The Cytotoxicity of RNase-Derived Peptides

 ,
,
Abstract
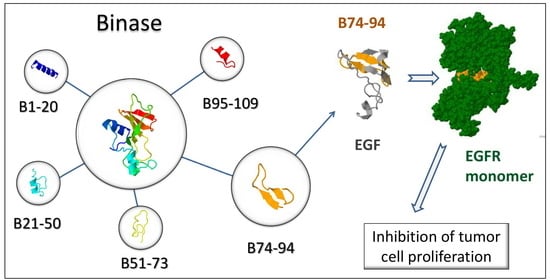
1. Introduction
2. Materials and Methods
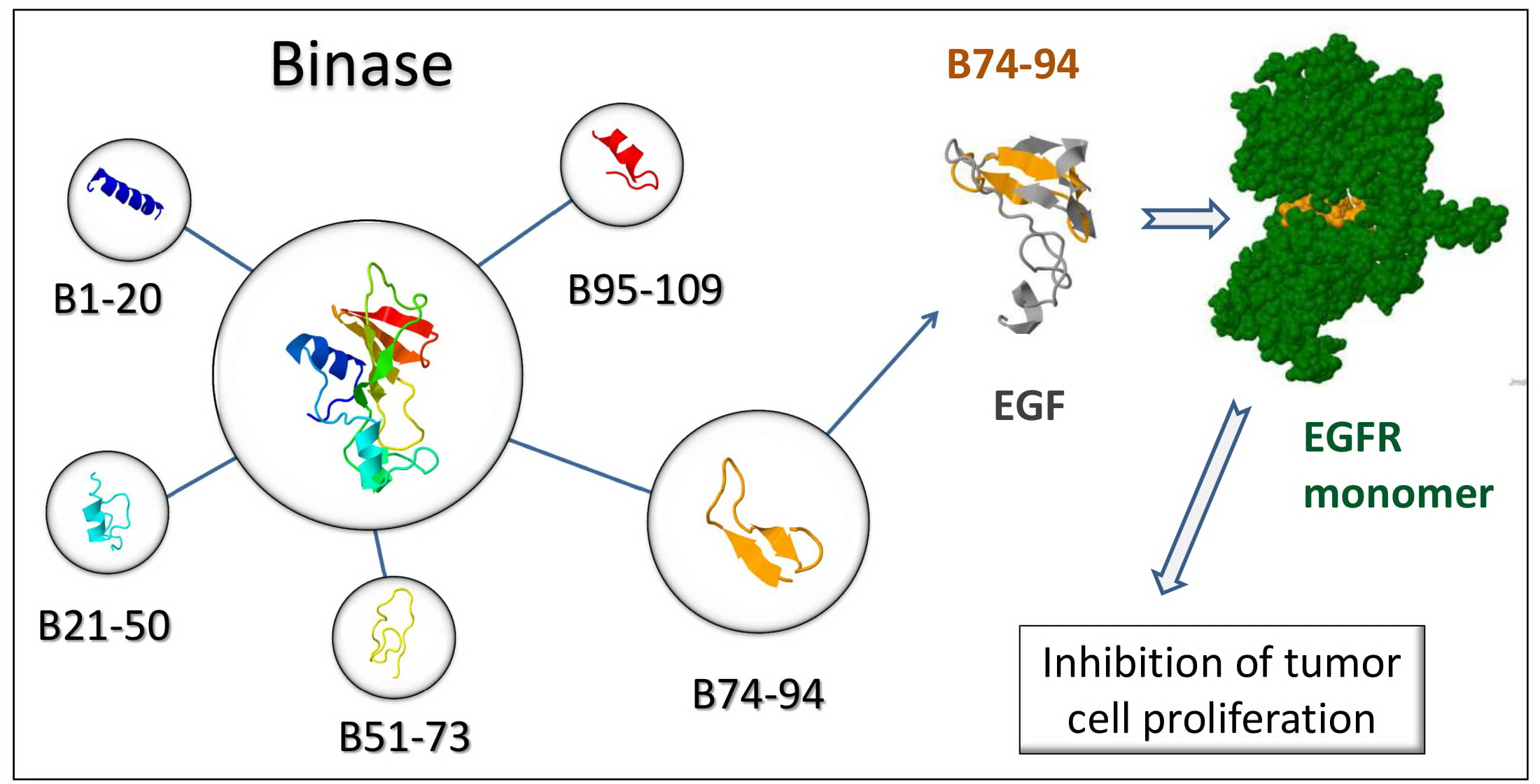
2.1. Binase and Binase-Derived Peptides
2.2. Cell Cultures
2.3. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Assay
2.4. Peptide Characterization and Modeling
2.5. Immunofluorescence Microscopy
2.6. Statistical Analysis
3. Results and Discussion
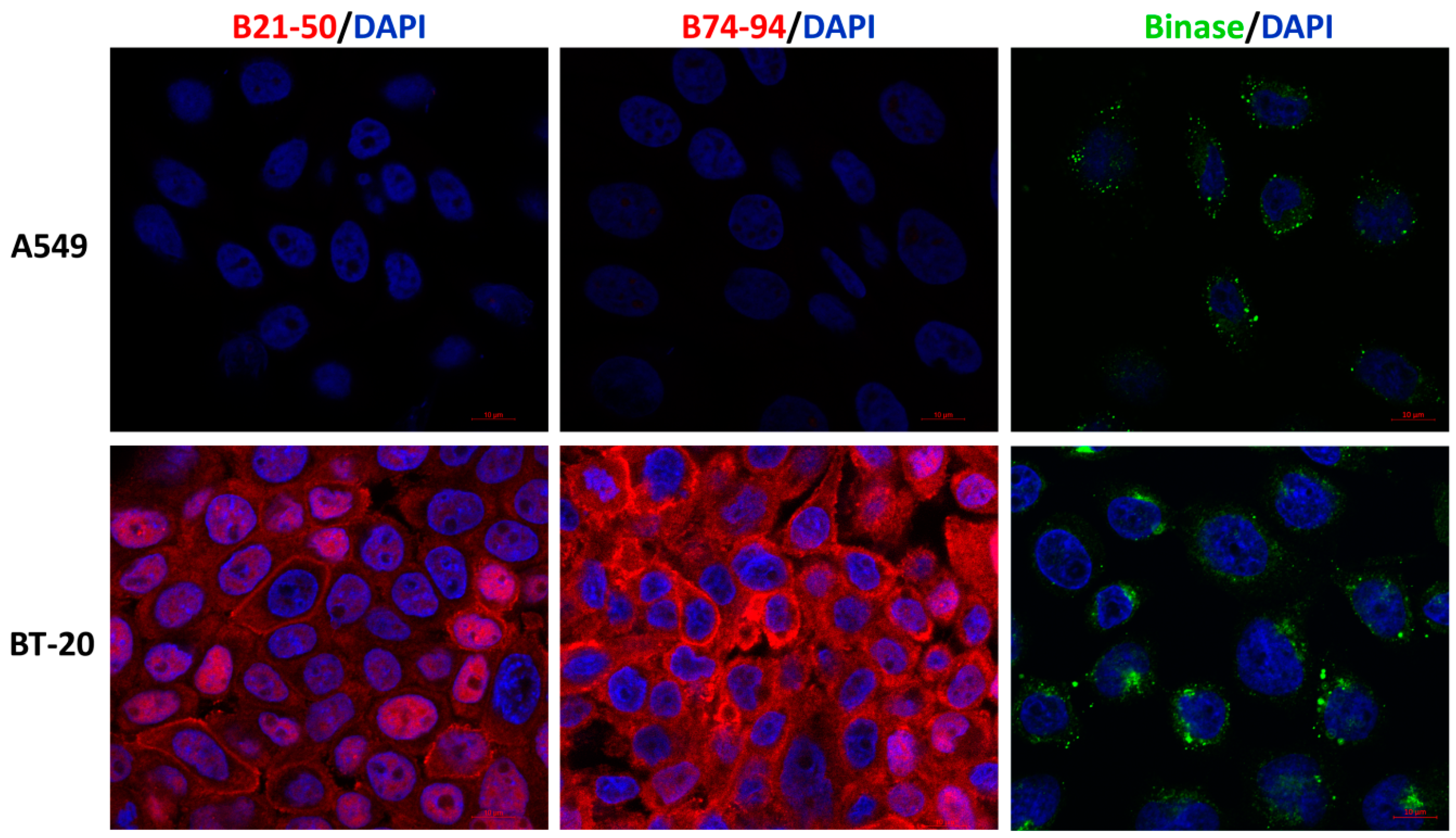
3.1. Characterization of Anticancer Potential of Binase-Derived Peptides
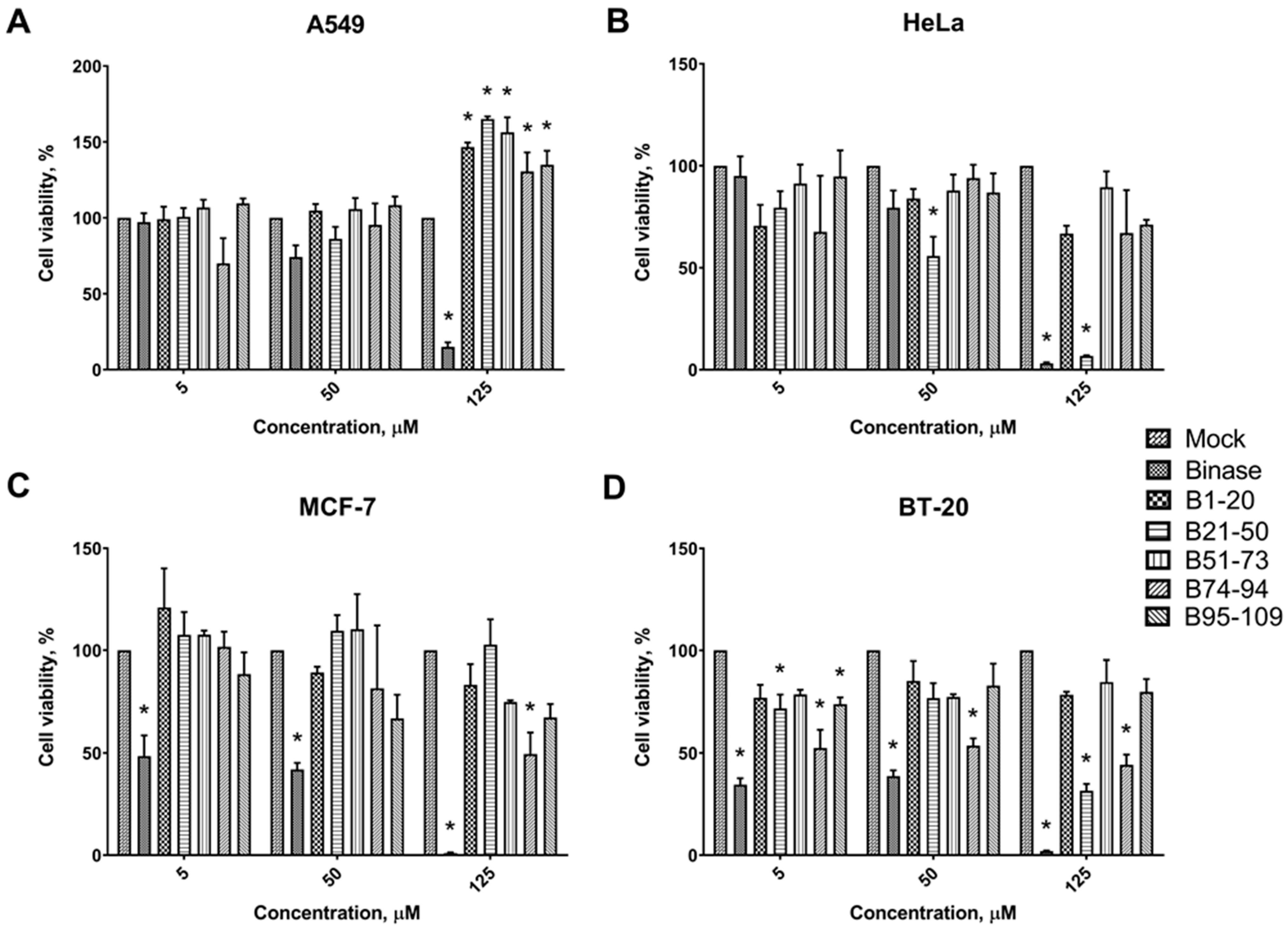
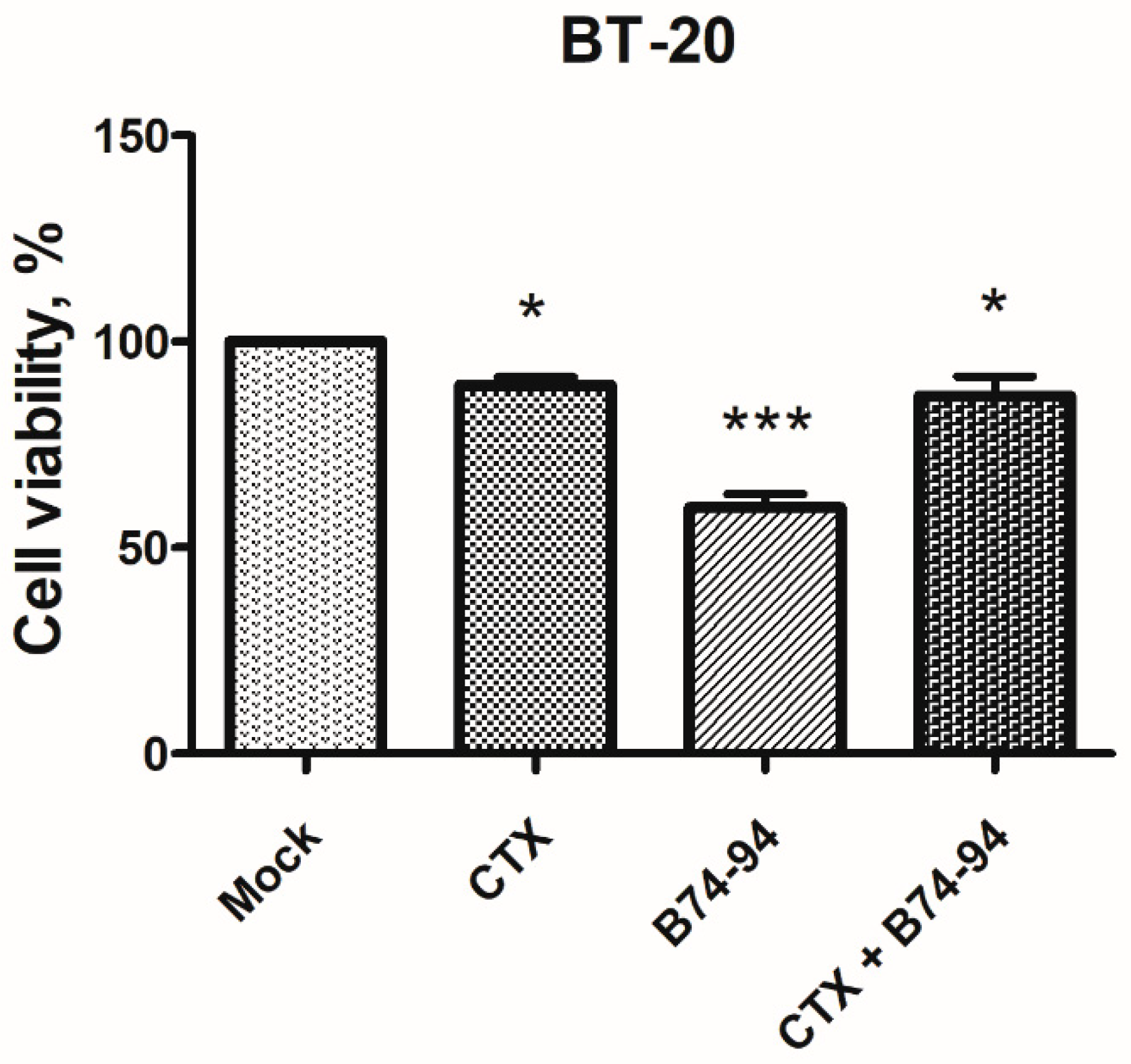
3.2. Cytotoxic Effects of Binase-Derived Peptides toward HeLa, A549, BT-20, and MCF-7 Cells
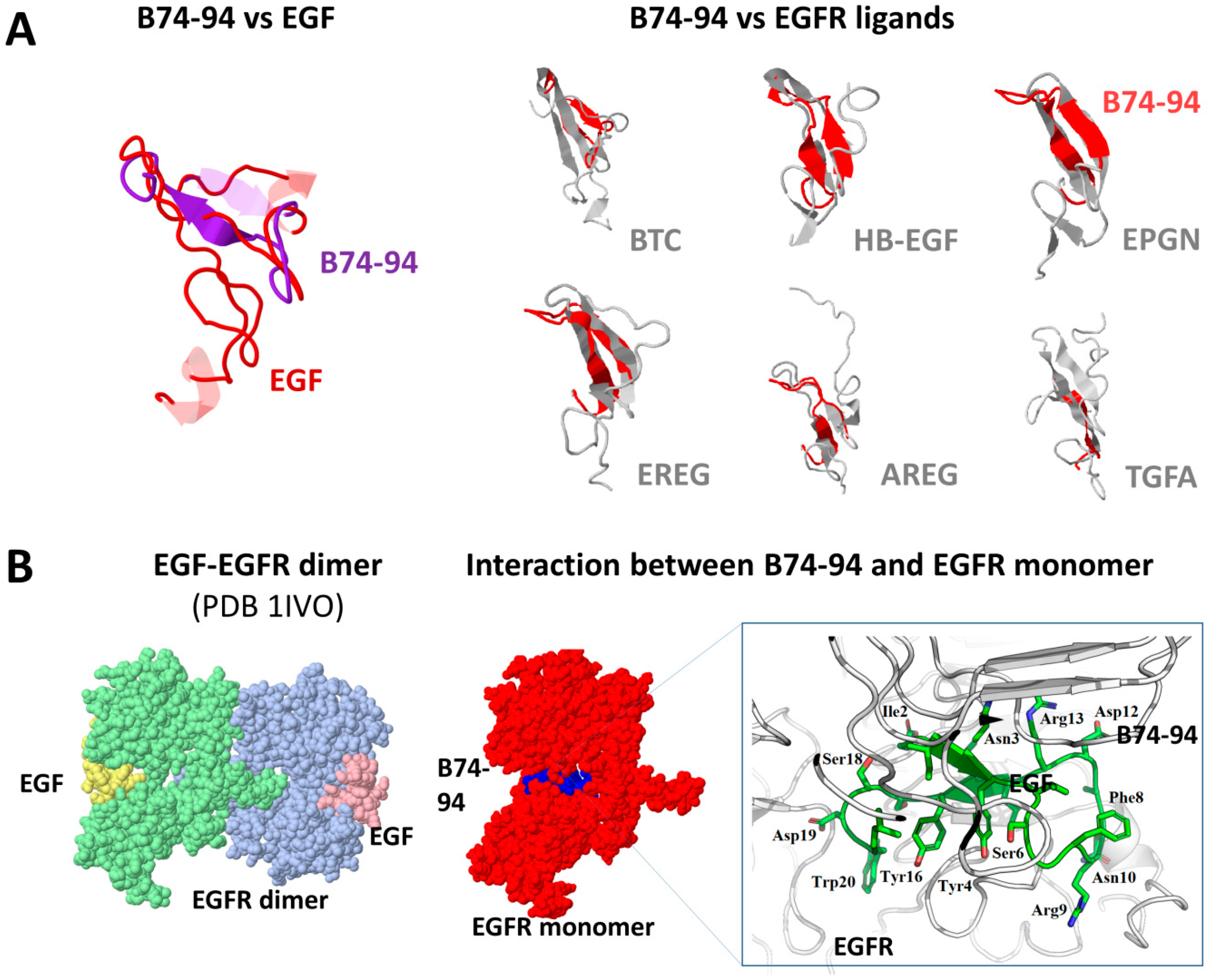
3.3. Assessment of Putative Interaction between EGFR and Binase-Derived Peptides
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A.R.B. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed]
- Domalaon, R.; Findlay, B.; Ogunsina, M.; Arthur, G.; Schweizer, F. Ultrashort cationic lipopeptides and lipopeptoids: Evaluation and mechanistic insights against epithelial cancer cells. Peptides 2016, 84, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Uggerhøj, L.E.; Poulsen, T.J.; Munk, J.K.; Fredborg, M.; Sondergaard, T.E.; Frimodt-Moller, N.; Hansen, P.R.; Wimmer, R. Rational Design of Alpha-Helical Antimicrobial Peptides: Do’s and Don’ts. ChemBioChem 2015, 16, 242–253. [Google Scholar] [CrossRef]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget 2017, 28, 46635–46651. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2011, 33, 190–234. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Wang, X.-F.; Wang, H.-Y.; Liu, Y.; Chen, Y. Studies on Mechanism of Action of Anticancer Peptides by Modulation of Hydrophobicity Within a Defined Structural Framework. Mol. Cancer Ther. 2011, 10, 416–426. [Google Scholar] [CrossRef]
- Makarov, A.A.; Kolchinsky, A.; Ilinskaya, O.N. Binase and other microbial RNases as potential anticancer agents. BioEssays 2008, 30, 781–790. [Google Scholar] [CrossRef]
