Targeting Receptors on Cancer Cells with Protein Toxins

 , ,
, , 
{kind=link}
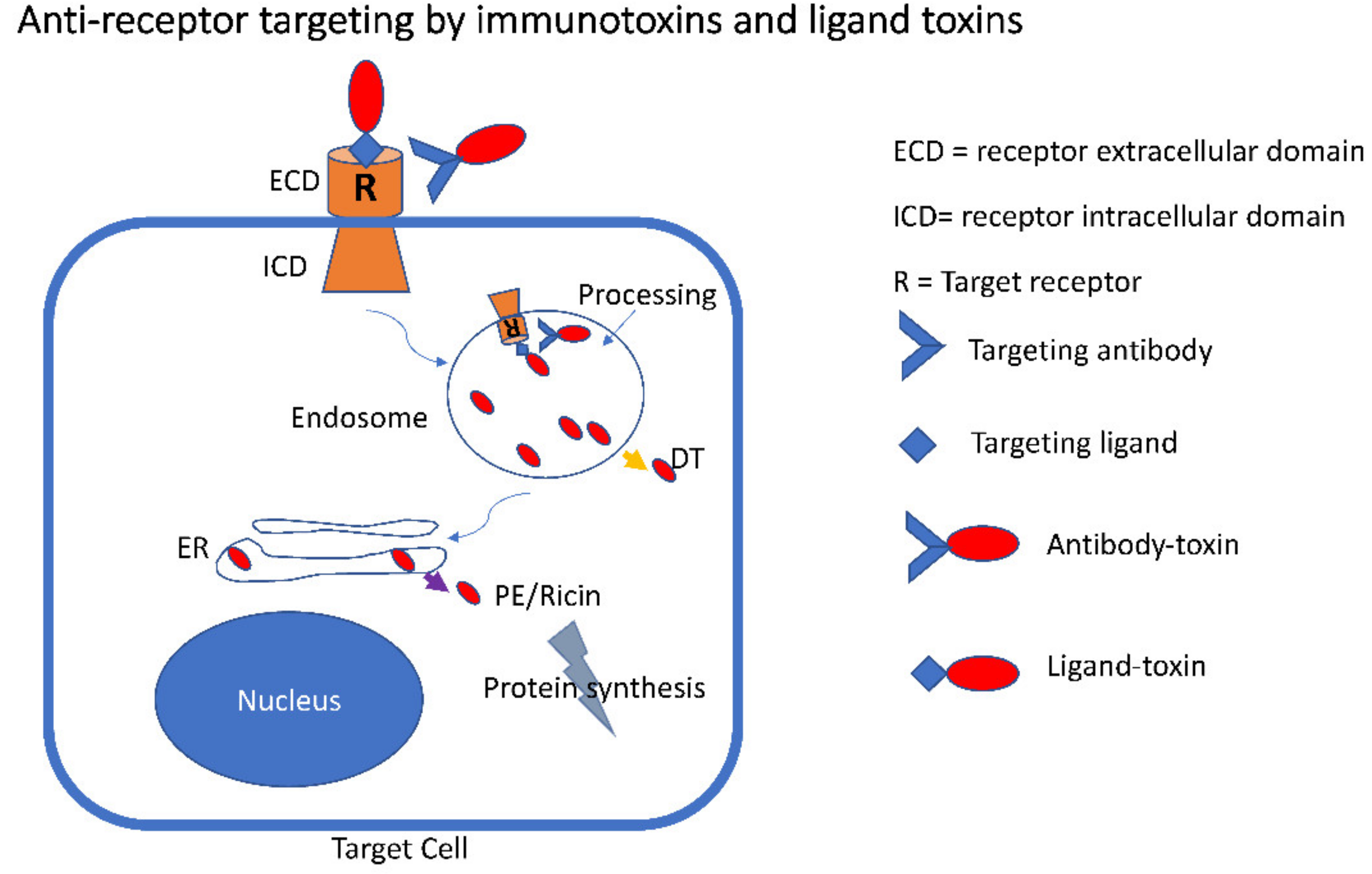{kind=link}
Abstract
1. Introduction
2. Immunotoxin Production
3. Immunotoxins Targeting EGFR
4. EGFRvIII and “Cancer-Expressed” EGFR
5. HER2
6. HER3
7. UPAR and Bispecific Targeting of EGFR
8. Immunotoxins Targeting the Transferrin Receptor
9. Immunotoxins Targeting Cytokine Receptors
10. Targeting the Interleukin-2 Receptor (IL2R)
11. Targeting Interleukin-3 Receptor (IL3R)
12. Targeting the Interleukin-4 (IL4R) and Interleukin-13 (IL13R) Receptors
13. Targeting Interleukin-7 Receptor (IL-7R)
14. Targeting Interleukin-6 Receptor (IL6R)
15. Other Receptors
15.1. Targeting C-C Chemokine Receptor Type 9 (CCR9)
15.2. The EPH Receptors
15.3. MSH Receptors
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DT | diphtheria toxin |
| PE | pseudomonas exotoxin |
| EGFR | epidermal growth factor receptor |
| EGFRvIII | EGFR with a deletion of exons 2–7 |
| DT388/DAB388 | truncated DT including the first 388 amino acids |
| PE40 | truncated PE including domains 2 and 3 |
| PE38 | truncated PE including domains 2 and 3 without a disulfide loop |
| TP40 | Transforming growth factor alpha-PE40 |
| scFv | antibody, single-chain Fv |
| ETA | exotoxin A (alternative name for PE) |
| ATF | amino-terminal fragment |
| uPAR | urokinase plasminogen activator receptor |
| LMB-2 | immunotoxin from the scFv of the antiTac antibody and PE40 |
References
- Onda, M. Recombinant immunotoxins with low endotoxins for clinical and animal studies. Antib. Eng. 2012, 907, 627–643. [Google Scholar] [CrossRef]
- Barth, S.; Huhn, M.; Wels, W.; Diehl, V.; Engert, A. Construction and in vitro evaluation of RFT5(scFv)-ETA’ a new recombinant single-chain immunotoxin with specific Cytotoxicity toward CD25+ Hodgkin-derived cell lines. Int. J. Mol. Med. 1998, 1, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Vitetta, E.S.; Thorpe, P.E. Immunotoxins containing ricin or its a chain. Semin. Cell Biol. 1991, 2, 47–58. [Google Scholar] [PubMed]
- Cao, Y.; Marks, J.W.; Liu, Z.; Cheung, L.H.; Hittelman, W.N.; Rosenblum, M.G. Design optimization and characterization of Her2/neu-targeted immunotoxins: Comparative in vitro and in vivo efficacy studies. Oncogene 2014, 33, 429–439. [Google Scholar] [CrossRef]
- Cohen, S. The epidermal growth factor (EGF). Cancer 1983, 51, 1787–1791. [Google Scholar] [CrossRef]
- Secq, V.; Villeret, J.; Fina, F.; Carmassi, M.; Carcopino, X.; Garcia, S.; Metellus, I.; Boubli, L.; Iovanna, J.; Charpin, C. Triple negative breast carcinoma EGFR amplification is not associated with EGFR, Kras or ALK mutations. Br. J. Cancer 2014, 110, 1045–1052. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Red Brewer, M.; Yun, C.H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef]
- Chaudhary, V.K.; FitzGerald, D.J.; Adhya, S.; Pastan, I. Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc. Natl. Acad. Sci. USA 1987, 84, 4538–4542. [Google Scholar] [CrossRef]
- Simon, N.; FitzGerald, D. Immunotoxin therapies for the treatment of epidermal growth factor receptor-dependent cancers. Toxins 2016, 8, 137. [Google Scholar] [CrossRef]
- Heimbrook, D.C.; Stirdivant, S.M.; Ahern, J.D.; Balishin, N.L.; Patrick, D.R.; Edwards, G.M.; Defeo-Jones, D.; FitzGerald, D.J.; Pastan, I.; Oliff, A. Transforming growth factor alpha-Pseudomonas exotoxin fusion protein prolongs survival of nude mice bearing tumor xenografts. Proc. Natl. Acad. Sci. USA 1990, 87, 4697–4701. [Google Scholar] [CrossRef]
- Mellon, J.K.; Cook, S.; Chambers, P.; Neal, D.E. Transforming growth factor alpha and epidermal growth factor levels in bladder cancer and their relationship to epidermal growth factor receptor. Br. J. Cancer 1996, 73, 654–658. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Berger, M.S.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Greer, K.; Herndon, J.E., 2nd; Kunwar, S.; et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro. Oncol. 2008, 10, 320–329. [Google Scholar] [CrossRef]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Bigner, D.D.; Berger, M.S.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E., 2nd; Kunwar, S.; Marcus, S.; et al. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neurooncol. 2003, 65, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.P.; Akiyoshi, D.E.; Arrigo, D.A.; Rhoad, A.E.; Sullivan, B.; Thomas, J.; Genbauffe, F.S.; Bacha, P.; Nichols, J.C. Cytotoxic properties of DAB486EGF and DAB389EGF, epidermal growth factor (EGF) receptor-targeted fusion toxins. J. Biol. Chem. 1991, 266, 21118–21124. [Google Scholar] [PubMed]
