Role of TGF-Beta and Smad7 in Gut Inflammation, Fibrosis and Cancer



{kind=link}
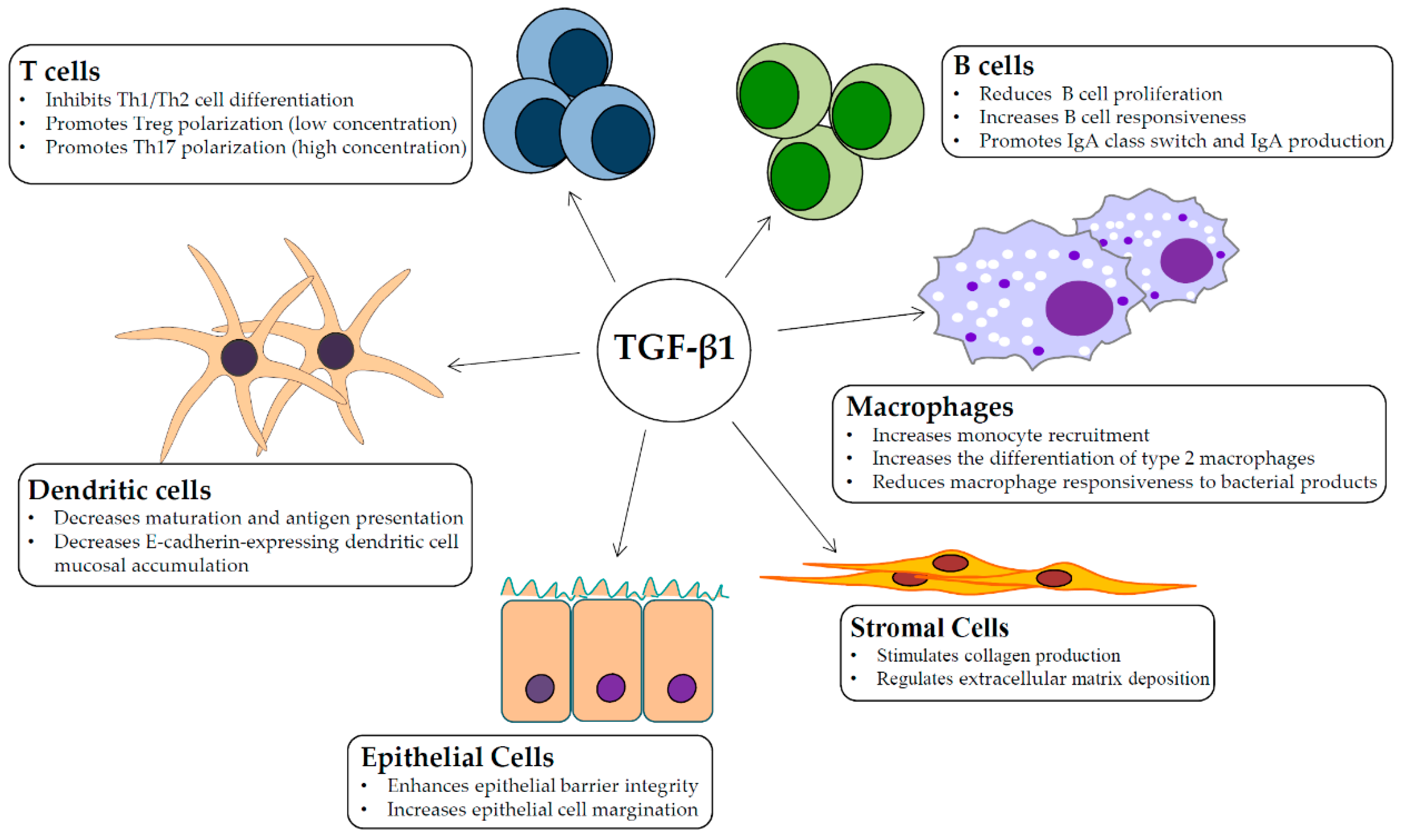
Abstract
1. Introduction
2. TGF-β1 Signaling and Intestinal Homeostasis
3. TGF-β1/Smad Signaling and Intestinal Fibrosis
4. Role of TGF-β1/Smad Signaling in Colorectal Cancer
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- MacDonald, T.T.; Monteleone, G. Immunity, inflammation, and allergy in the gut. Science 2005, 307, 1920–1925. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.T.; Monteleone, I.; Fantini, M.C.; Monteleone, G. Regulation of Homeostasis and Inflammation in the Intestine. Gastroenterology 2011, 140, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Bauche, D.; Marie, J.C. Transforming growth factor beta: A master regulator of the gut microbiota and immune cell interactions. Clin. Transl. Immunol. 2017, 6, e136. [Google Scholar] [CrossRef] [PubMed]
- Skeen, V.R.; Paterson, I.; Paraskeva, C.; Williams, A.C. TGF-beta 1 Signalling, Connecting Aberrant Inflammation and Colorectal Tumorigenesis. Curr. Pharm. Des. 2012, 18, 3874–3888. [Google Scholar] [CrossRef]
- Piek, E.; Heldin, C.H.; Ten Dijke, P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999, 13, 2105–2124. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Smads as transcriptional co-modulators. Curr. Opin. Cell Biol. 2000, 12, 235–243. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Gene Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef]
- Hayashi, H.; Abdollah, S.; Qiu, Y.B.; Cai, J.X.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGF beta receptor and functions as an antagonist of TGF beta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, N.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGF beta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Shi, W.B.; Sun, C.X.; He, B.; Xiong, W.C.; Shi, X.M.; Yao, D.C.; Cao, X. GADD34-PP1c recruited by Smad7 dephosphorylates TGF beta type 1 receptor. J. Cell Biol. 2004, 164, 291–300. [Google Scholar] [CrossRef]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.T.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Zhang, S.P.; Fei, T.; Zhang, L.X.; Zhang, R.; Chen, F.; Ning, Y.H.; Han, Y.N.; Feng, X.H.; Meng, A.M.; Chen, Y.G. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell Biol. 2007, 27, 4488–4499. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-beta Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Abrogation of TGF beta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Fang, D.F.; Zhu, J.F. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J. Exp. Med. 2017, 214, 1861–1876. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef]
- Gorelik, L.; Constant, S.; Flavell, R.A. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.K.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Gorham, J.D.; Guler, M.L.; Fenoglio, D.; Gubler, U.; Murphy, K.M. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J. Immunol. 1998, 161, 1664–1670. [Google Scholar] [PubMed]
- Gorelik, L.; Fields, P.E.; Flavell, R.A. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J. Immunol. 2000, 165, 4773–4777. [Google Scholar] [CrossRef] [PubMed]
- Heath, V.L.; Murphy, E.E.; Crain, C.; Tomlinson, M.G.; O’Garra, A. TGF-beta 1 down-regulates Th2 development and results in decreased IL-4-induced STAT6 activation and GATA-3 expression. Eur. J. Immunol. 2000, 30, 2639–2649. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4(+)CD25(-) T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef]