- Mitkevich, V.A.; Burnysheva, K.M.; Ilinskaya, O.N.; Pace, C.N.; Makarov, A.A. Cytotoxicity of RNase Sa to the acute myeloid leukemia Kasumi1 cells depends on the net charge. Oncoscience 2014, 11, 738–744. [Google Scholar] [CrossRef]
- Ardelt, W.; Ardelt, B.; Darzynkiewicz, Z. Ribonucleases as potential modalities in anticancer therapy. Eur. J. Pharmacol. 2009, 625, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Gotte, G.; Helmy, A.M.; Ercole, C.; Spadaccini, R.; Laurents, D.V.; Donadelli, M.; Picone, D. Double Domain Swapping in Bovine Seminal RNase: Formation of Distinct N- and C-swapped Tetramers and Multimers with Increasing Biological Activities. PLoS ONE 2012, 7, e46804. [Google Scholar] [CrossRef] [PubMed]
- Attery, A.; Dey, P.; Tripathi, P.; Batra, J.K. A ribonuclease inhibitor resistant dimer of human pancreatic ribonuclease displays specific antitumor activity. Int. J. Biol. Macromol. 2018, 107, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Mironova, N.L.; Petrushanko, I.Y.; Patutina, O.A.; Sen’kova, A.V.; Simonenko, O.V.; Mitkevich, V.A.; Markov, O.B.; Zenkova, M.A.; Makarov, A.A. Ribonuclease binase inhibits primary tumor growth and metastases via apoptosis induction in tumor cells. Cell Cycle 2013, 12, 2130–2131. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Spirin, P.V.; Fedorova, T.; Kretova, O.V.; Tchurikov, N.A.; Prassolov, V.S.; Ilinskaya, O.; Makarov, A.A. Sensitivity of acute myeloid leukemia Kasumi-1 cells to binase toxic action depends on the expression of KIT and AML1-ETO oncogenes. Cell Cycle 2011, 10, 4090–4097. [Google Scholar] [CrossRef]
- Makarov, A.A.; Ilinskaya, O.N. Cytotoxic ribonucleases: Molecular weapons and their targets. FEBS Lett. 2003, 540, 15–20. [Google Scholar] [CrossRef]
- Dudkina, E.; Kayumov, A.; Ulyanova, V.; Ilinskaya, O. New insight into secreted ribonuclease structure: Binase is a natural dimer. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Gotte, G.; Laurents, D.V.; Merlino, A.; Picone, D.; Spadaccini, R. Structural and functional relationships of natural and artificial dimeric bovine ribonucleases: New scaffolds for potential antitumor drugs. FEBS Lett. 2013, 587, 3601–3608. [Google Scholar] [CrossRef]