- Theodoulou, M.; Baselga, J.; Scher, H.; Trainor, K.; Mendelsohn, J. Phase I dose-escalation study of the safety, tolerability, pharmacokinetics and biologic effects of DAB389EGF in patients with solid malignancies that express EGF receptors (EGFR). Proc. Am. Soc. Clin. Oncol. 1995, 14, 480. [Google Scholar]
- Niesen, J.; Stein, C.; Brehm, H.; Hehmann-Titt, G.; Fendel, R.; Melmer, G.; Fischer, R.; Barth, S. Novel EGFR-specific immunotoxins based on panitumumab and cetuximab show in vitro and ex vivo activity against different tumor entities. J. Cancer Res. Clin. Oncol. 2015, 141, 2079–2095. [Google Scholar] [CrossRef]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS. J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Paolillo, M.; Boselli, C.; Schinelli, S. Glioblastoma under Siege: An Overview of current therapeutic strategies. Brain Sci. 2018, 8, 15. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Kuan, C.T.; Wikstrand, C.J.; Archer, G.; Beers, R.; Pastan, I.; Zalutsky, M.R.; Bigner, D.D. Increased binding affinity enhances targeting of glioma xenografts by EGFRvIII-specific scFv. Int. J. Cancer 2000, 88, 962–969. [Google Scholar] [CrossRef]
- Archer, G.E.; Sampson, J.H.; Lorimer, I.A.; McLendon, R.E.; Kuan, C.T.; Friedman, A.H.; Friedman, H.S.; Pastan, I.H.; Bigner, D.D. Regional treatment of epidermal growth factor receptor vIII-expressing neoplastic meningitis with a single-chain immunotoxin, MR-1. Clin. Cancer Res. 1999, 5, 2646–2652. [Google Scholar] [PubMed]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, 586. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Boskovitz, A.; Kuan, C.T.; Pegram, C.N.; Ayriss, J.; Wikstrand, C.J.; Buckley, A.F.; Lipp, E.S.; Herndon, J.E., 2nd; McLendon, R.E.; et al. Radioimmunotargeting of malignant glioma by monoclonal antibody D2C7 reactive against both wild-type and variant III mutant epidermal growth factor receptors. Nucl. Med. Biol. 2012, 39, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, V.; Bao, X.; Keir, S.T.; Pegram, C.N.; Szafranski, S.E.; Piao, H.; Wikstrand, C.J.; McLendon, R.E.; Kuan, C.T.; Pastan, I.H.; et al. Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin. Cancer Res. 2013, 19, 4717–4727. [Google Scholar] [CrossRef]
- Bao, X.; Chandramohan, V.; Reynolds, R.P.; Norton, J.N.; Wetsel, W.C.; Rodriguiz, R.M.; Aryal, D.K.; McLendon, R.E.; Levin, E.D.; Petry, N.A.; et al. Preclinical toxicity evaluation of a novel immunotoxin, D2C7-(scdsFv)-PE38KDEL, administered via intracerebral convection-enhanced delivery in rats. Invest. New Drugs 2016, 34, 149–158. [Google Scholar] [CrossRef]
- Bao, X.; Pastan, I.; Bigner, D.D.; Chandramohan, V. EGFR/EGFRvIII-targeted immunotoxin therapy for the treatment of glioblastomas via convection-enhanced delivery. Recept. Clin. Investig. 2016, 3. [Google Scholar] [CrossRef]
- Gan, H.K.; Lappas, M.; Cao, D.X.; Cvrljevdic, A.; Scott, A.M.; Johns, T.G. Targeting a unique EGFR epitope with monoclonal antibody 806 activates NF-kappaB and initiates tumour vascular normalization. J. Cell Mol. Med. 2009, 13, 3993–4001. [Google Scholar] [CrossRef]
- Luwor, R.B.; Johns, T.G.; Murone, C.; Huang, H.J.; Cavenee, W.K.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 2001, 61, 5355–5361. [Google Scholar] [PubMed]
- Orellana, L.; Thorne, A.H.; Lema, R.; Gustavsson, J.; Parisian, A.D.; Hospital, A.; Cordeiro, T.N.; Bernadó, P.; Scott, A.M.; Brun-Heath, I.; et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl. Acad. Sci. USA 2019, 116, 10009–10018. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.; Antignani, A.; Sarnovsky, R.; Hewitt, S.M.; FitzGerald, D. Targeting a cancer-specific epitope of the epidermal growth factor receptor in triple-negative breast cancer. J. Natl. Cancer Inst. 2016, 108, djw028. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.C.H.; Antignani, A.; Sarnovsky, R.; FitzGerald, D. Characterization of monoclonal antibodies generated to the 287-302 amino acid loop of the human epidermal growth factor receptor. Antib. Ther. 2019, 2, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Pai-Scherf, L.H.; Villa, J.; Pearson, D.; Watson, T.; Liu, E.; Willingham, M.C.; Pastan, I. Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin. Cancer Res. 1999, 5, 2311–2315. [Google Scholar]
- Reiter, Y.; Brinkmann, U.; Jung, S.H.; Lee, B.; Kasprzyk, P.G.; King, C.R.; Pastan, I. Improved binding and antitumor activity of a recombinant anti-erbB2 immunotoxin by disulfide stabilization of the Fv fragment. J. Biol. Chem. 1994, 269, 18327–18331. [Google Scholar]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef]
- Wels, W.; Harwerth, I.M.; Mueller, M.; Groner, B.; Hynes, N.E. Selective inhibition of tumor cell growth by a recombinant single-chain antibody-toxin specific for the erbB-2 receptor. Cancer Res. 1992, 52, 6310–6317. [Google Scholar]
