Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease




Abstract
1. Introduction
2. CKD-Associated High Cardiovascular Risk
Cholesterol-Lowering Drugs in Patients with CKD
3. What is the Physiological Function of HDL?
3.1. HDL Biosynthesis and Remodeling
3.2. HDL Structure and Composition
3.3. HDL Subclasses
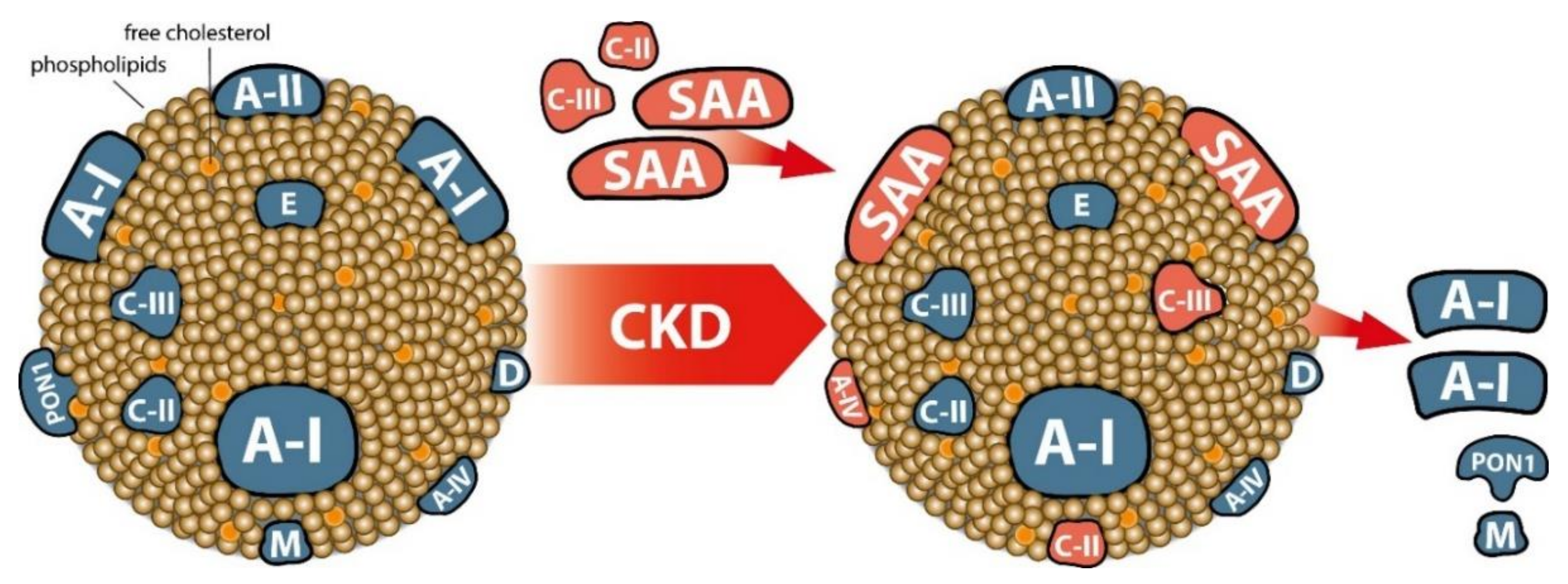
4. CKD Profoundly Changes HDL Maturation and Metabolism
4.1. CKD-Associated Changes in HDL Subclasses Distribution
4.2. CKD-Induced Changes in the HDL Proteome
5. HDL-Cholesterol Efflux Capacity, a Key Functional Metric of HDL
5.1. HDL Cholesterol Efflux Capacity Is a Robust Predictor of Cardiovascular Events in the General Population
5.2. HDL-Cholesterol Efflux Capacity in CKD Patients
6. CKD-Associated Changes of Other HDL Functions
7. Conclusions
Funding
Conflicts of Interest
References
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef]
- Rader, D.J.; Alexander, E.T.; Weibel, G.L.; Billheimer, J.; Rothblat, G.H. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 2009, 50, S189–S194. [Google Scholar] [CrossRef] [PubMed]
- Heine, G.H.; Eller, K.; Stadler, J.T.; Rogacev, K.S.; Marsche, G. Lipid-modifying therapy in chronic kidney disease: Pathophysiological and clinical considerations. Pharm. Ther. 2020, 207, 107459. [Google Scholar] [CrossRef]
- Moradi, H.; Streja, E.; Vaziri, N.D. ESRD-induced dyslipidemia-Should management of lipid disorders differ in dialysis patients? Semin. Dial. 2018, 31, 398–405. [Google Scholar] [CrossRef]
- Annema, W.; von Eckardstein, A. High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ. J. 2013, 77, 2432–2448. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Saemann, M.D.; Heinemann, A.; Holzer, M. Inflammation alters HDL composition and function: Implications for HDL-raising therapies. Pharm. Ther. 2013, 137, 341–351. [Google Scholar] [CrossRef]
- Toth, P.P.; Barter, P.J.; Rosenson, R.S.; Boden, W.E.; Chapman, M.J.; Cuchel, M.; D’Agostino, R.B.S.; Davidson, M.H.; Davidson, W.S.; Heinecke, J.W.; et al. High-density lipoproteins: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 484–525. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef]
- Shah, A.S.; Tan, L.; Long, J.L.; Davidson, W.S. Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 2013, 54, 2575–2585. [Google Scholar] [CrossRef]
- Knuplez, E.; Curcic, S.; Theiler, A.; Barnthaler, T.; Trakaki, A.; Trieb, M.; Holzer, M.; Heinemann, A.; Zimmermann, R.; Sturm, E.M.; et al. Lysophosphatidylcholines inhibit human eosinophil activation and suppress eosinophil migration in vivo. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158686. [Google Scholar] [CrossRef]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia alters HDL composition and function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Kopecky, C.; Kubicek, M.; Haidinger, M.; Doller, D.; Katholnig, K.; Suarna, C.; Eller, P.; Tolle, M.; Gerner, C.; et al. Serum Amyloid A in Uremic HDL Promotes Inflammation. J. Am. Soc. Nephrol. 2012, 23, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Schilcher, G.; Curcic, S.; Trieb, M.; Ljubojevic, S.; Stojakovic, T.; Scharnagl, H.; Kopecky, C.M.; Rosenkranz, A.R.; Heinemann, A.; et al. Dialysis Modalities and HDL Composition and Function. J. Am. Soc. Nephrol. 2015, 26, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yancey, P.G.; Ikizler, T.A.; Jerome, W.G.; Kaseda, R.; Cox, B.; Bian, A.; Shintani, A.; Fogo, A.B.; Linton, M.F.; et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J. Am. Coll. Cardiol. 2012, 60, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Speer, T.; Colin, S.; Charakida, M.; Zewinger, S.; Staels, B.; Chinetti-Gbaguidi, G.; Hettrich, I.; Rohrer, L.; O’Neill, F.; et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J. Am. Soc. Nephrol. 2014, 25, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Gautier, T.; Nijstad, N.; Tolle, M.; Schuchardt, M.; van der Giet, M.; Tietge, U.J. High density lipoprotein (HDL) particles from end-stage renal disease patients are defective in promoting reverse cholesterol transport. Sci. Rep. 2017, 7, 41481. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Bauer, L.; Kern, S.; Rogacev, K.S.; Emrich, I.E.; Zawada, A.; Fliser, D.; Heinemann, A.; Heine, G.H.; Marsche, G. HDL Cholesterol Efflux Capacity and Cardiovascular Events in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2017, 69, 246–247. [Google Scholar] [CrossRef]
- Chindhy, S.; Joshi, P.; Khera, A.; Ayers, C.R.; Hedayati, S.S.; Rohatgi, A. Impaired Renal Function on Cholesterol Efflux Capacity, HDL Particle Number, and Cardiovascular Events. J. Am. Coll. Cardiol. 2018, 72, 698–700. [Google Scholar] [CrossRef]
- Kopecky, C.; Ebtehaj, S.; Genser, B.; Drechsler, C.; Krane, V.; Antlanger, M.; Kovarik, J.J.; Kaltenecker, C.C.; Parvizi, M.; Wanner, C.; et al. HDL Cholesterol Efflux Does Not Predict Cardiovascular Risk in Hemodialysis Patients. J. Am. Soc. Nephrol. 2017, 28, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Lopez, F.L.; Matsushita, K.; Loehr, L.R.; Agarwal, S.K.; Chen, L.Y.; Soliman, E.Z.; Astor, B.C.; Coresh, J. Chronic kidney disease is associated with the incidence of atrial fibrillation: The Atherosclerosis Risk in Communities (ARIC) study. Circulation 2011, 123, 2946–2953. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Russell, S.D.; Loehr, L.R.; Crainiceanu, C.M.; Rosamond, W.D.; Chang, P.P.; Chambless, L.E.; Coresh, J. Reduced kidney function as a risk factor for incident heart failure: The atherosclerosis risk in communities (ARIC) study. J. Am. Soc. Nephrol. 2007, 18, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019, 96, 1048–1050. [Google Scholar] [CrossRef]