- Ilinskaya, O.; Decker, K.; Koschinski, A.; Dreyer, F.; Repp, H. Bacillus intermedius ribonuclease as inhibitor of cell proliferation and membrane current. Toxicology 2001, 156, 101–107. [Google Scholar] [CrossRef]
- Ilinskaya, O.N.; Singh, I.; Dudkina, E.V.; Ulyanova, V.V.; Kayumov, A.; Barreto, G. Direct inhibition of oncogenic KRAS by Bacillus pumilus ribonuclease (binase). Biochim. Biophys. Acta, Mol. Cell Res. 2016, 1863, 1559–1567. [Google Scholar] [CrossRef]
- Ulyanova, V.; Vershinina, V.; Ilinskaya, O. Barnase and binase: Twins with distinct fates. FEBS J. 2011, 278, 3633–3643. [Google Scholar] [CrossRef] [PubMed]
- Ilinskaya, O.N.; Zelenikhin, P.V.; Petrushanko, I.Y.; Mitkevich, V.A.; Prassolov, V.S.; Makarov, A.A. Binase induces apoptosis of transformed myeloid cells and does not induce T-cell immune response. Biochem. Biophys. Res. Commun. 2007, 361, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Ilinskaya, O.N.; Makarov, A.A. Antitumor RNases: Killer’s secrets. Cell Cycle 2015, 14, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, W.; Shogen, K.; Darzynkiewicz, Z. Onconase and Amphinase, the Antitumor Ribonucleases from Rana pipiens Oocytes. Curr. Pharm. Biotechnol. 2008, 9, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Tchurikov, N.A.; Zelenikhin, P.V.; Petrushanko, I.Y.; Makarov, A.A.; Ilinskaya, O. Binase cleaves cellular noncoding RNAs and affects coding mRNAs. FEBS J. 2010, 277, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Kretova, O.V.; Zelenikhin, P.V.; Prassolov, V.S.; Tchurikov, N.A.; Ilinskaya, O.N.; Makarov, A.A. Oncogenic c-kit transcript is a target for binase. Cell Cycle 2010, 9, 2674–2678. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Orlova, N.N.; Petrushanko, I.Y.; Simonenko, O.V.; Spirin, P.V. Expression of the FLT3-ITD oncogene sensitizes murine progenitor B-cell line BAF3 to cytotoxic action of binase. Mol. Biol. 2013, 47, 248–251. [Google Scholar] [CrossRef]
- Mironova, N.; Patutina, O.; Brenner, E.; Kurilshikov, A.; Vlassov, V.; Zenkova, M. The systemic tumor response to RNase A treatment affects the expression of genes involved in maintaining cell malignancy. Oncotarget 2017, 8, 78796–78810. [Google Scholar] [CrossRef][Green Version]