- Wels, W.; Beerli, R.; Hellmann, P.; Schmidt, M.; Marte, B.M.; Kornilova, E.S.; Hekele, A.; Mendelsohn, J.; Groner, B.; Hynes, N.E. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int. J. Cancer 1995, 60, 137–144. [Google Scholar] [CrossRef]
- Azemar, M.; Schmidt, M.; Arlt, F.; Kennel, P.; Brandt, B.; Papadimitriou, A.; Groner, B.; Wels, W. Recombinant antibody toxins specific for ErbB2 and EGF receptor inhibit the in vitro growth of human head and neck cancer cells and cause rapid tumor regression in vivo. Int. J. Cancer 2000, 86, 269–275. [Google Scholar] [CrossRef]
- Maurer-Gebhard, M.; Schmidt, M.; Azemar, M.; Altenschmidt, U.; Stocklin, E.; Wels, W.; Groner, B. Systemic treatment with a recombinant erbB-2 receptor-specific tumor toxin efficiently reduces pulmonary metastases in mice injected with genetically modified carcinoma cells. Cancer Res. 1998, 58, 2661–2666. [Google Scholar] [PubMed]
- von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jager, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005, 7, R617–R626. [Google Scholar] [CrossRef] [PubMed]
- Waltzman, R.J.; Sarkar, A.; Williams, E.T.; Iberg, A.T.; Higgins, J.T.; Willert, E.K. MT-5111: A novel HER2 targeting engineered toxin body in clinical development. J. Clin. Oncol. 2020, 38, 433. [Google Scholar] [CrossRef]
- Higgins, J.P.; Sarkar, A.; Williams, E.T.; Iberg, A.; Waltzman, R.; Willert, E.K. Abstract P1-18-35: MT-5111, a novel HER2 targeting engineered toxin body, under clinical development to overcome mechanisms of resistance to existing HER2 targeted therapies. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Capone, E.; Giansanti, F.; Ponziani, S.; Lamolinara, A.; Iezzi, M.; Cimini, A.; Angelucci, F.; Sorda, R.; Laurenzi, V.; Natali, P.G.; et al. EV20-Sap, a novel anti-HER-3 antibody-drug conjugate, displays promising antitumor activity in melanoma. Oncotarget 2017, 8, 95412–95424. [Google Scholar] [CrossRef]
- Tsai, A.K.; Oh, S.; Chen, H.; Shu, Y.; Ohlfest, J.R.; Vallera, D.A. A novel bispecific ligand-directed toxin designed to simultaneously target EGFR on human glioblastoma cells and uPAR on tumor neovasculature. J. Neurooncol. 2011, 103, 255–266. [Google Scholar] [CrossRef]
- Oh, S.; Tsai, A.K.; Ohlfest, J.R.; Panoskaltsis-Mortari, A.; Vallera, D.A. Evaluation of a bispecific biological drug designed to simultaneously target glioblastoma and its neovasculature in the brain. J. Neurosurg. 2011, 114, 1662–1671. [Google Scholar] [CrossRef]
- Schappa, J.T.; Frantz, A.M.; Gorden, B.H.; Dickerson, E.B.; Vallera, D.A.; Modiano, J.F. Hemangiosarcoma and its cancer stem cell subpopulation are effectively killed by a toxin targeted through epidermal growth factor and urokinase receptors. Int. J. Cancer 2013, 133, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Borgatti, A.; Fieberg, A.; Winter, A.L.; Stuebner, K.; Taras, E.; Todhunter, D.; Masyr, A.; Rendhal, A.; Vallera, D.A.; Koopmeiners, J.S.; et al. Impact of repeated cycles of EGF bispecific angiotoxin (eBAT) administered at a reduced interval from doxorubicin chemotherapy in dogs with splenic haemangiosarcoma. Vet. Comp. Oncol. 2020. [Google Scholar] [CrossRef]
- Oh, S.; Ohlfest, J.R.; Todhunter, D.A.; Vallera, V.D.; Hall, W.A.; Chen, H.; Vallera, D.A. Intracranial elimination of human glioblastoma brain tumors in nude rats using the bispecific ligand-directed toxin, DTEGF13 and convection enhanced delivery. J. Neurooncol. 2009, 95, 331–342. [Google Scholar] [CrossRef]
- Waldron, N.N.; Oh, S.; Vallera, D.A. Bispecific targeting of EGFR and uPAR in a mouse model of head and neck squamous cell carcinoma. Oral Oncol. 2012, 48, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Errico Provenzano, A.; Posteri, R.; Giansanti, F.; Angelucci, F.; Flavell, S.U.; Flavell, D.J.; Fabbrini, M.S.; Porro, D.; Ippoliti, R.; Ceriotti, A.; et al. Optimization of construct design and fermentation strategy for the production of bioactive ATF-SAP, a saporin based anti-tumoral uPAR-targeted chimera. Microb. Cell Fact. 2016, 15, 194. [Google Scholar] [CrossRef] [PubMed]
- Zuppone, S.; Assalini, C.; Minici, C.; Bertagnoli, S.; Branduardi, P.; Degano, M.; Fabbrini, M.S.; Montorsi, F.; Salonia, A.; Vago, R. The anti-tumoral potential of the saporin-based uPAR-targeting chimera ATF-SAP. Sci. Rep. 2020, 10, 2521. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Bernabeu, E.; Rodriguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820, 291–317. [Google Scholar] [CrossRef]
- Tortorella, S.; Karagiannis, T.C. The significance of transferrin receptors in oncology: The development of functional nano-based drug delivery systems. Curr. Drug. Deliv. 2014, 11, 427–443. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef]