- Rudensky, A.Y. Regulatory T cells and Foxp3. Immunol. Rev. 2011, 241, 260–268. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef]
- Marie, J.C.; Letterio, J.J.; Gavin, M.; Rudensky, A.Y. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 2005, 201, 1061–1067. [Google Scholar] [CrossRef]
- Chen, W.J.; Jin, W.W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4(+)CD25(-) naive T cells to CD4(+)CD25(+) regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Davidson, T.S.; DiPaolo, R.J.; Andersson, J.; Shevach, E.M. Cutting edge: IL-2 is essential for TGF-beta-mediated induction of Foxp(3+) T regulatory cells. J. Immunol. 2007, 178, 4022–4026. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Zhang, P.; Li, J.; Kulkarni, A.B.; Perruche, S.; Chen, W.J. A critical function for TGF-beta signaling in the development of natural CD4(+)CD25(+)Foxp3(+) regulatory T cells. Nat. Immunol. 2008, 9, 632–640. [Google Scholar] [CrossRef] [PubMed]
- del Rio, M.L.; Bernhardt, G.; Rodriguez-Barbosa, J.I.; Forster, R. Development and functional specialization of CD103+ dendritic cells. Immunol. Rev. 2010, 234, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Siddiqui, K.R.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103(+) DCs induces Foxp3(+) regulatory T cells via a TGF-beta- and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.J.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007, 204, 1765–1774. [Google Scholar] [CrossRef]
- Kashiwagi, I.; Morita, R.; Schichita, T.; Komai, K.; Saeki, K.; Matsumoto, M.; Takeda, K.; Nomura, M.; Hayashi, A.; Kanai, T.; et al. Smad2 and Smad3 Inversely Regulate TGF-beta Autoinduction in Clostridium butyricum-Activated Dendritic Cells. Immunity 2015, 43, 65–79. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.L.; Du, J.G.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing ROR gamma t function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef]
- Mora, J.R.; von Andrian, U.H. T-cell homing specificity and plasticity: New concepts and future challenges. Trends Immunol. 2006, 27, 235–243. [Google Scholar] [CrossRef]
- Zhang, N.; Bevan, M.J. Transforming Growth Factor-beta Signaling Controls the Formation and Maintenance of Gut-Resident Memory T Cells by Regulating Migration and Retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef]
- Cazac, B.B.; Roes, J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity 2000, 13, 443–451. [Google Scholar] [CrossRef]
- Ruane, D.; Chorny, A.; Lee, H.; Faith, J.; Pandey, G.; Shan, M.; Simchoni, N.; Rahman, A.; Garg, A.; Weinstein, E.G.; et al. Microbiota regulate the ability of lung dendritic cells to induce IgA class-switch recombination and generate protective gastrointestinal immune responses. J. Exp. Med. 2016, 213, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Roes, J.; Choi, B.K.; Cazac, B.B. Redirection of B cell responsiveness by transforming growth factor beta receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 7241–7246. [Google Scholar] [CrossRef] [PubMed]
- Kubinak, J.L.; Petersen, C.; Stephens, W.Z.; Soto, R.; Bake, E.; O’Connell, R.M.; Round, J.L. MyD88 Signaling in T Cells Directs IgA-Mediated Control of the Microbiota to Promote Health. Cell Host Microbe 2015, 17, 153–163. [Google Scholar] [CrossRef]
- Reboldi, A.; Arnon, T.I.; Rodda, L.B.; Atakilit, A.; Sheppard, D.; Cyster, J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 2016, 352, aaf4822. [Google Scholar] [CrossRef]