- Ulyanova, V.; Mahmud, R.S.; Laikov, A.; Dudkina, E.; Markelova, M.; Mostafa, A.; Pleschka, S.; Ilinskaya, O. Anti-Influenza Activity of the Ribonuclease Binase: Cellular Targets Detected by Quantitative Proteomics. Int. J. Mol. Sci. 2020, 21, 8294. [Google Scholar] [CrossRef]
- Dudkina, E.; Ulyanova, V.; Shah Mahmud, R.; Khodzhaeva, V.; Dao, L.; Vershinina, V.; Kolpakov, A.; Ilinskaya, O. Three-step procedure for preparation of pure Bacillus altitudinis ribonuclease. FEBS Open Bio. 2016, 6, 24–32. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P. In silico models for designing and discovering novel anticancer peptides. Sci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.; Open Source Drug Discovery Consortium. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Chaudhary, K.; Kumar, R.; Raghava, G.P.S. Computer-Aided Virtual Screening and Designing of Cell-Penetrating Peptides. Methods Mol. Biol. 2015, 1324, 59–69. [Google Scholar] [PubMed]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, 449–454. [Google Scholar] [CrossRef]
- Li, Z.; Natarajan, P.; Ye, Y.; Hrabe, T.; Godzik, A. POSA: A user-driven, interactive multiple protein structure alignment server. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006. [Google Scholar] [CrossRef]
- Ulyanova, V.V.; Khodzhaeva, V.S.; Dudkina, E.V.; Laikov, A.V.; Vershinina, V.I.; Ilinskaya, O.N. Preparations of Bacillus pumilus secreted RNase: One enzyme or two? Microbiology 2015, 84, 491–497. [Google Scholar] [CrossRef]
- Reibarkh, M.Y.; Nolde, D.E.; Vasilieva, L.I.; Bocharov, E.V.; Shulga, A.A.; Kirpichnikov, M.P.; Arseniev, A.S. Three-dimensional structure of binase in solution. FEBS Lett. 1998, 431, 250–254. [Google Scholar] [CrossRef]
- Melo, M.N.; Ferre, R.; Feliu, L.; Bardají, E.; Planas, M.; Castanho, M.A.R.B. Prediction of antibacterial activity from physicochemical properties of antimicrobial peptides. PLoS ONE 2011, 6, e28549. [Google Scholar] [CrossRef] [PubMed]