- Matsushita, K.; Coresh, J.; Sang, Y.; Chalmers, J.; Fox, C.; Guallar, E.; Jafar, T.; Jassal, S.K.; Landman, G.W.; Muntner, P.; et al. CKD Prognosis Consortium Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: A collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol. 2015, 3, 514–525. [Google Scholar] [CrossRef]
- de Jager, D.J.; Grootendorst, D.C.; Jager, K.J.; van Dijk, P.C.; Tomas, L.M.; Ansell, D.; Collart, F.; Finne, P.; Heaf, J.G.; De Meester, J.; et al. Cardiovascular and noncardiovascular mortality among patients starting dialysis. JAMA 2009, 302, 1782–1789. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. ESC Scientific Document Group 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2019, 41, 111–188. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. ESC Scientific Document Group 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar]
- Wanner, C.; Krane, V.; Marz, W.; Olschewski, M.; Mann, J.F.; Ruf, G.; Ritz, E. German Diabetes and Dialysis Study Investigators Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef]
- Holdaas, H.; Fellstrom, B.; Jardine, A.G.; Holme, I.; Nyberg, G.; Fauchald, P.; Gronhagen-Riska, C.; Madsen, S.; Neumayer, H.H.; Cole, E.; et al. Assessment of LEscol in Renal Transplantation (ALERT) Study Investigators Effect of fluvastatin on cardiac outcomes in renal transplant recipients: A multicentre, randomised, placebo-controlled trial. Lancet 2003, 361, 2024–2031. [Google Scholar] [CrossRef]
- Fellstrom, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.W.; Chevaile, A.; Cobbe, S.M.; Gronhagen-Riska, C.; et al. AURORA Study Group Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. SHARP Investigators The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): A randomised placebo-controlled trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef]
- Heine, G.H.; Rogacev, K.S.; Weingartner, O.; Marsche, G. Still a reasonable goal: Targeting cholesterol in dialysis and advanced chronic kidney disease patients. Semin. Dial. 2017, 30, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Marz, W.; Genser, B.; Drechsler, C.; Krane, V.; Grammer, T.B.; Ritz, E.; Stojakovic, T.; Scharnagl, H.; Winkler, K.; Holme, I.; et al. German Diabetes and Dialysis Study Investigators Atorvastatin and low-density lipoprotein cholesterol in type 2 diabetes mellitus patients on hemodialysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Fulcher, J.; Abeysuriya, N.; Park, L.; Kumar, S.; Luca Di Tanna, G.; Wilcox, I.; Keech, A.; Rodgers, A.; Lal, S. Intensive LDL cholesterol-lowering treatment beyond current recommendations for the prevention of major vascular events: A systematic review and meta-analysis of randomised trials including 327 037 participants. Lancet Diabetes Endocrinol. 2020, 8, 36–49. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef]
- Feng, M.; Darabi, M.; Tubeuf, E.; Canicio, A.; Lhomme, M.; Frisdal, E.; Lanfranchi-Lebreton, S.; Matheron, L.; Rached, F.; Ponnaiah, M.; et al. Free cholesterol transfer to high-density lipoprotein (HDL) upon triglyceride lipolysis underlies the U-shape relationship between HDL-cholesterol and cardiovascular disease. Eur. J. Prev. Cardiol. 2019. [Google Scholar] [CrossRef]
- Bodzioch, M.; Orso, E.; Klucken, J.; Langmann, T.; Bottcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Buchler, C.; Porsch-Ozcurumez, M.; et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351. [Google Scholar] [CrossRef]
- Norum, K.R.; Remaley, A.T.; Miettinen, H.E.; Strom, E.H.; Balbo, B.E.; Sampaio, C.A.; Wiig, I.; Kuivenhoven, J.A.; Calabresi, L.; Tesmer, J.J.; et al. Lecithin: Cholesterol Acyltransferase: Symposium on 50-years of biomedical research from its discovery to latest findings. J. Lipid Res. 2020, 61, 1142–1149. [Google Scholar] [CrossRef]
- Morton, R.E.; Liu, Y. The lipid transfer properties of CETP define the concentration and composition of plasma lipoproteins. J. Lipid Res. 2020, 61, 1168–1179. [Google Scholar] [CrossRef]
- Bailey, D.; Ruel, I.; Hafiane, A.; Cochrane, H.; Iatan, I.; Jauhiainen, M.; Ehnholm, C.; Krimbou, L.; Genest, J. Analysis of lipid transfer activity between model nascent HDL particles and plasma lipoproteins: Implications for current concepts of nascent HDL maturation and genesis. J. Lipid Res. 2010, 51, 785–797. [Google Scholar] [CrossRef]