- Klein, J.; Ju, W.J.; Heyer, J.; Wittek, B.; Haneke, T.; Knaus, P.; Kucherlapati, R.; Bottinger, E.P.; Nitschke, L.; Kneitz, B. B cell-specific deficiency for smad2 in vivo leads to defects in TGF-beta-Directed IgA switching and changes in B cell fate. J. Immunol. 2006, 176, 2389–2396. [Google Scholar] [CrossRef]
- Park, S.R.; Lee, J.H.; Kim, P.H. Smad3 and Smad4 mediate transforming growth factor-beta 1-induced IgA expression in murine B lymphocytes. Eur. J. Immunol. 2001, 31, 1706–1715. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Cortez, V.S.; Cervantes-Barragan, L.; Robinette, M.L.; Bando, J.K.; Wang, Y.M.; Geiger, T.L.; Gilfillan, S.; Fuchs, A.; Vivier, E.; Sun, J.C.; et al. Transforming Growth Factor-beta Signaling Guides the Differentiation of Innate Lymphoid Cells in Salivary Glands. Immunity 2016, 44, 1127–1139. [Google Scholar] [CrossRef]
- Wang, L.; Tang, J.; Yang, X.; Zanvit, P.; Cui, K.R.; Ku, W.L.; Jin, W.W.; Zhang, D.F.; Goldberg, N.; Cain, A.; et al. TGF-beta induces ST2 and programs ILC2 development. Nat. Commun. 2020, 11, 35. [Google Scholar] [CrossRef]
- Ihara, S.; Hirata, Y.; Serizawa, T.; Suzuki, N.; Sakitani, K.; Kinoshita, H.; Hayakawa, Y.; Nakagawa, H.; Ijichi, H.; Tateishi, K.; et al. TGF-beta Signaling in Dendritic Cells Governs Colonic Homeostasis by Controlling Epithelial Differentiation and the Luminal Microbiota. J. Immunol. 2016, 196, 4603–4613. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, R.; Larmonier, C.B.; Thurston, R.D.; Midura-Kiela, M.T.; Zheng, S.G.; Ghishan, F.K.; Kiela, P.R. Dendritic Cell-Specific Disruption of TGF-beta Receptor II Leads to Altered Regulatory T Cell Phenotype and Spontaneous Multiorgan Autoimmunity. J. Immunol. 2012, 189, 3878–3893. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Reizis, B.; Melton, A.C.; Masteller, E.; Tang, Q.Z.; Proctor, J.M.; Wang, Y.L.; Bernstein, X.; Huang, X.Z.; Reichardt, L.F.; et al. Loss of integrin alpha(v)beta(8) on dendritic cells causes autoimmunity and colitis in mice. Nature 2007, 449, 361. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.R.R.; Laffont, S.; Powrie, F. E-Cadherin Marks a Subset of Inflammatory Dendritic Cells that Promote T Cell-Mediated Colitis. Immunity 2010, 32, 557–567. [Google Scholar] [CrossRef]
- Smythies, L.E.; Maheshwari, A.; Clements, R.; Eckhoff, D.; Novak, L.; Vu, H.L.; Mosteller-Barnum, L.M.; Sellers, M.; Smith, P.D. Mucosal IL-8 and TGF-beta recruit blood monocytes: Evidence for cross-talk between the lamina propria stroma and myeloid cells. J. Leukoc. Biol. 2006, 80, 492–499. [Google Scholar] [CrossRef]
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and Tissue Specialization. Annu. Rev. Immunol. 2015, 33, 643–675. [Google Scholar] [CrossRef]
- Rani, R.; Smulian, A.G.; Greaves, D.R.; Hogan, S.P.; Herbert, D.R. TGF-beta limits IL-33 production and promotes the resolution of colitis through regulation of macrophage function. Eur. J. Immunol. 2011, 41, 2000–2009. [Google Scholar] [CrossRef]
- Wang, H.M.; Radjendirane, V.; Wary, K.K.; Chakrabarty, S. Transforming growth factor beta regulates cell-cell adhesion through extracellular matrix remodeling and activation of focal adhesion kinase in human colon carcinoma Moser cells. Oncogene 2004, 23, 5558–5561. [Google Scholar] [CrossRef]
- Howe, K.L.; Reardon, C.; Wang, A.; Nazli, A.; McKay, D.M. Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157: H7-induced increased permeability. Am. J. Pathol. 2005, 167, 1587–1597. [Google Scholar] [CrossRef]
