Control of Transcription Initiation by Biased Thermal Fluctuations on Repetitive Genomic Sequences
 ,
,  , , and
, , and
Abstract
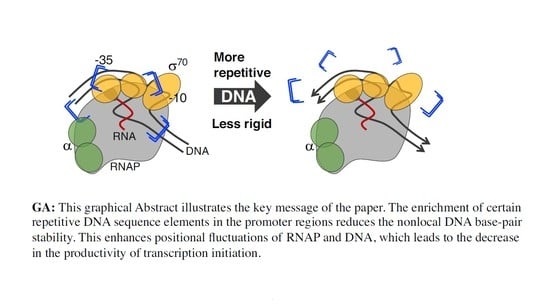
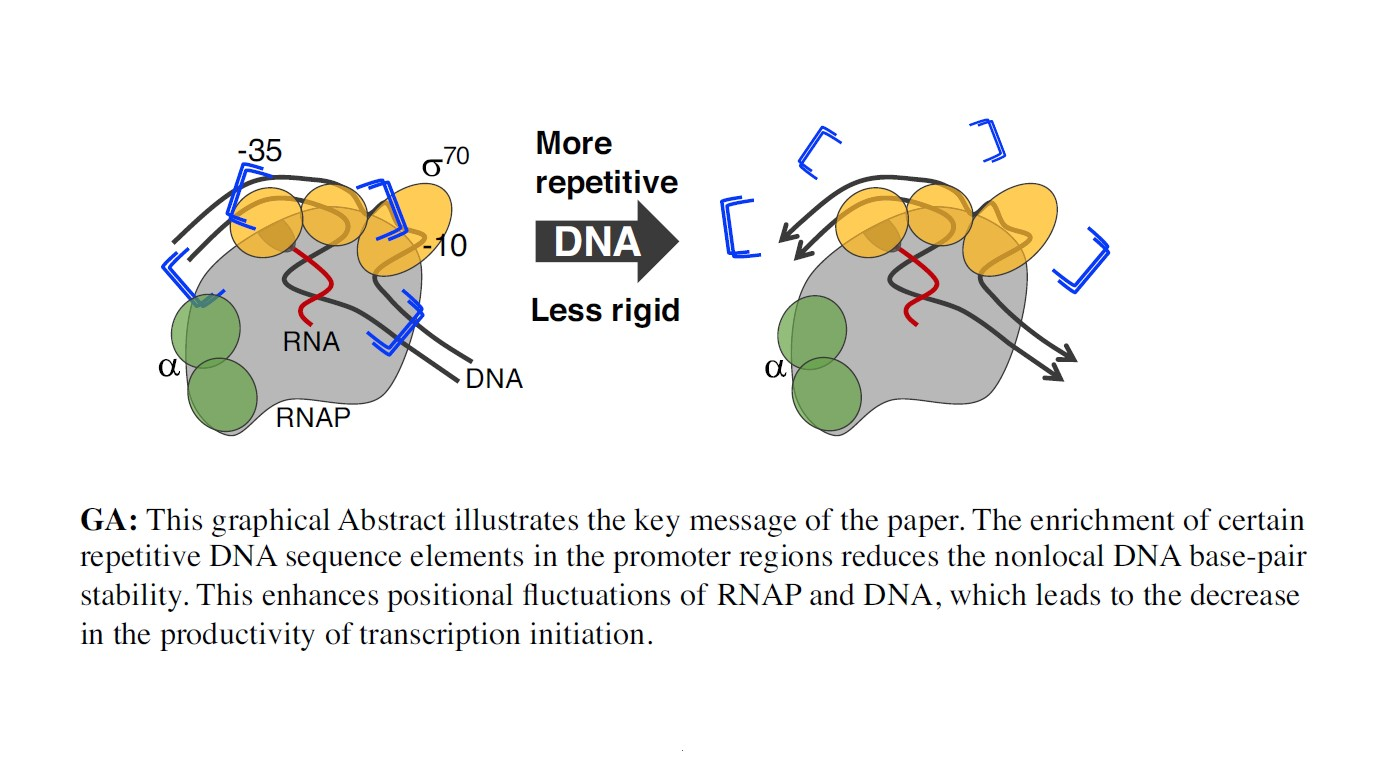{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
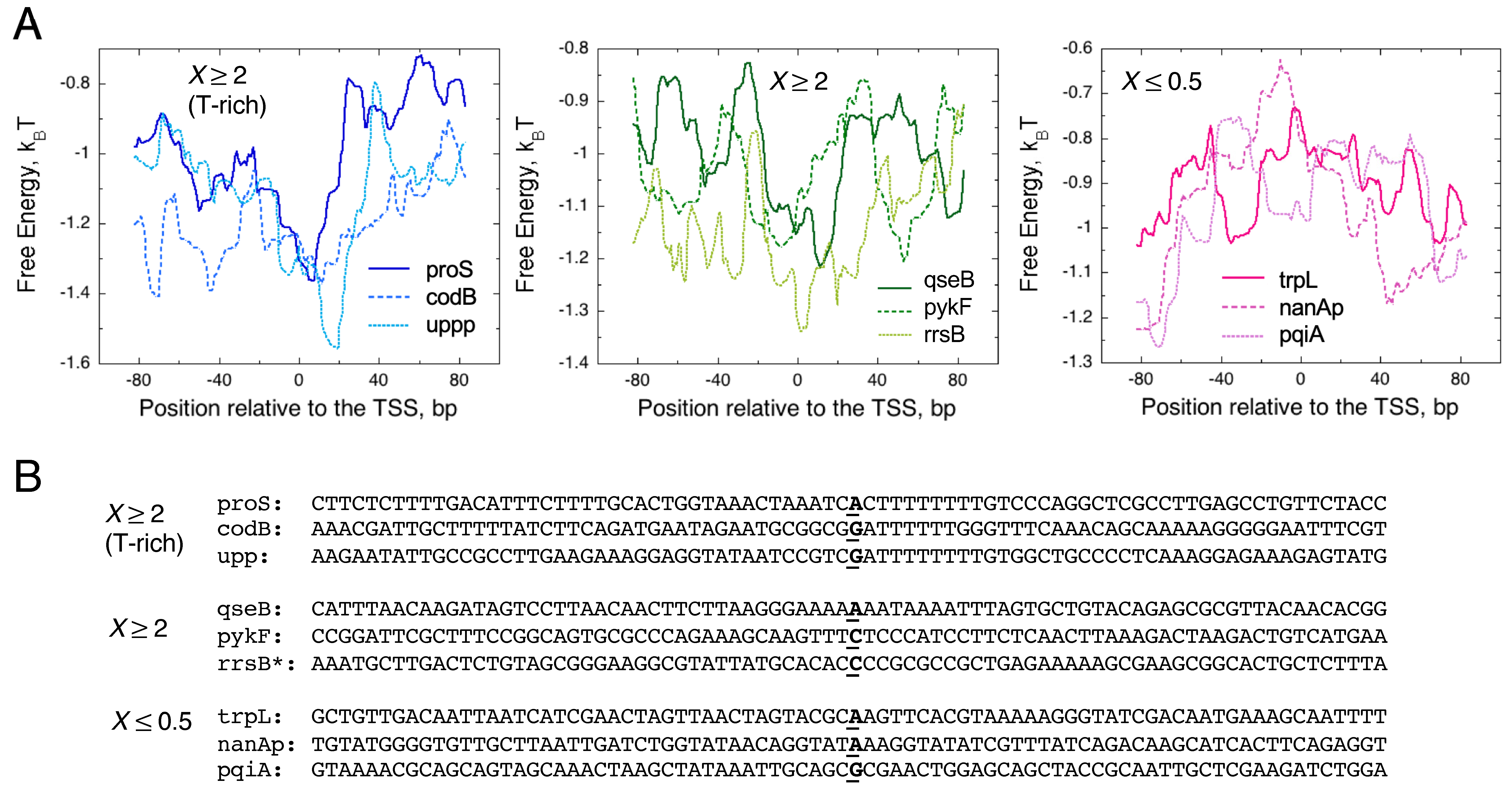{kind=link}
{kind=link}
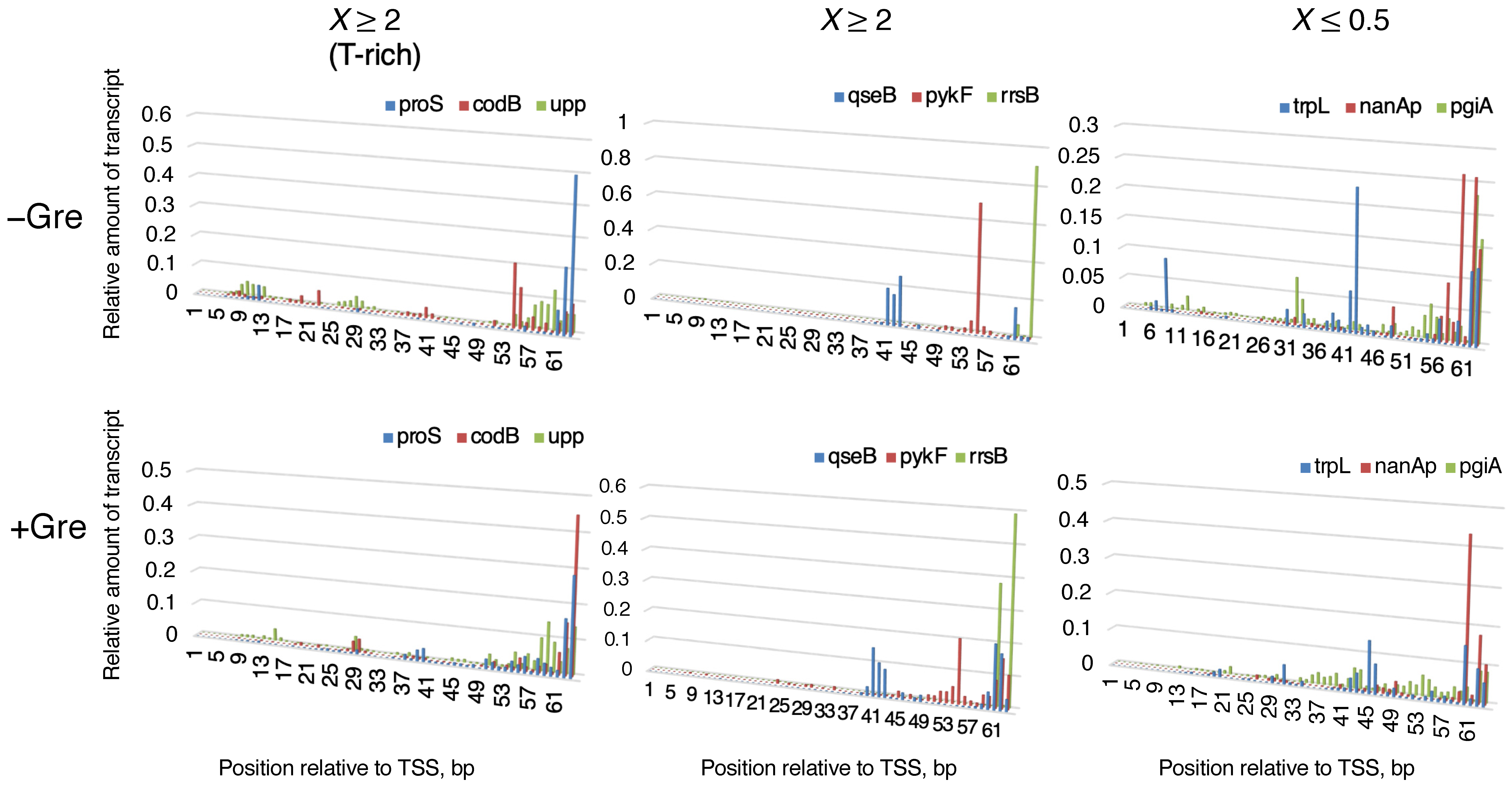
{kind=link}
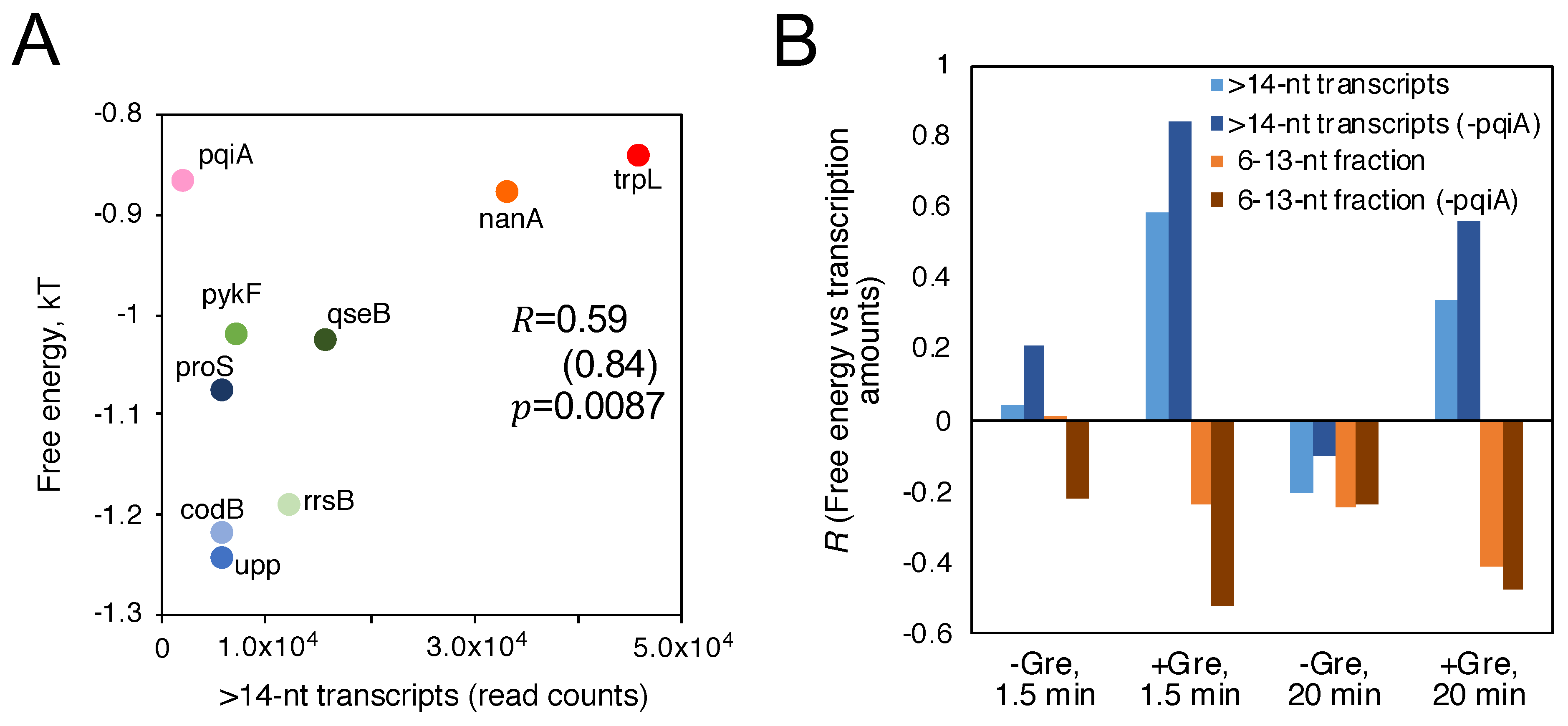
{kind=link}
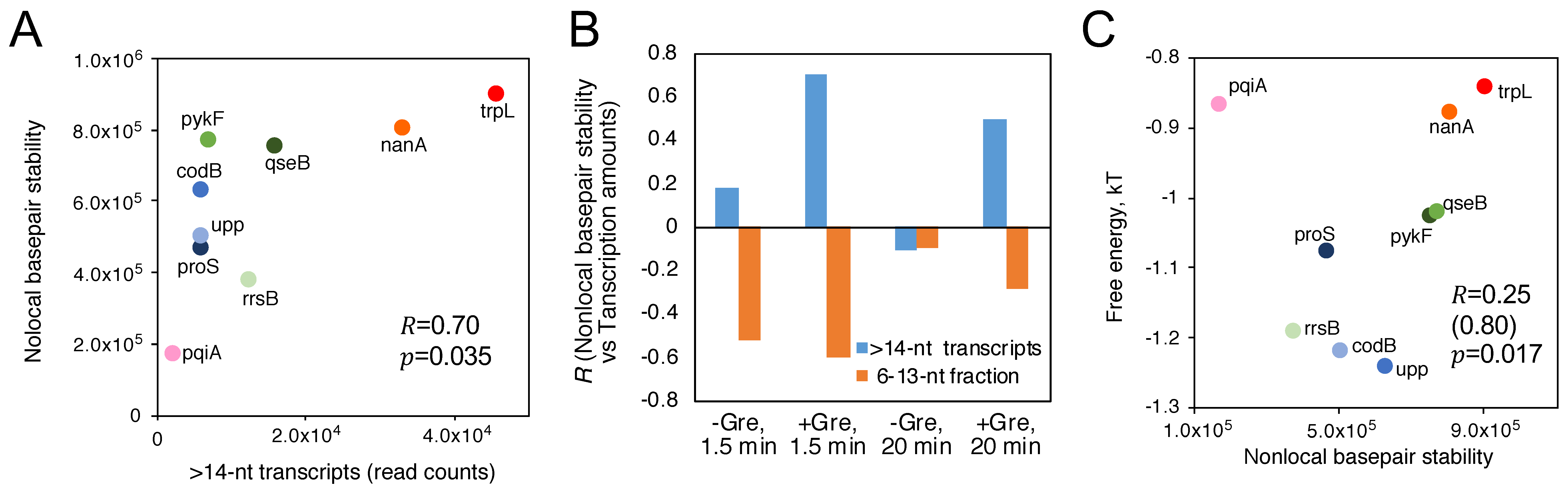{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
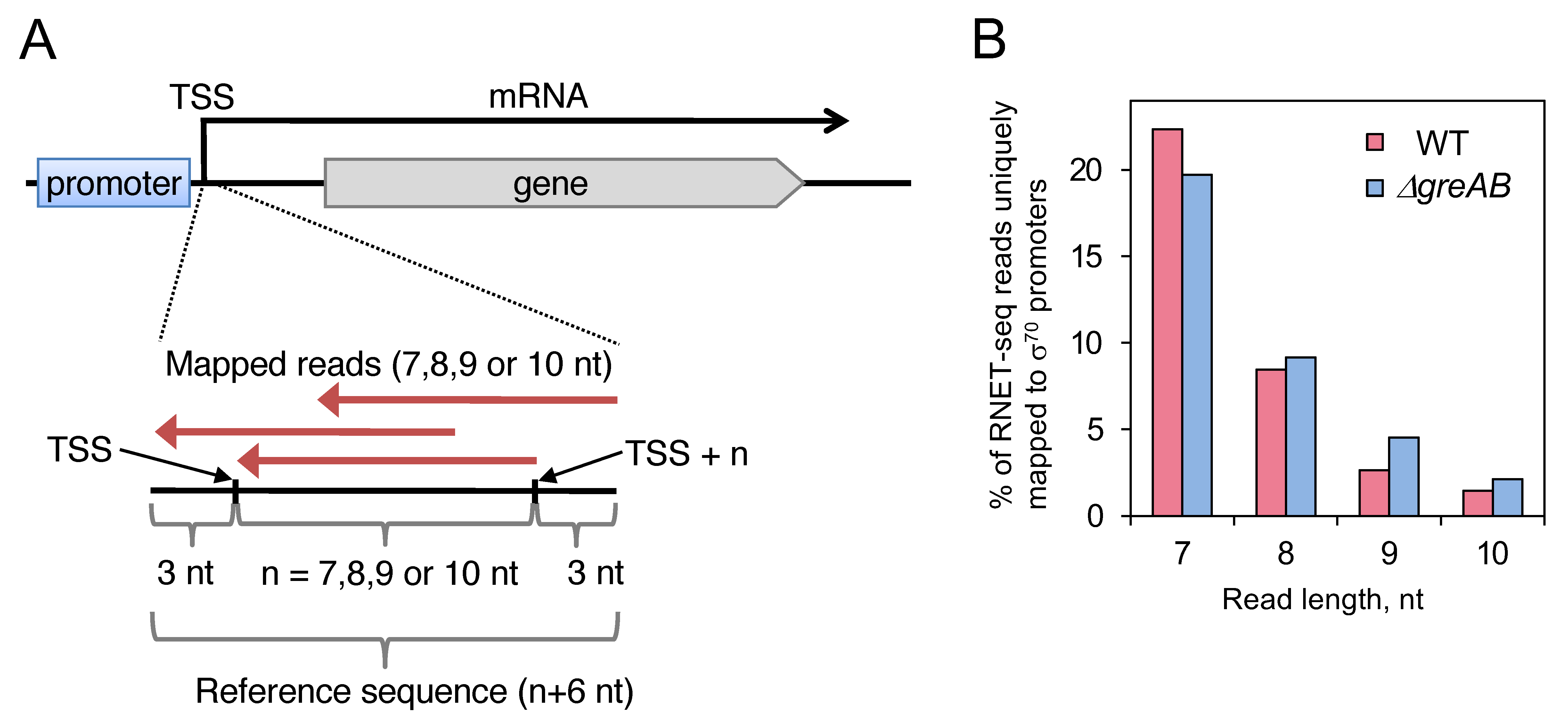
2.1. Mapping of Short Read Sequences to TSS
2.2. Calculation of FEINC
2.3. Calculation of the P-Value
2.4. In Vitro Transcription
2.5. Solution NMR
3. Results
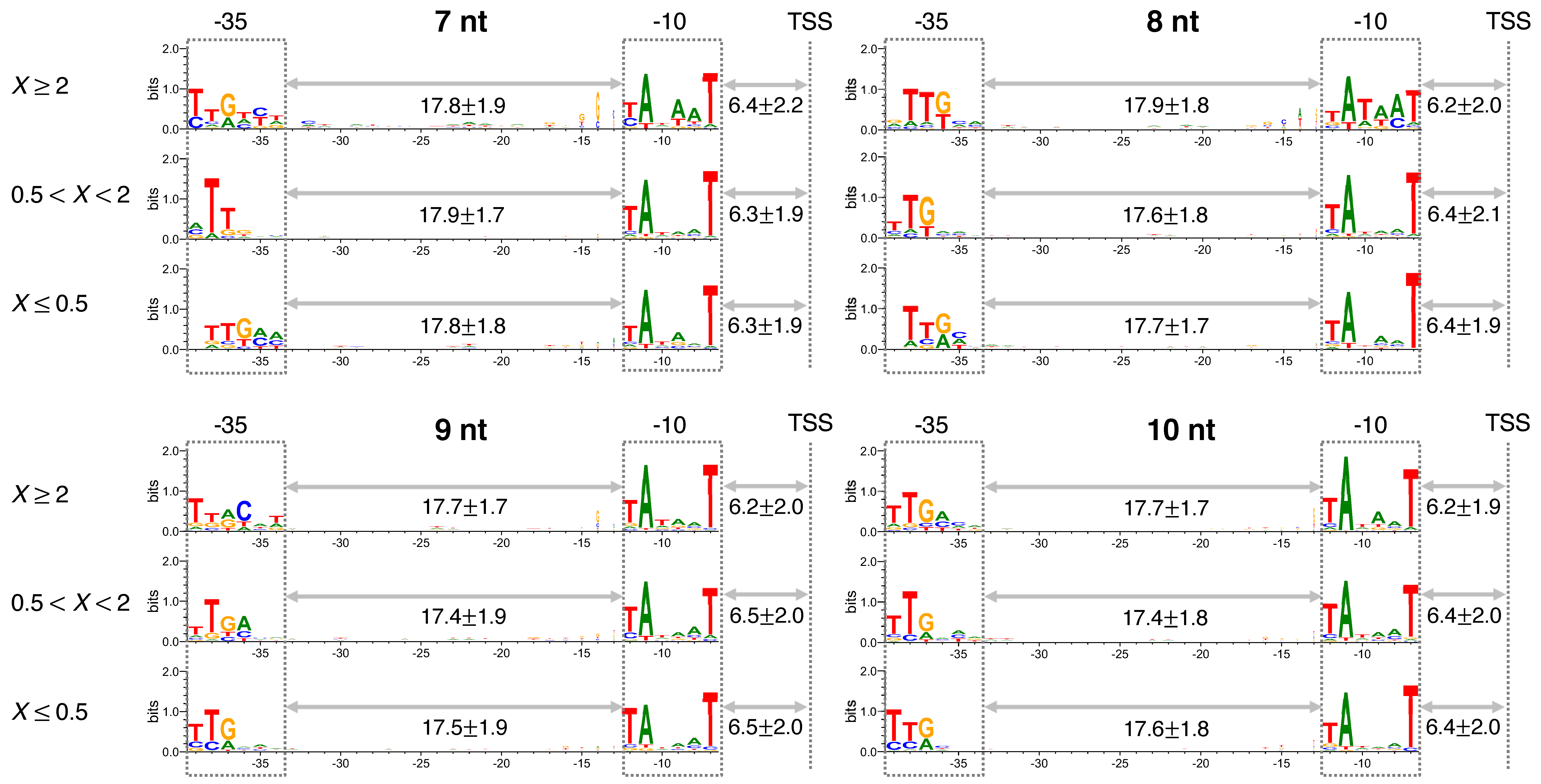
3.1. The Significance of a Consensus Mode of RNAP-DNA Binding
3.2. The Significance of a Nonconsensus Mode of RNAP-DNA (RNA-DNA Hybrid) Binding