- Schmitz, G.; Kaminski, W.E.; Orso, E. ABC transporters in cellular lipid trafficking. Curr. Opin. Lipidol. 2000, 11, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Kozarsky, K.; Krieger, M. Scavenger receptor class B, type I-mediated [3H]cholesterol efflux to high and low density lipoproteins is dependent on lipoprotein binding to the receptor. J. Biol. Chem. 2000, 275, 29993–30001. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Lund-Katz, S.; de la Llera-Moya, M.; Millar, J.S.; Chang, D.; Fuki, I.; Rothblat, G.H.; Phillips, M.C.; Rader, D.J. Pathways by which reconstituted high-density lipoprotein mobilizes free cholesterol from whole body and from macrophages. Arter. Thromb. Vasc. Biol. 2010, 30, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Rigotti, A.; Trigatti, B.L.; Penman, M.; Rayburn, H.; Herz, J.; Krieger, M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl. Acad. Sci. USA 1997, 94, 12610–12615. [Google Scholar] [CrossRef]
- Wiersma, H.; Gatti, A.; Nijstad, N.; Kuipers, F.; Tietge, U.J. Hepatic SR-BI, not endothelial lipase, expression determines biliary cholesterol secretion in mice. J. Lipid Res. 2009, 50, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- van der Velde, A.E.; Brufau, G.; Groen, A.K. Transintestinal cholesterol efflux. Curr. Opin. Lipidol. 2010, 21, 167–171. [Google Scholar] [CrossRef]
- Kostner, G.; Alaupovic, P. Studies of the composition and structure of plasma lipoproteins. Separation and quantification of the lipoprotein families occurring in the high density lipoproteins of human plasma. Biochemistry 1972, 11, 3419–3428. [Google Scholar] [CrossRef]
- Alaupovic, P.; Kostner, G.; Lee, D.M.; McConathy, W.J.; Magnani, H.N. Peptide composition of human plasma apolipoproteins A, B and C. Expos Annu. Biochim. Med. 1972, 31, 145–160. [Google Scholar]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharm. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharm. 2015, 224, 3–51. [Google Scholar]
- Davidson, W.S.; Silva, R.A.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arter. Thromb. Vasc. Biol. 2009, 29, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, B.F.; Tani, M.; Schaefer, E.J. Metabolic and functional relevance of HDL subspecies. Curr. Opin. Lipidol. 2011, 22, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.V.; Krauss, R.M.; Musliner, T.A. Nondenaturing polyacrylamide gradient gel electrophoresis. Methods Enzymol. 1986, 128, 417–431. [Google Scholar] [PubMed]
- Jeyarajah, E.J.; Cromwell, W.C.; Otvos, J.D. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin. Lab. Med. 2006, 26, 847–870. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Speer, T.; Kleber, M.E.; Scharnagl, H.; Woitas, R.; Lepper, P.M.; Pfahler, K.; Seiler, S.; Heine, G.H.; Marz, W.; et al. HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. J. Am. Soc. Nephrol. 2014, 25, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Silbernagel, G.; Genser, B.; Drechsler, C.; Scharnagl, H.; Grammer, T.B.; Stojakovic, T.; Krane, V.; Ritz, E.; Wanner, C.; Marz, W. HDL cholesterol, apolipoproteins, and cardiovascular risk in hemodialysis patients. J. Am. Soc. Nephrol. 2015, 26, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Moradi, H.; Streja, E.; Kashyap, M.L.; Vaziri, N.D.; Fonarow, G.C.; Kalantar-Zadeh, K. Elevated high-density lipoprotein cholesterol and cardiovascular mortality in maintenance hemodialysis patients. Nephrol. Dial. Transpl. 2014, 29, 1554–1562. [Google Scholar] [CrossRef]
- Calabresi, L.; Simonelli, S.; Conca, P.; Busnach, G.; Cabibbe, M.; Gesualdo, L.; Gigante, M.; Penco, S.; Veglia, F.; Franceschini, G. Acquired lecithin:cholesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J. Intern Med. 2015, 277, 552–561. [Google Scholar] [CrossRef]
- Miida, T.; Miyazaki, O.; Hanyu, O.; Nakamura, Y.; Hirayama, S.; Narita, I.; Gejyo, F.; Ei, I.; Tasaki, K.; Kohda, Y.; et al. LCAT-dependent conversion of prebeta1-HDL into alpha-migrating HDL is severely delayed in hemodialysis patients. J. Am. Soc. Nephrol. 2003, 14, 732–738. [Google Scholar] [CrossRef]
- Quaschning, T.; Krane, V.; Metzger, T.; Wanner, C. Abnormalities in uremic lipoprotein metabolism and its impact on cardiovascular disease. Am. J. Kidney. Dis. 2001, 38, S14–S19. [Google Scholar] [CrossRef] [PubMed]
- Kuchta, A.; Cwiklinska, A.; Czaplinska, M.; Wieczorek, E.; Kortas-Stempak, B.; Gliwinska, A.; Dabkowski, K.; Salaga-Zaleska, K.; Mickiewicz, A.; Debska-Slizien, A.; et al. Plasma Levels of Prebeta1-HDL Are Significantly Elevated in Non-Dialyzed Patients with Advanced Stages of Chronic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1202. [Google Scholar] [CrossRef] [PubMed]
- Kaysen, G.A. Lipid and lipoprotein metabolism in chronic kidney disease. J. Ren. Nutr. 2009, 19, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.T.; Dogra, G.K.; Irish, A.B.; Ooi, E.M.; Barrett, P.H.; Chan, D.C.; Watts, G.F. Chronic kidney disease delays VLDL-apoB-100 particle catabolism: Potential role of apolipoprotein C-III. J. Lipid Res. 2009, 50, 2524–2531. [Google Scholar] [CrossRef] [PubMed]
- Ooi, E.M.; Chan, D.T.; Watts, G.F.; Chan, D.C.; Ng, T.W.; Dogra, G.K.; Irish, A.B.; Barrett, P.H. Plasma apolipoprotein C-III metabolism in patients with chronic kidney disease. J. Lipid Res. 2011, 52, 794–800. [Google Scholar] [CrossRef]
- Gordts, P.L.; Nock, R.; Son, N.H.; Ramms, B.; Lew, I.; Gonzales, J.C.; Thacker, B.E.; Basu, D.; Lee, R.G.; Mullick, A.E.; et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Investig. 2016, 126, 2855–2866. [Google Scholar] [CrossRef]
- Witko-Sarsat, V.; Friedlander, M.; Capeillere-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef]