- Sinthuvanich, C.; Veiga, A.S.; Gupta, K.; Gaspar, D.; Blumenthal, R.; Schneider, J.P. Anticancer β-hairpin peptides: Membraneinduced folding triggers activity. J. Am. Chem. Soc. 2012, 134, 6210–6217. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Wong, S.W.; Ge, L.; Shaw, C.; Siu, S.W.; Kwok, H.F. In vitro and md simulation study to explore physicochemical parameters for antibacterial peptide to become potent anticancer peptide. Mol. Ther. Oncolytics 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Fuentes, H.A.; Aslam, M.; Saffarzadeh, M.; Kolpakov, A.; Zelenichin, P.; Praissner, K.T.; Ilinskaya, O.N. Internalization of Bacillus intermedius ribonuclease (BINASE) induces human alveolar adenocarcinoma cell death. Toxicon 2013, 69, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereiram, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.P.; Arami, H.; Banga, I.; Gupta, J.; Gandhi, S. Cell penetrating peptides in preclinical and clinical cancer diagnosis and therapy. Oncotarget 2018, 9, 37252–37267. [Google Scholar] [CrossRef]
- Habault, J.; Poyet, J.-L. Recent advances in cell penetrating peptide-based anticancer therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef]
- Meyer, M.; Jaspers, I. Respiratory protease/antiprotease balance determines susceptibility to viral infection and can be modified by nutritional antioxidants. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1189–L1201. [Google Scholar] [CrossRef]
- Hayashi, H.; Kubo, Y.; Izumida, M. Enterokinase enhances influenza a virus infection by activating trypsinogen in human cell lines. Front. Cell Infect. Microbiol. 2018, 8, 91. [Google Scholar] [CrossRef]
- Valero-Jiménez, A.; Zúñiga, J.; Cisneros, J.; Takahashi, E.; Kido, H.; Sato, K.; Yamaya, M.; Nishimura, H.; Nakayama, K.; Matsuyama, T. Transmembrane protease, serine 4 (TMPRSS4) is upregulated in IPF lungs and increases the fibrotic response in bleomycin-induced lung injury. PLoS ONE 2018, 13, e0192963. [Google Scholar] [CrossRef]
- Larzabal, L.; Nguewa, P.A.; Pio, R.; Blanco, D.; Sanchez, B.; Rodríguez, M.J.; Pajares, M.J.; Catena, R.; Montuenga, L.M.; Calvo, A. Overexpression of TMPRSS4 in non-small cell lung cancer is associated with poor prognosis in patients with squamous histology. Br. J. Cancer 2011, 105, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Luengo-Gil, G.; Calvo, M.I.; Martín-Villar, E.; Águila, S.; Bohdan, N.; Antón, A.I.; Espín, S.; Ayala de la Peña, F.; Vicente, V.; Corral, J.; et al. Antithrombin controls tumor migration, invasion and angiogenesis by inhibition of enteropeptidase. Sci. Rep. 2016, 6, 27544. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Liu, W.L.; Chen, X.; Wang, Y.W.; Shi, D.B.; Zhang, H.; Ma, R.R.; Liu, H.T.; Guo, X.Y.; Hou, F.; et al. Overexpression of TMPRSS4 promotes tumor proliferation and aggressiveness in breast cancer. Int. J. Mol. Med. 2017, 39, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.S.; Varela, F.A.; Hyland, T.E.; Schoenbeck, A.J.; White, J.M.; Tanabe, L.M.; Todi, S.V.; List, K. Phosphorylation of the type II transmembrane serine protease, TMPRSS13, in hepatocyte growth factor activator inhibitor-1 and -2-mediated cell-surface localization. J. Biol. Chem. 2017, 292, 14867–14884. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Shin, J.H.; Shim, H.S.; Lee, C.Y.; Kim, D.J.; Kim, Y.S.; Chung, K.Y. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, G.; Jiang, J. Profiling of apoptosis- and autophagy-associated molecules in human lung cancer A549 cells in response to cisplatin treatment using stable isotope labeling with amino acids in cell culture. Int. J. Oncol. 2019, 54, 1071–1085. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Zhivotovsky, B. Proteases in autophagy. Biochim. Biophys. Acta. 2012, 1824, 44–50. [Google Scholar] [CrossRef]
- Garbar, C.; Mascaux, C.; Giustiniani, J.; Merrouche, Y.; Bensussan, A. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple-negative MDA-MB231. Sci. Rep. 2017, 7, 7201. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Wang, Y.; Paueksakon, P.; Harris, R.C. Epidermal growth factor receptorinhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes 2014, 63, 2063–2072. [Google Scholar] [CrossRef]
