- Terracciano, S.; Russo, A.; Chini, M.G.; Vaccaro, M.C.; Potenza, M.; Vassallo, A.; Riccio, R.; Bifulco, G.; Bruno, I. Discovery of new molecular entities able to strongly interfere with Hsp90 C-terminal domain. Sci. Rep. 2018, 8, 1709. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Lavery, L.A.; Partridge, J.R.; Ramelot, T.A.; Elnatan, D.; Kennedy, M.A.; Agard, D.A. Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol. Cell 2014, 53, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Bagatell, R.; Falsey, R. The stress response: Implications for the clinical development of hsp90 inhibitors. Curr. Cancer Drug Targets 2003, 3, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. In Proceedings of the Annals of the New York Academy of Sciences; Blackwell Publishing Inc.: Oxford, UK, 2007; Volume 1113, pp. 202–216. [Google Scholar]
- Kimura, Y.; Rutherford, S.L.; Miyata, Y.; Yahara, I.; Freeman, B.C.; Yue, L.; Morimoto, R.I.; Lindquist, S. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997, 11, 1775–1785. [Google Scholar] [CrossRef]
- Klaić, L.; Morimoto, R.I.; Silverman, R.B. Celastrol analogues as inducers of the heat shock response. Design and synthesis of affinity probes for the identification of protein targets. ACS Chem. Biol. 2012, 7, 928–937. [Google Scholar] [CrossRef]
- Chadli, A.; Felts, S.J.; Wang, Q.; Sullivan, W.P.; Botuyan, M.V.; Fauq, A.; Ramirez-Alvarado, M.; Mer, G. Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone p23. J. Biol. Chem. 2010, 285, 4224–4231. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit Hsp90 and Cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef]
- Sreeramulu, S.; Gande, S.L.; Göbel, M.; Schwalbe, H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. Int. Ed. 2009, 48, 5853–5855. [Google Scholar] [CrossRef]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.-G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Wang, H.J.; Bao, Q.C.; Wang, L.; Jin, Y.H.; Zhang, Q.; Jiang, D.; You, Q.D.; Xu, X.L. Optimization and biological evaluation of celastrol derivatives as Hsp90–Cdc37 interaction disruptors with improved druglike properties. Bioorg. Med. Chem. 2016, 24, 5431–5439. [Google Scholar] [CrossRef]
- Akabas, M.H.; Stauffer, D.A.; Xu, M.; Karlin, A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science 1992, 258, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Akabas, M.H.; Karlin, A. Identification of Acetylcholine Receptor Channel-Lining Residues in the M1 Segment of the α-Subunit. Biochemistry 1995, 34, 12496–12500. [Google Scholar] [CrossRef]
- Kenyon, G.L.; Bruice, T.W. Novel Sulfhydryl Reagents. Methods Enzymol. 1977, 47, 407–430. [Google Scholar] [CrossRef] [PubMed]
- Silva, K.P.; Seraphim, T.V.; Borges, J.C. Structural and functional studies of Leishmania braziliensis Hsp90. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 351–361. [Google Scholar] [CrossRef]
- Seraphim, T.V.; Silva, K.P.; Dores-Silva, P.R.; Barbosa, L.R.S.; Borges, J.C. Insights on the structural dynamics of Leishmania braziliensis Hsp90 molecular chaperone by small angle X-ray scattering. Int. J. Biol. Macromol. 2017, 97, 503–512. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Baiocco, P.; Ilari, A.; Ceci, P.; Orsini, S.; Gramiccia, M.; Di Muccio, T.; Colotti, G. Inhibitory effect of silver nanoparticles on trypanothione reductase activity and Leishmania infantum proliferation. ACS Med. Chem. Lett. 2011, 2, 230–233. [Google Scholar] [CrossRef]
- Bassanini, I.; Parapini, S.; Basilico, N.; Sparatore, A. Novel Hydrophilic Riminophenazines as Potent Antiprotozoal Agents. ChemMedChem 2019, 14, 1940–1949. [Google Scholar] [CrossRef]