- Marsche, G.; Frank, S.; Hrzenjak, A.; Holzer, M.; Dirnberger, S.; Wadsack, C.; Scharnagl, H.; Stojakovic, T.; Heinemann, A.; Oettl, K. Plasma-advanced oxidation protein products are potent high-density lipoprotein receptor antagonists in vivo. Circ. Res. 2009, 104, 750–757. [Google Scholar] [CrossRef]
- Binder, V.; Ljubojevic, S.; Haybaeck, J.; Holzer, M.; El-Gamal, D.; Schicho, R.; Pieske, B.; Heinemann, A.; Marsche, G. The myeloperoxidase product hypochlorous acid generates irreversible high-density lipoprotein receptor inhibitors. Arter. Thromb. Vasc. Biol. 2013, 33, 1020–1027. [Google Scholar] [CrossRef]
- Samuelsson, O.; Attman, P.O.; Knight-Gibson, C.; Mulec, H.; Weiss, L.; Alaupovic, P. Fluvastatin improves lipid abnormalities in patients with moderate to advanced chronic renal insufficiency. Am. J. Kidney Dis. 2002, 39, 67–75. [Google Scholar] [CrossRef]
- Gille, A.; Duffy, D.; Tortorici, M.A.; Wright, S.D.; Deckelbaum, L.I.; D’Andrea, D.M. Moderate Renal Impairment Does Not Impact the Ability of CSL112 (Apolipoprotein A-I [Human]) to Enhance Cholesterol Efflux Capacity. J. Clin. Pharm. 2019, 59, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Liang, K.; Parks, J.S. Acquired lecithin-cholesterol acyltransferase deficiency in nephrotic syndrome. Am. J. Physiol. Ren. Physiol. 2001, 280, F823–F828. [Google Scholar] [CrossRef]
- Mekki, K.; Bouchenak, M.; Lamri, M.; Remaoun, M.; Belleville, J. Changes in plasma lecithin: Cholesterol acyltransferase activity, HDL(2), HDL(3) amounts and compositions in patients with chronic renal failure after different times of hemodialysis. Atherosclerosis 2002, 162, 409–417. [Google Scholar] [CrossRef]
- McLeod, R.; Reeve, C.E.; Frohlich, J. Plasma lipoproteins and lecithin:cholesterol acyltransferase distribution in patients on dialysis. Kidney Int. 1984, 25, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Miljkovic, M.; Stefanovic, A.; Vekic, J.; Zeljkovic, A.; Gojkovic, T.; Simic-Ogrizovic, S.; Bogavac-Stanojevic, N.; Cerne, D.; Ilic, J.; Stefanovic, I.; et al. Activity of paraoxonase 1 (PON1) on HDL2 and HDL3 subclasses in renal disease. Clin. Biochem. 2018, 60, 52–58. [Google Scholar] [CrossRef]
- Homma, K.; Homma, Y.; Shiina, Y.; Wakino, S.; Suzuki, M.; Fujishima, S.; Hayashi, K.; Hori, S.; Itoh, H. Skew of plasma low- and high-density lipoprotein distributions to less dense subfractions in normotriglyceridemic chronic kidney disease patients on maintenance hemodialysis treatment. Nephron Clin. Pract. 2013, 123, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Soto-Miranda, E.; Carreon-Torres, E.; Lorenzo, K.; Bazan-Salinas, B.; Garcia-Sanchez, C.; Franco, M.; Posadas-Romero, C.; Fragoso, J.M.; Lopez-Olmos, V.; Madero, M.; et al. Shift of high-density lipoprotein size distribution toward large particles in patients with proteinuria. Clin. Chim. Acta 2012, 414, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, A.; Ristovski-Kornic, D.; Kotur-Stevuljevic, J.; Spasojevic-Kalimanovska, V.; Vekic, J.; Miljkovic, M.; Paripovic, D.; Peco-Antic, A.; Jelic-Ivanovic, Z.; Zeljkovic, A. Alterations of HDL Particles in Children with End-stage Renal Disease. J. Med. Biochem. 2017, 36, 358–365. [Google Scholar] [CrossRef]
- Alabakovska, S.B.; Todorova, B.B.; Labudovic, D.D.; Tosheska, K.N. LDL and HDL subclass distribution in patients with end-stage renal diseases. Clin. Biochem. 2002, 35, 211–216. [Google Scholar] [CrossRef]
- Attman, P.O.; Alaupovic, P.; Tavella, M.; Knight-Gibson, C. Abnormal lipid and apolipoprotein composition of major lipoprotein density classes in patients with chronic renal failure. Nephrol. Dial. Transpl. 1996, 11, 63–69. [Google Scholar] [CrossRef]
- Tolle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prufer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar] [CrossRef]
- Kopecky, C.; Haidinger, M.; Birner-Grunberger, R.; Darnhofer, B.; Kaltenecker, C.C.; Marsche, G.; Holzer, M.; Weichhart, T.; Antlanger, M.; Kovarik, J.J.; et al. Restoration of renal function does not correct impairment of uremic HDL properties. J. Am. Soc. Nephrol. 2015, 26, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Rubinow, K.B.; Henderson, C.M.; Robinson-Cohen, C.; Himmelfarb, J.; de Boer, I.H.; Vaisar, T.; Kestenbaum, B.; Hoofnagle, A.N. Kidney function is associated with an altered protein composition of high-density lipoprotein. Kidney Int. 2017, 92, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Mange, A.; Goux, A.; Badiou, S.; Patrier, L.; Canaud, B.; Maudelonde, T.; Cristol, J.P.; Solassol, J. HDL proteome in hemodialysis patients: A quantitative nanoflow liquid chromatography-tandem mass spectrometry approach. PLoS ONE 2012, 7, e34107. [Google Scholar] [CrossRef]
- Shao, B.; de Boer, I.; Tang, C.; Mayer, P.S.; Zelnick, L.; Afkarian, M.; Heinecke, J.W.; Himmelfarb, J. A Cluster of Proteins Implicated in Kidney Disease Is Increased in High-Density Lipoprotein Isolated from Hemodialysis Subjects. J. Proteome Res. 2015, 14, 2792–2806. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zelnick, L.R.; Hoofnagle, A.N.; Vaisar, T.; Henderson, C.M.; Imrey, P.B.; Robinson-Cohen, C.; de Boer, I.H.; Shiu, Y.T.; Himmelfarb, J.; et al. HFM Study Alteration of HDL Protein Composition with Hemodialysis Initiation. Clin. J. Am. Soc. Nephrol. 2018, 13, 1225–1233. [Google Scholar] [CrossRef]