- Oshima, H.; Nakayama, M.; Han, T.S.; Naoi, K.; Ju, X.L.; Maeda, Y.; Robine, S.; Tsuchiya, K.; Sato, T.; Sato, H.; et al. Suppressing TGF beta Signaling in Regenerating Epithelia in an Inflammatory Microenvironment Is Sufficient to Cause Invasive Intestinal Cancer. Cancer Res. 2015, 75, 766–776. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Biancheri, P.; Giuffrida, P.; Docena, G.H.; MacDonald, T.T.; Corazza, G.R.; Di Sabatino, A. The role of transforming growth factor (TGF)-beta in modulating the immune response and fibrogenesis in the gut. Cytokine Growth F R. 2014, 25, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [PubMed]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H.; et al. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P., Jr.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; Consortium, I.-L. TGF-beta signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-beta: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6. [Google Scholar] [CrossRef]
- Angelov, N.; Moutsopoulos, N.; Jeong, M.J.; Nares, S.; Ashcroft, G.; Wahl, S.M. Aberrant mucosal wound repair in the absence of secretory leukocyte protease inhibitor. Thromb. Haemost. 2004, 92, 288–297. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Lei, K.; Jin, W.; Longenecker, G.; Kulkarni, A.B.; Greenwell-Wild, T.; Hale-Donze, H.; McGrady, G.; Song, X.Y.; Wahl, S.M. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for normal wound healing. Nat. Med. 2000, 6, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-beta and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-beta-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Salmela, M.T.; Pender, S.L.; Karjalainen-Lindsberg, M.L.; Puolakkainen, P.; Macdonald, T.T.; Saarialho-Kere, U. Collagenase-1 (MMP-1), matrilysin-1 (MMP-7), and stromelysin-2 (MMP-10) are expressed by migrating enterocytes during intestinal wound healing. Scand. J. Gastroenterol. 2004, 39, 1095–1104. [Google Scholar] [CrossRef]
- Burke, J.P.; Mulsow, J.J.; O’Keane, C.; Docherty, N.G.; Watson, R.W.; O’Connell, P.R. Fibrogenesis in Crohn’s disease. Am. J. Gastroenterol. 2007, 102, 439–448. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Jackson, C.L.; Pickard, K.M.; Buckley, M.; Rovedatti, L.; Leakey, N.A.; Picariello, L.; Cazzola, P.; Monteleone, G.; Tonelli, F.; et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut 2009, 58, 777–789. [Google Scholar] [CrossRef]
- Stallmach, A.; Schuppan, D.; Riese, H.H.; Matthes, H.; Riecken, E.O. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology 1992, 102, 1920–1929. [Google Scholar] [CrossRef]
- Li, G.; Zhou, R.; Zhang, Q.; Jiang, B.; Wu, Q.; Wang, C. Fibroproliferative effect of microRNA-21 in hypertrophic scar derived fibroblasts. Exp. Cell Res. 2016, 345, 93–99. [Google Scholar] [CrossRef]
- Speca, S.; Rousseaux, C.; Dubuquoy, C.; Rieder, F.; Vetuschi, A.; Sferra, R.; Giusti, I.; Bertin, B.; Dubuquoy, L.; Gaudio, E.; et al. Novel PPARgamma Modulator GED-0507-34 Levo Ameliorates Inflammation-driven Intestinal Fibrosis. Inflamm. Bowel Dis. 2016, 22, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Vallance, B.A.; Gunawan, M.I.; Hewlett, B.; Bercik, P.; Van Kampen, C.; Galeazzi, F.; Sime, P.J.; Gauldie, J.; Collins, S.M. TGF-beta1 gene transfer to the mouse colon leads to intestinal fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G116–G128. [Google Scholar] [CrossRef] [PubMed]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in IBD—A dynamic, multifactorial process. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Theiss, A.L.; Simmons, J.G.; Jobin, C.; Lund, P.K. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J. Biol. Chem. 2005, 280, 36099–36109. [Google Scholar] [CrossRef]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J. Clin. Investig. 2001, 108, 601–609. [Google Scholar] [CrossRef]