- Boohaker, R.J.; Lee, M.W.; Vishnubhotla, P.; Perez, J.M.; Khaled, A.R. The use of therapeutic peptides to target and to kill cancer cells. Curr. Med. Chem. 2012, 19, 3794–3804. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiao, X.; Shang, Y.; Zhang, S.; Li, Y.; He, H.; Chen, S.Z. RGD and NGR modified TRAIL protein exhibited potent anti-metastasis effects on TRAIL-insensitive cancer cells in vitro and in vivo. Amino Acids. 2017, 49, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L.; Raucher, D. Therapeutic peptides for cancer therapy. Part I—peptide inhibitors of signal transduction cascades. Expert Opin. Drug Deliv. 2009, 10, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Gembitsky, D.S.; Bozsó, Z.; O’Flaharty, M.; Ötvös, F.; Richard, M.; Lovas, S. A specific binding site for a fragment of the B-loop of epidermal growth factor and related peptides. Peptides 2002, 23, 97–102. [Google Scholar] [CrossRef]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell. 2017, 171, 683–695. [Google Scholar] [CrossRef]
- Mehrabi, M.; Mansouri, K.; Soleymani, B.; Hoseinkhani, Z.; Shahlaie, M.; Khodarahmi, R. Development of a human epidermal growth factor derivative with EGFR-blocking and depleted biological activities: A comparative in vitro study using EGFR-positive breast cancer cells. Int. J. Biol. Macromol. 2017, 103, 275–285. [Google Scholar] [CrossRef]
- Hossein-Nejad-Ariani, H.; Althagafi, E.; Kaur, K. Small peptide ligands for targeting EGFR in triple negative breast cancer cells. Sci. Rep. 2019, 9, 2723. [Google Scholar] [CrossRef]
- Tavakoli, F.; Ganjalikhany, M.R. Structure-based inhibitory peptide design targeting peptide-substrate binding site in EGFR tyrosine kinase. PLoS ONE 2019, 14, e0217031. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | MW, kDa | L 1, aa | Content of aa 2, % | pI | NCh at pH 7 3 | IIn 4 | Ain 5 | GRAVY 6 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hyd | Ac | Bas | Neu | |||||||||
| A | Binase | 12.2 | 109 | 38.5 | 10.1 | 14.7 | 36.7 | 9.4 | +3.90 | 27 | 79 | −0.42 |
| B1–20 | 2.3 | 20 | 50.0 | 10.0 | 15.0 | 25.0 | 8.5 | +0.79 | 36 | 117 | 0.14 | |
| B21–50 | 3.1 | 30 | 43.3 | 6.7 | 10.0 | 40.0 | 8.4 | +0.83 | 15 | 88 | −0.14 | |
| B51–73 | 2.5 | 23 | 30.4 | 13.0 | 17.4 | 39.1 | 9.7 | +0.77 | 25 | 38 | −1.08 | |
| B74–94 | 2.5 | 21 | 38.1 | 14.3 | 9.5 | 38.1 | 4.4 | −1.17 | 19 | 88 | −0.37 | |
| B95–109 | 1.9 | 15 | 26.7 | 6.7 | 26.7 | 40.0 | 9.5 | +1.86 | 63 | 59 | −0.76 | |