- Florens, N.; Calzada, C.; Delolme, F.; Page, A.; Guebre Egziabher, F.; Juillard, L.; Soulage, A.C.O. Proteomic Characterization of High-Density Lipoprotein Particles from Non-Diabetic Hemodialysis Patients. Toxins 2019, 11, 671. [Google Scholar] [CrossRef]
- Kronenberg, F. HDL in CKD-The Devil Is in the Detail. J. Am. Soc. Nephrol. 2018, 29, 1356–1371. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Hama, S.Y.; de Beer, F.C.; Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M.; La Du, B.N.; Fogelman, A.M.; Navab, M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Investig. 1995, 96, 2758–2767. [Google Scholar] [CrossRef]
- Malle, E.; De Beer, F.C. Human serum amyloid A (SAA) protein: A prominent acute-phase reactant for clinical practice. Eur. J. Clin. Investig. 1996, 26, 427–435. [Google Scholar] [CrossRef]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J. Serum amyloid A is a chemoattractant: Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Song, C.; Endoh, I.; Goyette, J.; Jessup, W.; Freedman, S.B.; McNeil, H.P.; Geczy, C.L. Serum amyloid A induces monocyte tissue factor. J. Immunol. 2007, 178, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Lee, S.Y.; Lee, H.; Cho, K.H.; Yun, J.; Bae, Y.S. Serum amyloid A induces CCL2 production via formyl peptide receptor-like 1-mediated signaling in human monocytes. J. Immunol. 2008, 181, 4332–4339. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Chang, M.Y.; Wang, S.; Wight, T.N.; McMillen, T.S.; Oram, J.F.; Vaisar, T.; Heinecke, J.W.; De Beer, F.C.; De Beer, M.C.; et al. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arter. Thromb. Vasc. Biol. 2011, 31, 1326–1332. [Google Scholar] [CrossRef] [PubMed]
- Christenson, K.; Bjorkman, L.; Ahlin, S.; Olsson, M.; Sjoholm, K.; Karlsson, A.; Bylund, J. Endogenous Acute Phase Serum Amyloid A Lacks Pro-Inflammatory Activity, Contrasting the Two Recombinant Variants That Activate Human Neutrophils through Different Receptors. Front. Immunol. 2013, 4, 92. [Google Scholar] [CrossRef]
- Bjorkman, L.; Karlsson, J.; Karlsson, A.; Rabiet, M.J.; Boulay, F.; Fu, H.; Bylund, J.; Dahlgren, C. Serum amyloid A mediates human neutrophil production of reactive oxygen species through a receptor independent of formyl peptide receptor like-1. J. Leukoc. Biol. 2008, 83, 245–253. [Google Scholar] [CrossRef]
- Cheng, N.; Liang, Y.; Du, X.; Ye, R.D. Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Murdoch, C.C.; Espenschied, S.T.; Matty, M.A.; Mueller, O.; Tobin, D.M.; Rawls, J.F. Intestinal Serum amyloid A suppresses systemic neutrophil activation and bactericidal activity in response to microbiota colonization. PLoS Pathog. 2019, 15, e1007381. [Google Scholar] [CrossRef]
- Frame, N.M.; Jayaraman, S.; Gantz, D.L.; Gursky, O. Serum amyloid A self-assembles with phospholipids to form stable protein-rich nanoparticles with a distinct structure: A hypothetical function of SAA as a "molecular mop" in immune response. J. Struct. Biol. 2017, 200, 293–302. [Google Scholar] [CrossRef]
- Wyler von Ballmoos, M.C.; Haring, B.; Sacks, F.M. The risk of cardiovascular events with increased apolipoprotein CIII: A systematic review and meta-analysis. J. Clin. Lipidol. 2015, 9, 498–510. [Google Scholar] [CrossRef]
- Scheffer, P.G.; Teerlink, T.; Dekker, J.M.; Bos, G.; Nijpels, G.; Diamant, M.; Kostense, P.J.; Stehouwer, C.D.; Heine, R.J. Increased plasma apolipoprotein C-III concentration independently predicts cardiovascular mortality: The Hoorn Study. Clin. Chem. 2008, 54, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Lamprea-Montealegre, J.A.; McClelland, R.L.; Otvos, J.D.; Mora, S.; Koch, M.; Jensen, M.K.; de Boer, I.H. Association of High-Density Lipoprotein Particles and High-Density Lipoprotein Apolipoprotein C-III Content With Cardiovascular Disease Risk According to Kidney Function: The Multi-Ethnic Study of Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e013713. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, A.; Wang, S.; Wang, T.; Hu, D.; Wu, S.; Peng, D. ApoCIII enrichment in HDL impairs HDL-mediated cholesterol efflux capacity. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2020, 21, 30–41. [Google Scholar] [CrossRef]
- Kawakami, A.; Aikawa, M.; Alcaide, P.; Luscinskas, F.W.; Libby, P.; Sacks, F.M. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation 2006, 114, 681–687. [Google Scholar] [CrossRef]
- Kawakami, A.; Aikawa, M.; Nitta, N.; Yoshida, M.; Libby, P.; Sacks, F.M. Apolipoprotein CIII-induced THP-1 cell adhesion to endothelial cells involves pertussis toxin-sensitive G protein- and protein kinase C alpha-mediated nuclear factor-kappaB activation. Arter. Thromb. Vasc. Biol. 2007, 27, 219–225. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Qi, R.; Wang, Y.; Zhang, X.; Yu, M.; Tang, Y.; Wang, M.; Shu, Y.N.; Huang, W.; et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: The effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc. Res. 2015, 107, 579–589. [Google Scholar] [CrossRef]