- Boirivant, M.; Pallone, F.; Di Giacinto, C.; Fina, D.; Monteleone, I.; Marinaro, M.; Caruso, R.; Colantoni, A.; Palmieri, G.; Sanchez, M.; et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology 2006, 131, 1786–1798. [Google Scholar] [CrossRef]
- Monteleone, G.; Fantini, M.C.; Onali, S.; Zorzi, F.; Sancesario, G.; Bernardini, S.; Calabrese, E.; Viti, F.; Monteleone, I.; Biancone, L.; et al. Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn’s disease. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 870–876. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sands, B.E.; Rossiter, G.; Li, X.; Usiskin, K.; Zhan, X.; Colombel, J.F. Effects of Mongersen (GED-0301) on Endoscopic and Clinical Outcomes in Patients with Active Crohn’s Disease. Gastroenterology 2018, 154, 61–64. [Google Scholar] [CrossRef]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef]
- Sands, B.E.; Feagan, B.G.; Sandborn, W.J.; Schreiber, S.; Peyrin-Biroulet, L.; Frederic Colombel, J.; Rossiter, G.; Usiskin, K.; Ather, S.; Zhan, X.; et al. Mongersen (GED-0301) for Active Crohn’s Disease: Results of a Phase 3 Study. Am. J. Gastroenterol. 2020, 115, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, F.; Calabrese, E.; Monteleone, I.; Fantini, M.; Onali, S.; Biancone, L.; Pallone, F.; Monteleone, G. A phase 1 open-label trial shows that smad7 antisense oligonucleotide (GED0301) does not increase the risk of small bowel strictures in Crohn’s disease. Aliment. Pharmacol. Ther. 2012, 36, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, P.; Pinzani, M.; Corazza, G.R.; Di Sabatino, A. Biomarkers of intestinal fibrosis—One step towards clinical trials for stricturing inflammatory bowel disease. United Eur. Gastroenterol. J. 2016, 4, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Izzo, R.; Bevivino, G.; De Simone, V.; Sedda, S.; Monteleone, I.; Marafini, I.; Di Giovangiulio, M.; Rizzo, A.; Franze, E.; Colantoni, A.; et al. Knockdown of Smad7 With a Specific Antisense Oligonucleotide Attenuates Colitis and Colitis-Driven Colonic Fibrosis in Mice. Inflamm. Bowel Dis. 2018, 24, 1213–1224. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Stolfi, C.; Marafini, I.; De Simone, V.; Pallone, F.; Monteleone, G. The dual role of Smad7 in the control of cancer growth and metastasis. Int. J. Mol. Sci. 2013, 14, 23774–23790. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef]
- Jess, T.; Gamborg, M.; Matzen, P.; Munkholm, P.; Sorensen, T.I. Increased risk of intestinal cancer in Crohn’s disease: A meta-analysis of population-based cohort studies. Am. J. Gastroenterol. 2005, 100, 2724–2729. [Google Scholar] [CrossRef]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.B.; Harpaz, N.; Itzkowitz, S.; Hossain, S.; Matula, S.; Kornbluth, A.; Bodian, C.; Ullman, T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: A cohort study. Gastroenterology 2007, 133, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Buckhaults, P.; Rago, C.; St Croix, B.; Romans, K.E.; Saha, S.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Secreted and cell surface genes expressed in benign and malignant colorectal tumors. Cancer Res. 2001, 61, 6996–7001. [Google Scholar] [PubMed]