| B | EGF | 6.2 | 53 | 30.2 | 17.0 | 13.2 | 40.0 | 4.8 | −4.45 | 51 | 72 | −0.43 |
| TGFA | 5.6 | 50 | 36.0 | 12.0 | 16.0 | 36.0 | 5.93 | −3.02 | 20 | 60 | −0.08 | |
| HBEGF | 9.7 | 86 | 29.1 | 12.8 | 27.9 | 30.2 | 8.98 | +8.07 | 49 | 70 | −0.91 | |
| AREG | 10.1 | 87 | 20.7 | 12.6 | 27.6 | 39.1 | 9.35 | +10.34 | 36 | 32 | −1.54 | |
| EREG | 5.3 | 46 | 28.3 | 8.7 | 10.9 | 52.2 | 607 | −1.35 | 8 | 68 | 0.06 | |
| EPGN | 14.7 | 132 | 38.6 | 9.1 | 13.6 | 38.6 | 7.32 | +0.89 | 33 | 91 | 0.02 | |
| BTC | 9.0 | 80 | 25.0 | 12.5 | 17.5 | 45.0 | 7.65 | +1.66 | 51 | 44 | −0.79 | |
| Peptide | ACP 1 | CPP 2 | TP 3 | HPho 4 | APath 5 | BI 6, kcal/mol | |
|---|---|---|---|---|---|---|---|
| A | B1–20 | 0.63 | −0.28 | −0.80 | −0.10 | 0.43 | 1.35 |
| B21–50 | 0.73 | −0.53 | −1.53 | −0.07 | 0.45 | 0.84 | |
| B51–73 | 0.47 | −0.46 | −0.60 | −0.33 | 0.54 | 3.46 | |
| B74–94 | 0.73 | −0.56 | −0.94 | −0.16 | 0.23 | 2.32 | |
| B95–109 | 0.69 | −0.31 | −0.89 | −0.27 | 0.67 | 2.98 | |
| B | LL–37 | 0.75 | −0.2 | −1.58 | −0.34 | 1.06 | 3.00 |
| NRC–07 | 0.8 | −0.09 | −0.9 | −0.14 | 0.95 | 0.87 | |
| Roseltide rT7 | 0.72 | −0.47 | 1.49 | −0.03 | 0.51 | 0.5 | |
| LfcinB | 0.8 | 0.18 | −0.92 | −0.34 | 0.98 | 2.75 | |
| HNp–1 | 0.83 | −0.13 | 0.18 | −0.10 | 0.41 | 1.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulyanova, V.; Dudkina, E.; Nadyrova, A.; Kalashnikov, V.; Surchenko, Y.; Ilinskaya, O. The Cytotoxicity of RNase-Derived Peptides. Biomolecules 2021, 11, 16. https://doi.org/10.3390/biom11010016
Ulyanova V, Dudkina E, Nadyrova A, Kalashnikov V, Surchenko Y, Ilinskaya O. The Cytotoxicity of RNase-Derived Peptides. Biomolecules. 2021; 11(1):16. https://doi.org/10.3390/biom11010016
Chicago/Turabian StyleUlyanova, Vera, Elena Dudkina, Alsu Nadyrova, Vladimir Kalashnikov, Yulia Surchenko, and Olga Ilinskaya. 2021. "The Cytotoxicity of RNase-Derived Peptides" Biomolecules 11, no. 1: 16. https://doi.org/10.3390/biom11010016
APA StyleUlyanova, V., Dudkina, E., Nadyrova, A., Kalashnikov, V., Surchenko, Y., & Ilinskaya, O. (2021). The Cytotoxicity of RNase-Derived Peptides. Biomolecules, 11(1), 16. https://doi.org/10.3390/biom11010016