- Trieb, M.; Wolf, P.; Knuplez, E.; Weger, W.; Schuster, C.; Peinhaupt, M.; Holzer, M.; Trakaki, A.; Eichmann, T.; Lass, A.; et al. Abnormal composition and function of high-density lipoproteins in atopic dermatitis patients. Allergy 2019, 74, 398–402. [Google Scholar] [CrossRef]
- Trakaki, A.; Sturm, G.J.; Pregartner, G.; Scharnagl, H.; Eichmann, T.O.; Trieb, M.; Knuplez, E.; Holzer, M.; Stadler, J.T.; Heinemann, A.; et al. Allergic rhinitis is associated with complex alterations in high-density lipoprotein composition and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1280–1292. [Google Scholar] [CrossRef]
- Mahrooz, A.; Mackness, M.; Bagheri, A.; Ghaffari-Cherati, M.; Masoumi, P. The epigenetic regulation of paraoxonase 1 (PON1) as an important enzyme in HDL function: The missing link between environmental and genetic regulation. Clin. Biochem. 2019, 73, 1–10. [Google Scholar] [CrossRef]
- Furlong, C.E.; Marsillach, J.; Jarvik, G.P.; Costa, L.G. Paraoxonases-1, -2 and -3: What are their functions? Chem. Biol. Interact. 2016, 259, 51–62. [Google Scholar] [CrossRef]
- Cabana, V.G.; Reardon, C.A.; Feng, N.; Neath, S.; Lukens, J.; Getz, G.S. Serum paraoxonase: Effect of the apolipoprotein composition of HDL and the acute phase response. J. Lipid Res. 2003, 44, 780–792. [Google Scholar] [CrossRef]
- Camps, J.; Marsillach, J.; Joven, J. The paraoxonases: Role in human diseases and methodological difficulties in measurement. Crit. Rev Clin. Lab. Sci. 2009, 46, 83–106. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, Y.; Goto, M.; Park, C.; Nunes, A.C.F.; Jing, W.; Streja, E.; Rhee, C.M.; Cruz, S.; Kashyap, M.L.; Vaziri, N.D.; et al. Association of Serum Paraoxonase/Arylesterase Activity With All-Cause Mortality in Maintenance Hemodialysis Patients. J. Clin. Endocrinol. Metab. 2019, 104, 4848–4856. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A.; Kotani, K.; Kimura, S. Paraoxonase 1 in chronic kidney failure. J. Lipids 2012, 2012, 726048. [Google Scholar] [CrossRef] [PubMed]
- Untersteller, K.; Meissl, S.; Trieb, M.; Emrich, I.E.; Zawada, A.M.; Holzer, M.; Knuplez, E.; Fliser, D.; Heine, G.H.; Marsche, G. HDL functionality and cardiovascular outcome among nondialysis chronic kidney disease patients. J. Lipid Res. 2018, 59, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Mackness, M.I.; Durrington, P.N.; Mackness, B. The role of paraoxonase 1 activity in cardiovascular disease: Potential for therapeutic intervention. Am. J. Cardiovasc. Drugs 2004, 4, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Mackness, B.; Quarck, R.; Verreth, W.; Mackness, M.; Holvoet, P. Human paraoxonase-1 overexpression inhibits atherosclerosis in a mouse model of metabolic syndrome. Arter. Thromb. Vasc. Biol. 2006, 26, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, M.; Vaya, J.; Shih, D.; Aviram, M. Paraoxonase 1 (PON1) enhances HDL-mediated macrophage cholesterol efflux via the ABCA1 transporter in association with increased HDL binding to the cells: A possible role for lysophosphatidylcholine. Atherosclerosis 2005, 179, 69–77. [Google Scholar] [CrossRef]
- Yancey, P.G.; Bortnick, A.E.; Kellner-Weibel, G.; de la Llera-Moya, M.; Phillips, M.C.; Rothblat, G.H. Importance of different pathways of cellular cholesterol efflux. Arter. Thromb. Vasc. Biol. 2003, 23, 712–719. [Google Scholar] [CrossRef]
- Davidson, W.S.; Rodrigueza, W.V.; Lund-Katz, S.; Johnson, W.J.; Rothblat, G.H.; Phillips, M.C. Effects of acceptor particle size on the efflux of cellular free cholesterol. J. Biol. Chem. 1995, 270, 17106–17113. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Yu, S.; Yvan-Charvet, L.; Wang, N.; Mzhavia, N.; Langlois, R.; Pagler, T.; Li, R.; Welch, C.L.; Goldberg, I.J.; et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J. Clin. Investig. 2008, 118, 3701–3713. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Kern, S.; Trieb, M.; Trakaki, A.; Marsche, G. HDL structure and function is profoundly affected when stored frozen in the absence of cryoprotectants. J. Lipid Res. 2017, 58, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- de la Llera-Moya, M.; Drazul-Schrader, D.; Asztalos, B.F.; Cuchel, M.; Rader, D.J.; Rothblat, G.H. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arter. Thromb. Vasc. Biol. 2010, 30, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y.; Ding, D.; Li, X.; Yang, Y.; Li, Q.; Zheng, Y.; Wang, D.; Ling, W. Cholesterol efflux capacity is an independent predictor of all-cause and cardiovascular mortality in patients with coronary artery disease: A prospective cohort study. Atherosclerosis 2015, 249, 116–124. [Google Scholar] [CrossRef]
- Szili-Torok, T.; Annema, W.; Anderson, J.L.C.; Bakker, S.J.L.; Tietge, U.J.F. High Density Lipoprotein Cholesterol Efflux Predicts Incident New Onset Diabetes After Transplantation (NODAT) in Renal Transplant Recipients Independent of High Density Lipoprotein Cholesterol Levels. Diabetes 2019, 68, 1915–1923. [Google Scholar] [CrossRef]
- Sankaranarayanan, S.; Oram, J.F.; Asztalos, B.F.; Vaughan, A.M.; Lund-Katz, S.; Adorni, M.P.; Phillips, M.C.; Rothblat, G.H. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. J. Lipid Res. 2009, 50, 275–284. [Google Scholar] [CrossRef]