- Hopkins, C.R.; Trowbridge, I.S. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell Biol. 1983, 97, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef]
- Klausner, R.D.; Ashwell, G.; van Renswoude, J.; Harford, J.B.; Bridges, K.R. Binding of apotransferrin to K562 cells: Explanation of the transferrin cycle. Proc. Natl. Acad. Sci. USA 1983, 80, 2263–2266. [Google Scholar] [CrossRef]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. [Google Scholar] [CrossRef]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F. Ribosome-inactivating proteins. Toxicon 2004, 44, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, I.S.; Domingo, D.L. Anti-transferrin receptor monoclonal antibody and toxin-antibody conjugates affect growth of human tumour cells. Nature 1981, 294, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Zovickian, J.; Johnson, V.G.; Youle, R.J. Potent and specific killing of human malignant brain tumor cells by an anti-transferrin receptor antibody-ricin immunotoxin. J. Neurosurg. 1987, 66, 850–861. [Google Scholar] [CrossRef]
- Laske, D.W.; Muraszko, K.M.; Oldfield, E.H.; DeVroom, H.L.; Sung, C.; Dedrick, R.L.; Simon, T.R.; Colandrea, J.; Copeland, C.; Katz, D.; et al. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 1997, 41, 1039–1049. [Google Scholar] [CrossRef]
- Pirker, R.; FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Enhancement of the activity of immunotoxins made with either ricin A chain or Pseudomonas exotoxin in human ovarian and epidermoid carcinoma cell lines. Cancer Res. 1988, 48, 3919–3923. [Google Scholar]
- Bergamaschi, G.; Cazzola, M.; Dezza, L.; Savino, E.; Consonni, L.; Lappi, D. Killing of K562 cells with conjugates between human transferrin and a ribosome-inactivating protein (SO-6). Br. J. Haematol. 1988, 68, 379–384. [Google Scholar] [CrossRef]
- Cimini, A.; Mei, S.; Benedetti, E.; Laurenti, G.; Koutris, I.; Cinque, B.; Cifone, M.G.; Galzio, R.; Pitari, G.; Di Leandro, L.; et al. Distinct cellular responses induced by saporin and a transferrin-saporin conjugate in two different human glioblastoma cell lines. J. Cell Physiol. 2012, 227, 939–951. [Google Scholar] [CrossRef]
- Ippoliti, R.; Lendaro, E.; D’Agostino, I.; Fiani, M.L.; Guidarini, D.; Vestri, S.; Benedetti, P.A.; Brunori, M. A chimeric saporin-transferrin conjugate compared to ricin toxin: Role of the carrier in intracellular transport and toxicity. FASEB J. 1995, 9, 1220–1225. [Google Scholar] [CrossRef]
- Scott, C.F., Jr.; Goldmacher, V.S.; Lambert, J.M.; Jackson, J.V.; McIntyre, G.D. An immunotoxin composed of a monoclonal antitransferrin receptor antibody linked by a disulfide bond to the ribosome-inactivating protein gelonin: Potent in vitro and in vivo effects against human tumors. J. Natl. Cancer Inst. 1987, 79, 1163–1172. [Google Scholar] [CrossRef]
- FitzGerald, D.J.; Trowbridge, I.S.; Pastan, I.; Willingham, M.C. Enhancement of toxicity of antitransferrin receptor antibody-Pseudomonas exotoxin conjugates by adenovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 4134–4138. [Google Scholar] [CrossRef] [PubMed]
- Pirker, R.; FitzGerald, D.J.P.; Willingham, M.C.; Pastan, I.; Hamilton, T.C.; Ozols, R.F. Anti-transferrin receptor antibody linked to pseudomonas exotoxin as a model immunotoxin in human ovarian carcinoma cell lines. Cancer Res. 1985, 45, 751–757. [Google Scholar] [PubMed]
- Batra, J.K.; Fitzgerald, D.J.; Chaudhary, V.K.; Pastan, I. Single-chain immunotoxins directed at the human transferrin receptor containing Pseudomonas exotoxin A or diphtheria toxin: Anti-TFR(Fv)-PE40 and DT388-anti-TFR(Fv). Mol. Cell Biol. 1991, 11, 2200–2205. [Google Scholar] [CrossRef] [PubMed][Green Version]
- FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Antitumor effects of an immunotoxin made with Pseudomonas exotoxin in a nude mouse model of human ovarian cancer. Proc. Natl. Acad. Sci. USA 1986, 83, 6627–6630. [Google Scholar] [CrossRef]
- Batra, J.K.; Jinno, Y.; Chaudhary, V.K.; Kondo, T.; Willingham, M.C.; FitzGerald, D.J.; Pastan, I. Antitumor activity in mice of an immunotoxin made with anti-transferrin receptor and a recombinant form of Pseudomonas exotoxin. Proc. Natl. Acad. Sci. USA 1989, 86, 8545–8549. [Google Scholar] [CrossRef]
- Shinohara, H.; Fan, D.; Ozawa, S.; Yano, S.; Van Arsdell, M.; Viner, J.L.; Beers, R.; Pastan, I.; Fidler, I.J. Site-specific expression of transferrin receptor by human colon cancer cells directly correlates with eradication by antitransferrin recombinant immunotoxin. Int. J. Oncol. 2000, 17, 643–651. [Google Scholar] [CrossRef]