- Friedman, E.; Gold, L.I.; Klimstra, D.; Zeng, Z.S.; Winawer, S.; Cohen, A. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol. Biomark. Prev. 1995, 4, 549–554. [Google Scholar]
- Schroy, P.; Rifkin, J.; Coffey, R.J.; Winawer, S.; Friedman, E. Role of transforming growth factor beta 1 in induction of colon carcinoma differentiation by hexamethylene bisacetamide. Cancer Res. 1990, 50, 261–265. [Google Scholar]
- Tsushima, H.; Kawata, S.; Tamura, S.; Ito, N.; Shirai, Y.; Kiso, S.; Imai, Y.; Shimomukai, H.; Nomura, Y.; Matsuda, Y.; et al. High levels of transforming growth factor beta 1 in patients with colorectal cancer: Association with disease progression. Gastroenterology 1996, 110, 375–382. [Google Scholar] [CrossRef]
- Turtoi, A.; Blomme, A.; Debois, D.; Somja, J.; Delvaux, D.; Patsos, G.; Di Valentin, E.; Peulen, O.; Mutijima, E.N.; De Pauw, E.; et al. Organized proteomic heterogeneity in colorectal cancer liver metastases and implications for therapies. Hepatology 2014, 59, 924–934. [Google Scholar] [CrossRef]
- Tsushima, H.; Ito, N.; Tamura, S.; Matsuda, Y.; Inada, M.; Yabuuchi, I.; Imai, Y.; Nagashima, R.; Misawa, H.; Takeda, H.; et al. Circulating transforming growth factor beta 1 as a predictor of liver metastasis after resection in colorectal cancer. Clin. Cancer Res. 2001, 7, 1258–1262. [Google Scholar]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming Growth Factor beta Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pasche, B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007, 16, R14–R20. [Google Scholar] [CrossRef]
- Grady, W.M.; Rajput, A.; Myeroff, L.; Liu, D.F.; Kwon, K.; Willis, J.; Markowitz, S. Mutation of the type II transforming growth factor-beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998, 58, 3101–3104. [Google Scholar] [PubMed]
- Grady, W.M.; Markowitz, S.D. Genetic and epigenetic alterations in colon cancer. Annu. Rev. Genom. Hum. Genet. 2002, 3, 101–128. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef]
- Pinheiro, M.; Ahlquist, T.; Danielsen, S.A.; Lind, G.E.; Veiga, I.; Pinto, C.; Costa, V.; Afonso, L.; Sousa, O.; Fragoso, M.; et al. Colorectal carcinomas with microsatellite instability display a different pattern of target gene mutations according to large bowel site of origin. BMC Cancer 2010, 10, 587. [Google Scholar] [CrossRef]
- De Miranda, N.F.; van Dinther, M.; van den Akker, B.E.; van Wezel, T.; ten Dijke, P.; Morreau, H. Transforming Growth Factor beta Signaling in Colorectal Cancer Cells With Microsatellite Instability Despite Biallelic Mutations in TGFBR2. Gastroenterology 2015, 148, 1427–1437. [Google Scholar] [CrossRef]
- Trobridge, P.; Knoblaugh, S.; Washington, M.K.; Munoz, N.M.; Tsuchiya, K.D.; Rojas, A.; Song, X.; Ulrich, C.M.; Sasazuki, T.; Shirasawa, S.; et al. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology 2009, 136, 1680–1688. [Google Scholar] [CrossRef]
- Geng, L.; Chaudhuri, A.; Talmon, G.; Wisecarver, J.L.; Wang, J. TGF-Beta suppresses VEGFA-mediated angiogenesis in colon cancer metastasis. PLoS ONE 2013, 8, e59918. [Google Scholar] [CrossRef]