- Artl, A.; Marsche, G.; Lestavel, S.; Sattler, W.; Malle, E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arter. Thromb. Vasc. Biol. 2000, 20, 763–772. [Google Scholar] [CrossRef]
- Annema, W.; Dikkers, A.; Freark de Boer, J.; Dullaart, R.P.; Sanders, J.S.; Bakker, S.J.; Tietge, U.J. HDL Cholesterol Efflux Predicts Graft Failure in Renal Transplant Recipients. J. Am. Soc. Nephrol. 2016, 27, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.M.; Tsuchida, Y.; Nowak, K.L.; Sarkar, S.; Chonchol, M.; Whitfield, V.; Salas, N.; Dikalova, A.; Yancey, P.G.; Huang, J.; et al. IL-1 Inhibition and Function of the HDL-Containing Fraction of Plasma in Patients with Stages 3 to 5 CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Krankel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chapman, M.J. Antiatherogenic function of HDL particle subpopulations: Focus on antioxidative activities. Curr. Opin. Lipidol. 2010, 21, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Dantoine, T.F.; Debord, J.; Charmes, J.P.; Merle, L.; Marquet, P.; Lachatre, G.; Leroux-Robert, C. Decrease of serum paraoxonase activity in chronic renal failure. J. Am. Soc. Nephrol. 1998, 9, 2082–2088. [Google Scholar] [PubMed]
- Capeillere-Blandin, C.; Gausson, V.; Nguyen, A.T.; Descamps-Latscha, B.; Drueke, T.; Witko-Sarsat, V. Respective role of uraemic toxins and myeloperoxidase in the uraemic state. Nephrol. Dial. Transpl. 2006, 21, 1555–1563. [Google Scholar] [CrossRef]
- Koeth, R.A.; Kalantar-Zadeh, K.; Wang, Z.; Fu, X.; Tang, W.H.; Hazen, S.L. Protein Carbamylation Predicts Mortality in ESRD. J. Am. Soc. Nephrol. 2013, 24, 853–861. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Horkko, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Sun, J.T.; Yang, K.; Lu, L.; Zhu, Z.B.; Zhu, J.Z.; Ni, J.W.; Han, H.; Chen, N.; Zhang, R.Y. Increased carbamylation level of HDL in end-stage renal disease: Carbamylated-HDL attenuated endothelial cell function. Am. J. Physiol. Ren. Physiol. 2016, 310, F511–F517. [Google Scholar] [CrossRef]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein Carbamylation Renders High-Density Lipoprotein Dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef]
- Holzer, M.; Zangger, K.; El-Gamal, D.; Binder, V.; Curcic, S.; Konya, V.; Schuligoi, R.; Heinemann, A.; Marsche, G. Myeloperoxidase-Derived Chlorinating Species Induce Protein Carbamylation Through Decomposition of Thiocyanate and Urea: Novel Pathways Generating Dysfunctional High-Density Lipoprotein. Antioxid. Redox Signal. 2012, 17, 1043–1052. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Density (Ultracentrifugation) | δ g/mL |
|---|---|
| HDL2 | 1.063–1.125 |
| HDL3 | 1.125–1.210 |
| Size (Electrophoresis) | nm |
| HDL2b | 9.7–12.0 |
| HDL2a | 8.8–9.7 |
| HDL3a | 8.2–8.8 |
| HDL3b | 7.8–8.2 |
| HDL3c | 7.2–7.8 |
| Charge and Size (2D Electrophoresis) | particles |
| Preβ-HDL | preβ1, preβ2 |
| α-HDL | α1, α2, α3, α4 |
| Preα-HDL | preα1, preα2, preα3 |
| Composition (Antibody-Based) | |
| LpA-I | apoA-I |
| LpA-I:A-II | apoA-I + apoA-II |
| Size + Particle Number (Nuclear Magnetic Resonance (NMR)) | nm |
| Large HDL | 8.8–13.0 |
| Medium HDL | 8.2–8.8 |
| Small HDL | 7.3–8.2 |
| Study | Cohort | Method | Control | Chronic Kidney Disease (CKD) | Hemodialysis (HD) |
|---|---|---|---|---|---|
| Samuelsson et al. 2002 [70] | CKD, n = 45 Controls, n = 45 | Immuno-absorption | mg/dL LpA-I = 34.7 ± 7.1 LpA-I:A-II=103.4 ± 18.0 | mg/dL LpA-I = 32.6 ± 5.3 LpA-I:A-II = 93.6 ± 14.5 | - |
| Calabresi et al. 2015 [59] | CKD, n = 50 HD, n = 198 Controls, n = 40 | Immuno-absorption + Native gel electrophoresis | mg/dL LpA-I = 50.1 ± 13.2 LpA-I:A-II = 84.6 ± 12.7 % of total HDL protein preβ-HDL = 13.1 ± 3.2 size (nm) HDL2 = 11.2 ± 0.3 HDL3 = 8.8 ± 0.3 | mg/dL LpA-I = 55.2 ± 15.9 LpA-I:A-II = 64.0 ± 14.4 % of total HDL protein preβ-HDL = 15.8 ± 4.7 size (nm) HDL2 = 11.1 ± 0.2 HDL3 = 8.9 ± 0.2 | mg/dL LpA-I = 43.1 ± 12.8 LpA-I:A-II = 49.2 ± 13.6 % of total HDL protein preβ-HDL = 17.1 ± 4.7 size (nm) HDL2 = 11.2 ± 0.4 HDL3 = 8.8 ± 0.4 |
| Holzer et al. 2015 [13] | CKD, n = 24 Controls, n = 20 | Ultracentrifugation + Native gel electrophoresis | % of total protein HDL2 = 44.8 ± 5.7 HDL3 = 55.2 ± 5.8 | - | % of total protein HDL2 = 40.7 ± 6.1 HDL3 = 59.4 ± 6.0 |
| Homma et al. 2013 [76] | CKD, n = 40 Controls, n = 40 | sequential ultracentrifugation | mg/dL cholesterol HDL2 = 21.8 ± 6.9 HDL3 = 21.5 ± 4.8 | - | mg/dL cholesterol HDL2 = 30.6 ± 12.3 HDL3 = 17.6 ± 4.5 |
| Kuchta et al. 2019 [62] | 3 CKD groups Stage 3a, n = 17 Stage 3b, n = 34 Stage 4, n = 17 | ELISA | - | pre-β1 HDL (mg/dL) Stage 3a, 1.85 ± 0.20 Stage 3b, 2.20 ± 0.21 Stage 4, 2.70 ± 0.22 | - |
| Gille et al. 2019 [71] | CKD, n = 16 Controls, n = 16 | ELISA | mg/mL preβ1-HDL = 16 ± 3 | mg/mL preβ1-HDL = 23 ± 1 | - |
| Miida et al. 2003 [60] | CKD, n = 45 Controls, n = 45 | 2D Native gel electrophoresis | % of apoA-I preβ1-HDL = 5.0 ± 2.0 preβ2-HDL = 4.6 ± 2.5 preβ3-HDL = 0.9 ± 0.5 HDL2b = 20.0 ± 13.5 HDL2a = 37.4 ± 10.0 HDL3 = 27.0 ± 9.7 | - | % of apoA-I preβ1-HDL = 13.5 ± 3.5 preβ2-HDL = 6.0 ± 2.1 preβ3-HDL = 1.0 ± 0.5 HDL2b = 22.5 ± 5.5 HDL2a = 35.5 ± 5.8 HDL3 = 21.5 ± 4.1 |