- Antignani, A.; Segal, D.; Simon, N.; Kreitman, R.J.; Huang, D.; FitzGerald, D.J. Essential role for Bim in mediating the apoptotic and antitumor activities of immunotoxins. Oncogene 2017, 36, 4953–4962. [Google Scholar] [CrossRef]
- Greenfield, L.; Johnson, V.G.; Youle, R.J. Mutations in diphtheria toxin separate binding from entry and amplify immunotoxin selectivity. Science 1987, 238, 536–539. [Google Scholar] [CrossRef]
- Johnson, V.G.; Wilson, D.; Greenfield, L.; Youle, R.J. The role of the diphtheria toxin receptor in cytosol translocation. J. Biol. Chem. 1988, 263, 1295–1300. [Google Scholar]
- Laske, D.W.; Youle, R.J.; Oldfield, E.H. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat. Med. 1997, 3, 1362–1368. [Google Scholar] [CrossRef]
- Weaver, M.; Laske, D.W. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003, 65, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. The structure, function, and expression of interleukin-2 receptors on normal and malignant lymphocytes. Science 1986, 232, 727–732. [Google Scholar] [CrossRef]
- FitzGerald, D.J.; Waldmann, T.A.; Willingham, M.C.; Pastan, I. Pseudomonas exotoxin-anti-TAC. Cell-specific immunotoxin active against cells expressing the human T cell growth factor receptor. J. Clin. Investig. 1984, 74, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.K.; FitzGerald, D.; Gately, M.; Chaudhary, V.K.; Pastan, I. Anti-Tac(Fv)-PE40, a single chain antibody Pseudomonas fusion protein directed at interleukin 2 receptor bearing cells. J. Biol. Chem. 1990, 265, 15198–15202. [Google Scholar]
- Chaudhary, V.K.; Gallo, M.G.; FitzGerald, D.J.; Pastan, I. A recombinant single-chain immunotoxin composed of anti-Tac variable regions and a truncated diphtheria toxin. Proc. Natl. Acad. Sci. USA 1990, 87, 9491–9494. [Google Scholar] [CrossRef]
- Reiter, Y.; Brinkmann, U.; Kreitman, R.J.; Jung, S.H.; Lee, B.; Pastan, I. Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry 1994, 33, 5451–5459. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.M.; Saleh, M.N.; Krueger, J.G.; Nichols, J.C.; Murphy, J.R. Diphtheria toxin fusion proteins. Curr. Top. Microbiol. Immunol. 1998, 234, 63–81. [Google Scholar] [CrossRef]
- LeMaistre, C.F.; Saleh, M.N.; Kuzel, T.M.; Foss, F.; Platanias, L.C.; Schwartz, G.; Ratain, M.; Rook, A.; Freytes, C.O.; Craig, F.; et al. Phase I trial of a ligand fusion-protein (DAB389IL-2) in lymphomas expressing the receptor for interleukin-2. Blood 1998, 91, 399–405. [Google Scholar]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schon, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin’s lymphoma. Blood 1997, 89, 403–410. [Google Scholar] [CrossRef]
- Schnell, R.; Borchmann, P.; Staak, J.O.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Engert, A. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin’s lymphoma. Ann. Oncol. 2003, 14, 729–736. [Google Scholar] [CrossRef]
- Powell, D.J., Jr.; Attia, P.; Ghetie, V.; Schindler, J.; Vitetta, E.S.; Rosenberg, S.A. Partial reduction of human FOXP3+ CD4 T cells in vivo after CD25-directed recombinant immunotoxin administration. J. Immunother. 2008, 31, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Stetler-Stevenson, M.; Jaffe, E.S.; Conlon, K.C.; Steinberg, S.M.; Wilson, W.; Waldmann, T.A.; Pastan, I. Complete remissions of adult T-cell leukemia with Anti-CD25 recombinant immunotoxin LMB-2 and chemotherapy to block immunogenicity. Clin. Cancer Res. 2016, 22, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Kobayashi, K.; Pastan, I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 4575–4582. [Google Scholar] [CrossRef]
- Broughton, S.E.; Hercus, T.R.; Nero, T.L.; Kan, W.L.; Barry, E.F.; Dottore, M.; Cheung Tung Shing, K.S.; Morton, C.J.; Dhagat, U.; Hardy, M.P.; et al. A dual role for the N-terminal domain of the IL-3 receptor in cell signalling. Nat. Commun. 2018, 9, 386. [Google Scholar] [CrossRef]
- Hogge, D.E.; Yalcintepe, L.; Wong, S.H.; Gerhard, B.; Frankel, A.E. Variant diphtheria toxin-interleukin-3 fusion proteins with increased receptor affinity have enhanced cytotoxicity against acute myeloid leukemia progenitors. Clin. Cancer Res. 2006, 12, 1284–1291. [Google Scholar] [CrossRef]
- Liu, T.F.; Urieto, J.O.; Moore, J.E.; Miller, M.S.; Lowe, A.C.; Thorburn, A.; Frankel, A.E. Diphtheria toxin fused to variant interleukin-3 provides enhanced binding to the interleukin-3 receptor and more potent leukemia cell cytotoxicity. Exp. Hematol. 2004, 32, 277–281. [Google Scholar] [CrossRef]
- Li, M.; Liu, Z.S.; Liu, X.L.; Hui, Q.; Lu, S.Y.; Qu, L.L.; Li, Y.S.; Zhou, Y.; Ren, H.L.; Hu, P. Clinical targeting recombinant immunotoxins for cancer therapy. Onco. Targets. Ther. 2017, 10, 3645–3665. [Google Scholar] [CrossRef]
- Syed, Y.Y. Tagraxofusp: First global approval. Drugs 2019, 79, 579–583. [Google Scholar] [CrossRef]