- Ku, J.L.; Park, S.H.; Yoon, K.A.; Shin, Y.K.; Kim, K.H.; Choi, J.S.; Kang, H.C.; Kim, I.J.; Han, I.O.; Park, J.G. Genetic alterations of the TGF-beta signaling pathway in colorectal cancer cell lines: A novel mutation in Smad3 associated with the inactivation of TGF-beta-induced transcriptional activation. Cancer Lett. 2007, 247, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Morgan, W.; Lewis, S.; Willis, J.; Elston, R.C.; Markowitz, S.D.; Wiesner, G.L. Is TGFBR1*6A a susceptibility allele for nonsyndromic familial colorectal neoplasia? Cancer Epidemiol. Biomark. Prev. 2007, 16, 892–894. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carvajal-Carmona, L.G.; Churchman, M.; Bonilla, C.; Walther, A.; Lefevre, J.H.; Kerr, D.; Dunlop, M.; Houlston, R.; Bodmer, W.F.; Tomlinson, I. Comprehensive assessment of variation at the transforming growth factor beta type 1 receptor locus and colorectal cancer predisposition. Proc. Natl. Acad. Sci. USA 2010, 107, 7858–7862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Huang, Y.; Cheng, B.; Wang, Y.; Xiong, B. TGFBR1*6A is a potential modifier of migration and invasion in colorectal cancer cells. Oncol. Lett. 2018, 15, 3971–3976. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Cossu, C.; Mancarella, S.; Giannelli, G. The Interactivity between TGFbeta and BMP Signaling in Organogenesis, Fibrosis, and Cancer. Cells 2019, 8, 1130. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; de Rooij, K.; et al. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208. [Google Scholar] [CrossRef]
- Means, A.L.; Freeman, T.J.; Zhu, J.; Woodbury, L.G.; Marincola-Smith, P.; Wu, C.; Meyer, A.R.; Weaver, C.J.; Padmanabhan, C.; An, H.; et al. Epithelial Smad4 Deletion Up-Regulates Inflammation and Promotes Inflammation-Associated Cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 257–276. [Google Scholar] [CrossRef]
- Takagi, Y.; Koumura, H.; Futamura, M.; Aoki, S.; Ymaguchi, K.; Kida, H.; Tanemura, H.; Shimokawa, K.; Saji, S. Somatic alterations of the SMAD-2 gene in human colorectal cancers. Br. J. Cancer 1998, 78, 1152–1155. [Google Scholar] [CrossRef]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2015, 60, 762–772. [Google Scholar] [CrossRef]
- Houlston, R.; Bevan, S.; Williams, A.; Young, J.; Dunlop, M.; Rozen, P.; Eng, C.; Markie, D.; Woodford-Richens, K.; Rodriguez-Bigas, M.A.; et al. Mutations in DPC4 (SMAD4) cause juvenile polyposis syndrome, but only account for a minority of cases. Hum. Mol. Genet. 1998, 7, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Jarvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Boulay, J.L.; Mild, G.; Lowy, A.; Reuter, J.; Lagrange, M.; Terracciano, L.; Laffer, U.; Herrmann, R.; Rochlitz, C. SMAD7 is a prognostic marker in patients with colorectal cancer. Int. J. Cancer 2003, 104, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Broderick, P.; Carvajal-Carmona, L.; Pittman, A.M.; Webb, E.; Howarth, K.; Rowan, A.; Lubbe, S.; Spain, S.; Sullivan, K.; Fielding, S.; et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat. Genet. 2007, 39, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Tenesa, A.; Farrington, S.M.; Prendergast, J.G.; Porteous, M.E.; Walker, M.; Haq, N.; Barnetson, R.A.; Theodoratou, E.; Cetnarskyj, R.; Cartwright, N.; et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet. 2008, 40, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Herrick, J.; Curtin, K.; Samowitz, W.; Wolff, R.K.; Caan, B.J.; Duggan, D.; Potter, J.D.; Peters, U. Increased risk of colon cancer associated with a genetic polymorphism of SMAD7. Cancer Res. 2010, 70, 1479–1485. [Google Scholar] [CrossRef]
- Thompson, C.L.; Plummer, S.J.; Acheson, L.S.; Tucker, T.C.; Casey, G.; Li, L. Association of common genetic variants in SMAD7 and risk of colon cancer. Carcinogenesis 2009, 30, 982–986. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, W.; Nie, M.; Li, C.; Wang, L. SMAD7 polymorphisms and colorectal cancer risk: A meta-analysis of case-control studies. Oncotarget 2016, 7, 75561–75570. [Google Scholar] [CrossRef]