- Tonelli, M.; Gabriele, E.; Piazza, F.; Basilico, N.; Parapini, S.; Tasso, B.; Loddo, R.; Sparatore, F.; Sparatore, A. Benzimidazole derivatives endowed with potent antileishmanial activity. J. Enzym. Inhib. Med. Chem. 2018, 33, 210–226. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Bassanini, I.; Parapini, S.; Galli, C.; Vaiana, N.; Pancotti, A.; Basilico, N.; Taramelli, D.; Romeo, S. Discovery and Pharmacophore Mapping of a Low-Nanomolar Inhibitor of P. falciparum Growth. ChemMedChem 2019, 14, 1982–1994. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Ivy, S.P.; Kopil, C.; Sikorski, R.; Morris, M.J.; Slovin, S.F.; Kelly, W.K.; DeLaCruz, A.; Curley, T.; Heller, G.; et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin. Cancer Res. 2007, 13, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Glaze, E.R.; Lambert, A.L.; Smith, A.C.; Page, J.G.; Johnson, W.D.; McCormick, D.L.; Brown, A.P.; Levine, B.S.; Covey, J.M.; Egorin, M.J.; et al. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: Potential clinical relevance. Cancer Chemother. Pharmacol. 2005, 56, 637–647. [Google Scholar] [CrossRef]






| Leishmania Promastigotes (IC50, μM) | Healthy Cell Lines (IC50, μM) | ||||
|---|---|---|---|---|---|
| L. infantum | L. tropica | HMEC-1 | THP-1 | BMDM | |
| CE | 0.21 ± 0.07 | 0.49 ± 0.17 | 0.38 ± 0.08 | 0.90 ± 0.30 | 1.76 ± 0.43 |
| PR | 0.26 ± 0.02 | 0.24 ± 0.04 | N.T. 1 | 1.10 ± 0.35 | 1.27 ± 0.09 |
| 1 | 0.06 ± 0.02 | 0.09 ± 0.03 | 0.51 ± 0.08 | 2.08 ± 0.24 | 3.61 ± 0.38 |
| 2 | 0.26 ± 0.06 | 0.21 ± 0.04 | N.T. 1 | 1.78 ± 0.48 | N.T. 1 |
| 3 | 0.06 ± 0.01 | 0.08 ± 0.04 | 0.53 ± 0.17 | 2.79 ± 0.44 | 4.53 ± 0.05 |
| 4 | 0.25 ± 0.08 | 0.20 ± 0.04 | 0.35 ± 0.16 | 1.89 ± 0.43 | 2.59 ± 0.71 |
| 5 | 0.12 ± 0.02 | 0.12 ± 0.05 | 0.12 ± 0.03 | 1.88 ± 0.34 | 2.68 ± 0.81 |
| 6 | 0.09 ± 0.03 | 0.14 ± 0.03 | 0.40 ± 0.03 | 0.85 ± 0.24 | 2.15 ± 0.02 |
| 7 | 0.20 ± 0.01 | 0.15 ± 0.04 | 0.13 ± 0.01 | 0.48 ± 0.17 | 1.78 ± 0.53 |
| 8 | 4.05 ± 1.96 | 3.44 ± 1.12 | 3.03 ± 0.01 | 2.51 ± 0.87 | 13.88 ± 2.78 |
| 9 | 2.31 ± 0.28 | 3.07 ± 1.36 | 4.07 ± 0.05 | 3.28 ± 0.90 | 12.96 ± 2.48 |
| 10 | 0.46 ± 0.12 | 0.34 ± 0.21 | 0.17 ± 0.04 | 0.93 ± 0.27 | 2.86 ± 0.17 |
| 11 | 14.45 ± 1.96 | 15.15 ± 2.71 | >14.17 | >15.90 | >15.90 |
| AMP | 0.16 ± 0.05 | 0.17 ± 0.02 | >20 | >20 | >20 |
| L. infantum Amastigotes IC50 (μM) 1 | S.I. 2 | |
|---|---|---|
| 1 | 0.19 ± 0.11 | 11 |
| 3 | 0.13 ± 0.09 | 21 |
| 5 | 0.52 ± 0.28 | 3.6 |
| 6 | 0.66 ± 0.06 | 1.3 |
| AMP | 0.18 ± 0.03 | >109 |
| Compound | IC50 (μM) 1 |
|---|---|
| CE | 201 ± 10 |
| 1 | 83 ± 2 |
| 2 | >125 |
| 3 | 81 ± 1 |
| 4 | 48 ± 3 |
| 5 | 20 ± 2 |
| 6 | 65 ± 4 |
| 7 | >125 |
| 8 | 67 ± 3 |
| 9 | >150 |
| 10 | >125 |
| 11 | 47 ± 5 |
| PR | >500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassanini, I.; Parapini, S.; Ferrandi, E.E.; Gabriele, E.; Basilico, N.; Taramelli, D.; Sparatore, A. Design, Synthesis and In Vitro Investigation of Novel Basic Celastrol Carboxamides as Bio-Inspired Leishmanicidal Agents Endowed with Inhibitory Activity against Leishmania Hsp90. Biomolecules 2021, 11, 56. https://doi.org/10.3390/biom11010056
Bassanini I, Parapini S, Ferrandi EE, Gabriele E, Basilico N, Taramelli D, Sparatore A. Design, Synthesis and In Vitro Investigation of Novel Basic Celastrol Carboxamides as Bio-Inspired Leishmanicidal Agents Endowed with Inhibitory Activity against Leishmania Hsp90. Biomolecules. 2021; 11(1):56. https://doi.org/10.3390/biom11010056
Chicago/Turabian StyleBassanini, Ivan, Silvia Parapini, Erica E. Ferrandi, Elena Gabriele, Nicoletta Basilico, Donatella Taramelli, and Anna Sparatore. 2021. "Design, Synthesis and In Vitro Investigation of Novel Basic Celastrol Carboxamides as Bio-Inspired Leishmanicidal Agents Endowed with Inhibitory Activity against Leishmania Hsp90" Biomolecules 11, no. 1: 56. https://doi.org/10.3390/biom11010056
APA StyleBassanini, I., Parapini, S., Ferrandi, E. E., Gabriele, E., Basilico, N., Taramelli, D., & Sparatore, A. (2021). Design, Synthesis and In Vitro Investigation of Novel Basic Celastrol Carboxamides as Bio-Inspired Leishmanicidal Agents Endowed with Inhibitory Activity against Leishmania Hsp90. Biomolecules, 11(1), 56. https://doi.org/10.3390/biom11010056