- Obiri, N.I.; Hillman, G.G.; Haas, G.P.; Sud, S.; Puri, R.K. Expression of high affinity interleukin-4 receptors on human renal cell carcinoma cells and inhibition of tumor cell growth in vitro by interleukin-4. J. Clin. Invest. 1993, 91, 88–93. [Google Scholar] [CrossRef]
- Obiri, N.I.; Siegel, J.P.; Varricchio, F.; Puri, R.K. Expression of high-affinity IL-4 receptors on human melanoma, ovarian and breast carcinoma cells. Clin. Exp. Immunol. 1994, 95, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.K.; Leland, P.; Kreitman, R.J.; Pastan, I. Human neurological cancer cells express interleukin-4 (IL-4) receptors which are targets for the toxic effects of IL4-Pseudomonas exotoxin chimeric protein. Int. J. Cancer 1994, 58, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Leland, P.; Puri, R.K. Structure, function, and targeting of interleukin 4 receptors on human head and neck cancer cells. Cancer Res. 2000, 60, 2981–2987. [Google Scholar] [PubMed]
- Joshi, B.H.; Leland, P.; Lababidi, S.; Varrichio, F.; Puri, R.K. Interleukin-4 receptor alpha overexpression in human bladder cancer correlates with the pathological grade and stage of the disease. Cancer Med. 2014, 3, 1615–1628. [Google Scholar] [CrossRef]
- Husain, S.R.; Obiri, N.I.; Gill, P.; Zheng, T.; Pastan, I.; Debinski, W.; Puri, R.K. Receptor for interleukin 13 on AIDS-associated Kaposi’s sarcoma cells serves as a new target for a potent Pseudomonas exotoxin-based chimeric toxin protein. Clin. Cancer Res. 1997, 3, 151–156. [Google Scholar]
- May, R.D.; Fung, M. Strategies targeting the IL-4/IL-13 axes in disease. Cytokine 2015, 75, 89–116. [Google Scholar] [CrossRef]
- Murata, T.; Obiri, N.I.; Debinski, W.; Puri, R.K. Structure of IL-13 receptor: Analysis of subunit composition in cancer and immune cells. Biochem. Biophys. Res. Commun. 1997, 238, 90–94. [Google Scholar] [CrossRef]
- Shimamura, T.; Husain, S.R.; Puri, R.K. The IL-4 and IL-13 pseudomonas exotoxins: New hope for brain tumor therapy. Neurosurg. Focus 2006, 20, 1–7. [Google Scholar] [CrossRef]
- Ishige, K.; Shoda, J.; Kawamoto, T.; Matsuda, S.; Ueda, T.; Hyodo, I.; Ohkohchi, N.; Puri, R.K.; Kawakami, K. Potent in vitro and in vivo antitumor activity of interleukin-4-conjugated Pseudomonas exotoxin against human biliary tract carcinoma. Int. J. Cancer 2008, 123, 2915–2922. [Google Scholar] [CrossRef]
- Kreitman, R.J. Immunotoxins. Expert Opin. Pharmacother. 2000, 1, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Rainov, N.G.; Heidecke, V. Long term survival in a patient with recurrent malignant glioma treated with intratumoral infusion of an IL4-targeted toxin (NBI-3001). J. Neurooncol. 2004, 66, 197–201. [Google Scholar] [CrossRef]
- Rand, R.W.; Kreitman, R.J.; Patronas, N.; Varricchio, F.; Pastan, I.; Puri, R.K. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clin. Cancer Res. 2000, 6, 2157–2165. [Google Scholar] [PubMed]
- Kawakami, K.; Kawakami, M.; Husain, S.R.; Puri, R.K. Targeting interleukin-4 receptors for effective pancreatic cancer therapy. Cancer Res. 2002, 62, 3575–3580. [Google Scholar] [PubMed]
- Shimamura, T.; Royal, R.E.; Kioi, M.; Nakajima, A.; Husain, S.R.; Puri, R.K. Interleukin-4 cytotoxin therapy synergizes with gemcitabine in a mouse model of pancreatic ductal adenocarcinoma. Cancer Res. 2007, 67, 9903–9912. [Google Scholar] [CrossRef] [PubMed]
- Lakkis, F.; Steele, A.; Pacheco-Silva, A.; Rubin-Kelley, V.; Strom, T.B.; Murphy, J.R. Interleukin 4 receptor targeted cytotoxicity: Genetic construction and in vivo immunosuppressive activity of a diphtheria toxin-related murine interleukin 4 fusion protein. Eur. J. Immunol. 1991, 21, 2253–2258. [Google Scholar] [CrossRef]
- Hershey, G.K. IL-13 receptors and signaling pathways: An evolving web. J. Allergy. Clin. Immunol. 2003, 111, 677–690. [Google Scholar] [CrossRef]
- Debinski, W.; Obiri, N.I.; Powers, S.K.; Pastan, I.; Puri, R.K. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin. Cancer Res. 1995, 1, 1253–1258. [Google Scholar]
- Kunwar, S. Convection enhanced delivery of IL13-PE38QQR for treatment of recurrent malignant glioma: Presentation of interim findings from ongoing phase 1 studies. Acta Neurochir. Suppl. 2003, 88, 105–111. [Google Scholar] [CrossRef]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro. Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef]
- Prados, M.; Kunwar, S.; Lang, F.F.; Ram, Z.; Westphal, M.; Barnett, G.; Sampson, J.H.; Croteau, D.; Puri, R.K. Final results of phase I/II studies of IL13-PE38QQR administered intratumorally (IT) and/or peritumorally (PT) via convection-enhanced delivery (CED) in patients undergoing tumor resection for recurrent malignant glioma. J. Clin. Oncol. 2005, 23, 1506. [Google Scholar] [CrossRef]