| Alabakovska et al. 2002 [79] | CKD, n = 42 HD, n = 63 Controls, n = 345 | Native gel electrophoresis | percent distribution HDL2b = 50.0% HDL2a = 45.5% HDL3a = 4.5% HDL3b = 0.0% HDL3c = 0.0% | percent distribution HDL2b = 16.5% HDL2a = 62.0% HDL3a = 21.5% HDL3b = 0.0% HDL3c = 0.0% | percent distribution HDL2b = 30.0% HDL2a = 67.0% HDL3a = 3.0% HDL3b = 0.0% HDL3c = 0.0% |
| Stefanovic et al. 2017 [78] | CKD, n = 19 PT, n = 19 | Native gel electrophoresis | % of total HDL protein HDL2b = 48.6 ± 4.9 HDL2a = 22.6 ± 2.3 HDL3a = 14.0 ± 2.2 HDL3b = 7.5 ± 1.5 HDL3c = 7.2 ± 1.5 | % of total HDL protein HDL2b = 39.3 ± 5.4 HDL2a = 21.3 ± 2.1 HDL3a = 16.8 ± 2.6 HDL3b = 10.1 ± 2.1 HDL3c = 12.6 ± 6.5 | - |
| Miljkovic et al. 2018 [75] | CKD, n = 21 HD, n = 56 Controls, n = 20 | Native gel electrophoresis | % of total HDL protein HDL2b = 54.0 ± 9.7 HDL 2a = 18(16.1–21.5) HDL3a = 11.0 ± 3.2 HDL3b = 6.2(3.8–8.4) HDL3c = 10.4 (6.0–11.4) | % of total HDL protein HDL2b = 46.0 ± 9.8 HDL 2a = 20 (17.9-23.6) HDL3a = 13.0 ± 2.8 HDL3b = 7.4 (5.4–9.3) HDL3c = 13.9 (5.2–16.9) | % of total HDL protein HDL2b = 44.0 ± 11.6 HDL 2a = 22 (18.1-24.1) HDL3a = 14.0 ± 4.5 HDL3b = 7.7 (6.6–10.9) HDL3c = 10.2 (6.8–14.8) |
| Soto-Miranda et al. 2012 [77] | CKD, n = 40 Controls, n = 40 | Ultracentrifugation + Native gel electrophoresis | % of total HDL protein HDL2b = 15.7 ± 5.7 HDL2a = 8.8 ± 1.9 HDL3a = 24.1 ± 2.7 HDL3b = 20.6 ± 2.9 HDL3c = 31.9 ± 7.3 | % of total HDL protein HDL2b = 23.5 ± 5.9 HDL2a = 11.6 ± 1.9 HDL3a = 24.6 ± 2.4 HDL3b = 15.6 ± 2.5 HDL3c = 24.8 ± 5.7 | - |
| Study | Cohort | Isolation Method | Detected Proteins | Proteins Upregulated | Proteins Downregulated | Validation Test | Functional Assessment |
|---|---|---|---|---|---|---|---|
| Holzer et al. 2011 [12] | Control, n = 19 HD, n = 27 | density gradient ultracentrifugation | 35 | apoC-III, SAA1, SAA4, apoC-II, apoA-IV, A1At, RBP4, TTR, a2CAT | apoA-I, apoA-II, apoC-I, apoM | Results for albumin, Lp-PLA2, A1AT, ApoAIV, ApoA-I, RBP4, TTR and SAA1 confirmed by immunoblot. | Total cholesterol efflux ↓ scavenger receptor BI (SR-BI)-specific cholesterol efflux ↓ ATP-binding cassette transporter A1 (ABCA1)-specific cholesterol efflux ↔ Macrophage net cholesterol efflux ↓ HDL-associated Lp-PLA2 activity ↑ |
| Weichhart et al. 2012 [13] | Control, n = 10 HD, n = 10 | sequential ultracentrifugation | 49 | apoC-II, SAA, SP-B, AMBP | - | Replica cohort of 12 control and 14 HD used to confirm MS result by immunoblot for TF, Sp-B, PEDF, SAA, apoC-II, apoA-I | HDL anti-inflammatory activity ↓ HDL anti-oxidative activity ↓ |
| Mange et al. 2012 [84] | Control, n = 7 HD, n = 7 | sequential ultracentrifugation | 122 | apoA2, apoC3, AMBP, apoD, apoC2, B2MG, SAA4, apo(a), RBP4, ApoC1, LCAT, ApoA4, ApoE, SAA, ApoM, PON1, ApoC4, ApoL1, ApoB100 | ST, C3, FIB, HG, Igα, A2MG, CFH, Igμ, FIBR, HP, KIN1, PT, HRG, ITIH4, VTN, AT3, CLUS, Igλ | Results for apoC2, apoC3, ST, HG confirmed in validation cohort. | not performed |
| Shao et al. 2015 [85] | Control, n = 20 HD, n = 40 | sequential ultracentrifugation | 63 | AMBP, B2MG, CFD, CST3, PTGDS, RBP4, SAA1, CST3, AMBP, CFD, PTGDS, SAA4, TTR, ApoCII, apoCIII, A1GP2, apoAIV,Igk, SP-B, Igλ, SP-B | apoA-I, apoA-II, apoL-I, apoM, PON1, VTN. | Shotgun proteomics used for identification of proteins, followed by SRM to quantify and validate. | not performed |
| Kopecky et al. 2015 [82] | Controls, n = 15 HD, n = 14 KTxpoor, n = 14 KTxgood, n = 14 | density gradient ultracentrifugation | 80 | Ktxpoor and HD: AMBP, B2MG, RBP4, Igγ3, FIBR, CFD, ZA2GP. Ktxpoor: B2GP1, LRA2GP, apo(a), CAMP, A1CT, ANG, PC1, CYS, SHDP, VDBP, A1AGP | - | Enrichment of SAA and SP-B in Ktxgood, Ktxpoor and HD quantified with ELISA. | Cholesterol efflux ↓ vs. Ktxgood Arylesterase activity ↓ vs. Ktxgood, Ktxpoor, HD; Leukocyte cholesterol content ↑ vs. Ktxgood, Ktxpoor, HD |
| Rubinow et al. 2017 [83] | CKD, n = 538. 5 groups: eGFR >60, n = 92 eGFR = 45-60, n = 91 eGFR = 30-45, n = 106 eGFR = 15-30, n = 102 eGFR < 15, n = 34 | 2-step density gradient ultracentrifugation | 38 | RBP4, apoC3↑ | ApoL1, CETP, VN↓ | - | not performed |
| Wang et al. 2018 [86] | Pre-dialysis, n = 110 Hemodialysis, n = 143 | 2-step density gradient ultracentrifugation | 38 | SAA2, HBB, SAA1, HPR, CETP, PLTP, ApoE | - | - | Cholesterol efflux ↓ vs. pre-dialysis |
| Florens et al. 2019 [87] | Control, n = 8 HD, n = 9 | sequential ultracentrifugation | 326 | UDP 1, B2MG, SP-B, AMBP, IGF2, IGHA2, IGLC2, HLA-B, CFD, ITIH4 | GUCA, CAPN1, KRT16, RAB6B, GM2A, PTGDS, SCGB, PRDX3, SCF2 | - | not performed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsche, G.; Heine, G.H.; Stadler, J.T.; Holzer, M. Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules 2020, 10, 1348. https://doi.org/10.3390/biom10091348
Marsche G, Heine GH, Stadler JT, Holzer M. Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules. 2020; 10(9):1348. https://doi.org/10.3390/biom10091348
Chicago/Turabian StyleMarsche, Gunther, Gunnar H. Heine, Julia T. Stadler, and Michael Holzer. 2020. "Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease" Biomolecules 10, no. 9: 1348. https://doi.org/10.3390/biom10091348
APA StyleMarsche, G., Heine, G. H., Stadler, J. T., & Holzer, M. (2020). Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules, 10(9), 1348. https://doi.org/10.3390/biom10091348