- Li, J.; Zou, L.; Zhou, Y.; Li, L.; Zhu, Y.; Yang, Y.; Gong, Y.; Lou, J.; Ke, J.; Zhang, Y.; et al. A low-frequency variant in SMAD7 modulates TGF-beta signaling and confers risk for colorectal cancer in Chinese population. Mol. Carcinog. 2017, 56, 1798–1807. [Google Scholar] [CrossRef]
- Campbell, P.T.; Lin, Y.; Bien, S.A.; Figueiredo, J.C.; Harrison, T.A.; Guinter, M.J.; Berndt, S.I.; Brenner, H.; Chan, A.T.; Chang-Claude, J.; et al. Association of body mass index with colorectal cancer risk by genome-wide variants. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. Smad7 induces tumorigenicity by blocking TGF-beta-induced growth inhibition and apoptosis. Exp. Cell Res. 2005, 307, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Rachakonda, G.; Deane, N.G.; Datta, P.K. Smad7 induces hepatic metastasis in colorectal cancer. Br. J. Cancer 2008, 99, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Waldner, M.J.; Stolfi, C.; Sarra, M.; Fina, D.; Becker, C.; Neurath, M.F.; Macdonald, T.T.; Pallone, F.; Monteleone, G.; et al. Smad7 expression in T cells prevents colitis-associated cancer. Cancer Res. 2011, 71, 7423–7432. [Google Scholar] [CrossRef]
- Rizzo, A.; De Mare, V.; Rocchi, C.; Stolfi, C.; Colantoni, A.; Neurath, M.F.; Macdonald, T.T.; Pallone, F.; Monteleone, G.; Fantini, M.C. Smad7 induces plasticity in tumor-infiltrating Th17 cells and enables TNF-alpha-mediated killing of colorectal cancer cells. Carcinogenesis 2014, 35, 1536–1546. [Google Scholar] [CrossRef]
- Stolfi, C.; De Simone, V.; Colantoni, A.; Franze, E.; Ribichini, E.; Fantini, M.C.; Caruso, R.; Monteleone, I.; Sica, G.S.; Sileri, P.; et al. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell Death Dis. 2014, 5, e1073. [Google Scholar] [CrossRef]
- De Simone, V.; Bevivino, G.; Sedda, S.; Izzo, R.; Laudisi, F.; Dinallo, V.; Franze, E.; Colantoni, A.; Ortenzi, A.; Salvatori, S.; et al. Smad7 knockdown activates protein kinase RNA-associated eIF2alpha pathway leading to colon cancer cell death. Cell Death Dis. 2017, 8, e2681. [Google Scholar] [CrossRef]
- Polvani, S.; Pepe, S.; Milani, S.; Galli, A. COUP-TFII in Health and Disease. Cells 2019, 9, 101. [Google Scholar] [CrossRef]
- Wang, H.; Nie, L.; Wu, L.; Liu, Q.; Guo, X. NR2F2 inhibits Smad7 expression and promotes TGF-beta-dependent epithelial-mesenchymal transition of CRC via transactivation of miR-21. Biochem. Biophys. Res. Commun. 2017, 485, 181–188. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolfi, C.; Troncone, E.; Marafini, I.; Monteleone, G. Role of TGF-Beta and Smad7 in Gut Inflammation, Fibrosis and Cancer. Biomolecules 2021, 11, 17. https://doi.org/10.3390/biom11010017
Stolfi C, Troncone E, Marafini I, Monteleone G. Role of TGF-Beta and Smad7 in Gut Inflammation, Fibrosis and Cancer. Biomolecules. 2021; 11(1):17. https://doi.org/10.3390/biom11010017
Chicago/Turabian StyleStolfi, Carmine, Edoardo Troncone, Irene Marafini, and Giovanni Monteleone. 2021. "Role of TGF-Beta and Smad7 in Gut Inflammation, Fibrosis and Cancer" Biomolecules 11, no. 1: 17. https://doi.org/10.3390/biom11010017
APA StyleStolfi, C., Troncone, E., Marafini, I., & Monteleone, G. (2021). Role of TGF-Beta and Smad7 in Gut Inflammation, Fibrosis and Cancer. Biomolecules, 11(1), 17. https://doi.org/10.3390/biom11010017