- Kioi, M.; Kawakami, K.; Puri, R.K. Analysis of antitumor activity of an interleukin-13 (IL-13) receptor-targeted cytotoxin composed of IL-13 antagonist and Pseudomonas exotoxin. Clin. Cancer Res. 2004, 10, 6231–6238. [Google Scholar] [CrossRef]
- Puri, R.K.; Leland, P.; Obiri, N.I.; Husain, S.R.; Kreitman, R.J.; Haas, G.P.; Pastan, I.; Debinski, W. Targeting of interleukin-13 receptor on human renal cell carcinoma cells by a recombinant chimeric protein composed of interleukin-13 and a truncated form of Pseudomonas exotoxin A (PE38QQR). Blood 1996, 87, 4333–4339. [Google Scholar] [CrossRef] [PubMed]
- Maini, A.; Hillman, G.; Haas, G.P.; Wang, C.Y.; Montecillo, E.; Hamzavi, F.; Pontes, J.E.; Leland, P.; Pastan, I.; Debinski, W.; et al. Interleukin-13 receptors on human prostate carcinoma cell lines represent a novel target for a chimeric protein composed of IL-13 and a mutated form of Pseudomonas exotoxin. J. Urol. 1997, 158, 948–953. [Google Scholar] [CrossRef]
- Liu, T.F.; Cai, J.; Gibo, D.M.; Debinski, W. Reoxygenation of hypoxic glioblastoma multiforme cells potentiates the killing effect of an interleukin-13-based cytotoxin. Clin. Cancer Res. 2009, 15, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Rustamzadeh, E.; Hall, W.A.; Todhunter, D.A.; Low, W.C.; Liu, H.; Panoskaltsis-Mortari, A.; Vallera, D.A. Intracranial therapy of glioblastoma with the fusion protein DTIL13 in immunodeficient mice. Int. J. Cancer 2006, 118, 2594–2601. [Google Scholar] [CrossRef]
- Todhunter, D.A.; Hall, W.A.; Rustamzadeh, E.; Shu, Y.; Doumbia, S.O.; Vallera, D.A. A bispecific immunotoxin (DTAT13) targeting human IL-13 receptor (IL-13R) and urokinase-type plasminogen activator receptor (uPAR) in a mouse xenograft model. Protein. Eng. Des. Sel. 2004, 17, 157–164. [Google Scholar] [CrossRef]
- Oh, S.; Stish, B.J.; Vickers, S.M.; Buchsbaum, D.J.; Saluja, A.K.; Vallera, D.A. A new drug delivery method of bispecific ligand-directed toxins, which reduces toxicity and promotes efficacy in a model of orthotopic pancreatic cancer. Pancreas 2010, 39, 913–922. [Google Scholar] [CrossRef]
- Stish, B.J.; Chen, H.; Shu, Y.; Panoskaltsis-Mortari, A.; Vallera, D.A. A bispecific recombinant cytotoxin (DTEGF13) targeting human interleukin-13 and epidermal growth factor receptors in a mouse xenograft model of prostate cancer. Clin. Cancer Res. 2007, 13, 6486–6493. [Google Scholar] [CrossRef]
- McElroy, C.A.; Dohm, J.A.; Walsh, S.T. Structural and biophysical studies of the human IL-7/IL-7Ralpha complex. Structure 2009, 17, 54–65. [Google Scholar] [CrossRef]
- Barata, J.T.; Durum, S.K.; Seddon, B. Flip the coin: IL-7 and IL-7R in health and disease. Nat. Immunol. 2019, 20, 1584–1593. [Google Scholar] [CrossRef]
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K.; et al. A fully human anti-IL-7Ralpha antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 2155–2168. [Google Scholar] [CrossRef]
- Oliveira, M.L.; Akkapeddi, P.; Ribeiro, D.; Melao, A.; Barata, J.T. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia: An update. Adv. Biol. Regul. 2019, 71, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Long, B.W.; Witte, P.L.; Abraham, G.N.; Gregory, S.A.; Plate, J.M. Apoptosis and interleukin 7 gene expression in chronic B-lymphocytic leukemia cells. Proc. Natl. Acad. Sci. USA 1995, 92, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and specificity: Insights from the interleukin 6 family of cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef]
- Burger, R. Impact of interleukin-6 in hematological malignancies. Transfus. Med. Hemother. 2013, 40, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, J.; Muller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lucke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Klostermann, S.; Eggle, D.; Kruger, A. Interleukin 6/interleukin 6 receptor interaction and its role as a therapeutic target for treatment of cachexia and cancer. Cancer Genom. Proteom. 2010, 7, 287–302. [Google Scholar]
- Taher, M.Y.; Davies, D.M.; Maher, J. The role of the interleukin (IL)-6/IL-6 receptor axis in cancer. Biochem. Soc. Trans. 2018, 46, 1449–1462. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Purification and characterization of IL6-PE4E, a recombinant fusion of interleukin 6 with Pseudomonas exotoxin. Bioconjug. Chem. 1993, 4, 581–585. [Google Scholar] [CrossRef]
- Boayue, K.B.; Gu, L.; Yeager, A.M.; Kreitman, R.J.; Findley, H.W. Pediatric acute myelogenous leukemia cells express IL-6 receptors and are sensitive to a recombinant IL6-Pseudomonas exotoxin. Leukemia 1998, 12, 182–191. [Google Scholar] [CrossRef][Green Version]
- Gu, L.; Zhou, M.; Jurickova, I.; Yeager, A.M.; Kreitman, R.J.; Phillips, C.N.; Findley, H.W. Expression of interleukin-6 receptors by pediatric acute lymphoblastic leukemia cells with the t(4;11) translocation: A possible target for therapy with recombinant IL6-Pseudomonas exotoxin. Leukemia 1997, 11, 1779–1786. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, D.J.; Han, J.S.; Li, Y.S.; Liu, Z.S.; Lu, S.Y.; Ren, H.L. In vitro and in vivo antitumor effects of the recombinant immunotoxin IL6(T23)-PE38KDEL in multiple myeloma. Oncol. Lett. 2012, 4, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Grinberg, A.; Farber, J.M.; Love, P.E. A role for CCR9 in T lymphocyte development and migration. J. Immunol. 2002, 168, 2811–2819. [Google Scholar] [CrossRef]
- King, J.; Mir, H.; Singh, S. Association of cytokines and chemokines in pathogenesis of breast cancer. Prog. Mol. Biol. Transl. Sci. 2017, 151, 113–136. [Google Scholar] [CrossRef]
- Tu, Z.; Xiao, R.; Xiong, J.; Tembo, K.M.; Deng, X.; Xiong, M.; Liu, P.; Wang, M.; Zhang, Q. CCR9 in cancer: Oncogenic role and therapeutic targeting. J. Hematol. Oncol. 2016, 9, 10. [Google Scholar] [CrossRef]
- Mao, W.; Luis, E.; Ross, S.; Silva, J.; Tan, C.; Crowley, C.; Chui, C.; Franz, G.; Senter, P.; Koeppen, H.; et al. EphB2 as a therapeutic antibody drug target for the treatment of colorectal cancer. Cancer Res. 2004, 64, 781–788. [Google Scholar] [CrossRef][Green Version]
- Sun, X.L.; Xu, Z.M.; Ke, Y.Q.; Hu, C.C.; Wang, S.Y.; Ling, G.Q.; Yan, Z.J.; Liu, Y.J.; Song, Z.H.; Jiang, X.D.; et al. Molecular targeting of malignant glioma cells with an EphA2-specific immunotoxin delivered by human bone marrow-derived mesenchymal stem cells. Cancer Lett. 2011, 312, 168–177. [Google Scholar] [CrossRef]
- Ferluga, S.; Tome, C.M.; Herpai, D.M.; D’Agostino, R.; Debinski, W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget 2016, 7, 59860–59876. [Google Scholar] [CrossRef]
- Rezaie, E.; Amani, J.; Bidmeshki Pour, A.; Mahmoodzadeh Hosseini, H. A new scfv-based recombinant immunotoxin against EPHA2-overexpressing breast cancer cells; High in vitro anti-cancer potency. Eur. J. Pharmacol. 2020, 870, 172912. [Google Scholar] [CrossRef]
- Murphy, J.R.; Bishai, W.; Borowski, M.; Miyanohara, A.; Boyd, J.; Nagle, S. Genetic construction, expression, and melanoma-selective cytotoxicity of a diphtheria toxin-related alpha-melanocyte-stimulating hormone fusion protein. Proc. Natl. Acad. Sci. USA 1986, 83, 8258–8262. [Google Scholar] [CrossRef] [PubMed]
- Tatro, J.B.; Wen, Z.; Entwistle, M.L.; Atkins, M.B.; Smith, T.J.; Reichlin, S.; Murphy, J.R. Interaction of an alpha-melanocyte-stimulating hormone-diphtheria toxin fusion protein with melanotropin receptors in human melanoma metastases. Cancer Res. 1992, 52, 2545–2548. [Google Scholar] [PubMed]
- Hui, Q.; Ma, J.; Song, J.; Liu, Z.; Ren, H.; Jiang, W.; Wang, Y.; Xu, Y.; Guo, D.; Zhang, X.; et al. In vitro and in vivo studies of antitumor effects of the recombinant immunotoxin MSH-PE38KDEL on melanoma. Neoplasma 2014, 61, 392–400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kreitman, R.J. Hairy cell leukemia: Present and future directions. Leuk. Lymphoma. 2019, 60, 2869–2879. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Development of recombinant immunotoxins for hairy cell leukemia. Biomolecules 2020, 10, 1140. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; King, E.M.; Pastan, I. Strategies to reduce the immunogenicity of recombinant immunotoxins. Am. J. Pathol. 2018, 188, 1736–1743. [Google Scholar] [CrossRef]
- Antignani, A.; Mathews Griner, L.; Guha, R.; Simon, N.; Pasetto, M.; Keller, J.; Huang, M.; Angelus, E.; Pastan, I.; Ferrer, M.; et al. Chemical screens identify drugs that enhance or mitigate cellular responses to antibody-toxin fusion proteins. PLoS ONE 2016, 11, e0161415. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antignani, A.; Ho, E.C.H.; Bilotta, M.T.; Qiu, R.; Sarnvosky, R.; FitzGerald, D.J. Targeting Receptors on Cancer Cells with Protein Toxins. Biomolecules 2020, 10, 1331. https://doi.org/10.3390/biom10091331
Antignani A, Ho ECH, Bilotta MT, Qiu R, Sarnvosky R, FitzGerald DJ. Targeting Receptors on Cancer Cells with Protein Toxins. Biomolecules. 2020; 10(9):1331. https://doi.org/10.3390/biom10091331
Chicago/Turabian StyleAntignani, Antonella, Eric Chun Hei Ho, Maria Teresa Bilotta, Rong Qiu, Robert Sarnvosky, and David J. FitzGerald. 2020. "Targeting Receptors on Cancer Cells with Protein Toxins" Biomolecules 10, no. 9: 1331. https://doi.org/10.3390/biom10091331
APA StyleAntignani, A., Ho, E. C. H., Bilotta, M. T., Qiu, R., Sarnvosky, R., & FitzGerald, D. J. (2020). Targeting Receptors on Cancer Cells with Protein Toxins. Biomolecules, 10(9), 1331. https://doi.org/10.3390/biom10091331