3.3. Sequence and Nonlocal-Base pair-Stability-Related Contribution to Transcription Productivity
4. Discussion
FEINC Predicts Conformational Heterogeneity Of Moribund Complexes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saecker, R.M.; Record, M.T.; Dehaseth, P.L. Mechanism of bacterial transcription initiation: RNA polymerase—Promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011, 412, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.F.; Busby, S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004, 2, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Court, D. Regulatory sequences involved in the promotion and termination of RNA transcription. Annu. Rev. Genet. 1979, 13, 319–353. [Google Scholar] [CrossRef] [PubMed]
- Einav, T.; Phillips, R. How the avidity of polymerase binding to the -35/-10 promoter sites affects gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 13340–13345. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Kapanidis, A.N. Recent Advances in Understanding σ70-Dependent Transcription Initiation Mechanisms. J. Mol. Biol. 2019, 431, 3947–3959. [Google Scholar] [CrossRef]
- Johnston, D.E.; McClure, W.R. Abortive Initiation of in Vitro RNA Synthesis on Bacteriophage DNA; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1976; pp. 413–428. [Google Scholar]
- Carpousis, A.J.; Gralla, J.D. Cycling of ribonucleic acid polymerase to produce oligonucleotides during initiation in vitro at the lac UV5 promoter. Biochemistry 1980, 19, 3245–3253. [Google Scholar] [CrossRef] [PubMed]
- Kubori, T.; Shimamoto, N. A branched pathway in the early stage of transcription by Escherichia coli RNA polymerase. J. Mol. Biol. 1996, 256, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Nagai, H.; Shimamoto, N. Polymerase arrest at the lambdaP(R) promoter during transcription initiation. J. Biol. Chem. 2000, 275, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.; Chung, S.; Allen, B.L.; Wang, S.; Lee, J.; Lu, S.W.; Grimaud, L.W.; Ingargiola, A.; Michalet, X.; Alhadid, Y.; et al. Backtracked and paused transcription initiation intermediate of Escherichia coli RNA polymerase. Proc. Natl. Acad. Sci. USA 2016, 113, E6562–E6571. [Google Scholar] [CrossRef]
- Duchi, D.; Bauer, D.L.; Fernandez, L.; Evans, G.; Robb, N.; Hwang, L.C.; Gryte, K.; Tomescu, A.; Zawadzki, P.; Morichaud, Z.; et al. RNA Polymerase Pausing during Initial Transcription. Mol. Cell 2016, 63, 939–950. [Google Scholar] [CrossRef]
- Dulin, D.; Bauer, D.L.V.; Malinen, A.M.; Bakermans, J.J.W.; Kaller, M.; Morichaud, Z.; Petushkov, I.; Depken, M.; Brodolin, K.; Kulbachinskiy, A.; et al. Pausing controls branching between productive and non-productive pathways during initial transcription in bacteria. Nat. Commun. 2018, 9, 1478. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Martin, C.T. Insights into the mechanism of initial transcription in Escherichia coli RNA polymerase. J. Biol. Chem. 2013, 288, 31993–32003. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, N. Nanobiology of RNA polymerase: Biological consequence of inhomogeneity in reactant. Chem. Rev. 2013, 113, 8400–8422. [Google Scholar] [CrossRef] [PubMed]
- Susa, M.; Kubori, T.; Shimamoto, N. A pathway branching in transcription initiation in Escherichia coli. Mol. Microbiol. 2006, 59, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Henderson, K.L.; Felth, L.C.; Molzahn, C.M.; Shkel, I.; Wang, S.; Chhabra, M.; Ruff, E.F.; Bieter, L.; Kraft, J.E.; Record, M.T. Mechanism of transcription initiation and promoter escape by. Proc. Natl. Acad. Sci. USA 2017, 114, E3032–E3040. [Google Scholar] [CrossRef]
- Imashimizu, M.; Tanaka, K.; Shimamoto, N. Comparative Study of Cyanobacterial and E. coli RNA Polymerases: Misincorporation, Abortive Transcription, and Dependence on Divalent Cations. Genet. Res. Int. 2011, 2011, 572689. [Google Scholar] [CrossRef]
- Sen, R.; Nagai, H.; Shimamoto, N. Conformational switching of Escherichia coli RNA polymerase-promoter binary complex is facilitated by elongation factor GreA and GreB. Genes Cells 2001, 6, 389–401. [Google Scholar] [CrossRef]
- Borukhov, S.; Sagitov, V.; Goldfarb, A. Transcript cleavage factors from E. coli. Cell 1993, 72, 459–466. [Google Scholar] [CrossRef]
- Skancke, J.; Bar, N.; Kuiper, M.; Hsu, L.M. Sequence-Dependent Promoter Escape Efficiency Is Strongly Influenced by Bias for the Pretranslocated State during Initial Transcription. Biochemistry 2015, 54, 4267–4275. [Google Scholar] [CrossRef]
- Heyduk, E.; Heyduk, T. DNA template sequence control of bacterial RNA polymerase escape from the promoter. Nucleic Acids Res. 2018, 46, 4469–4486. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Turnbough, C.L. Transcription start site sequence and spacing between the -10 region and the start site affect reiterative transcription-mediated regulation of gene expression in Escherichia coli. J. Bacteriol. 2014, 196, 2912–2920. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lerner, E.; Ingargiola, A.; Lee, J.J.; Borukhov, S.; Michalet, X.; Weiss, S. Different types of pausing modes during transcription initiation. Transcription 2017, 8, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Winkelman, J.T.; Pukhrambam, C.; Vvedenskaya, I.O.; Zhang, Y.; Taylor, D.M.; Shah, P.; Ebright, R.H.; Nickels, B.E. XACT-Seq Comprehensively Defines the Promoter-Position and Promoter-Sequence Determinants for Initial-Transcription Pausing. Mol. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Imashimizu, M.; Takahashi, H.; Oshima, T.; McIntosh, C.; Bubunenko, M.; Court, D.L.; Kashlev, M. Visualizing translocation dynamics and nascent transcript errors in paused RNA polymerases in vivo. Genome Biol. 2015, 16, 98. [Google Scholar] [CrossRef]
- Imashimizu, M.; Afek, A.; Takahashi, H.; Lubkowska, L.; Lukatsky, D.B. Control of transcriptional pausing by biased thermal fluctuations on repetitive genomic sequences. Proc. Natl. Acad. Sci. USA 2016, 113, E7409–E7417. [Google Scholar] [CrossRef]
- Santos-Zavaleta, A.; Salgado, H.; Gama-Castro, S.; Sánchez-Pérez, M.; Gómez-Romero, L.; Ledezma-Tejeida, D.; García-Sotelo, J.S.; Alquicira-Hernández, K.; Muñiz-Rascado, L.J.; Peña-Loredo, P.; et al. RegulonDB v 10.5: Tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res. 2019, 47, D212–D220. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Afek, A.; Lukatsky, D.B. Positive and negative design for nonconsensus protein-DNA binding affinity in the vicinity of functional binding sites. Biophys. J. 2013, 105, 1653–1660. [Google Scholar] [CrossRef]
- Imashimizu, M.; Kireeva, M.L.; Lubkowska, L.; Kashlev, M.; Shimamoto, N. The Role of Pyrophosphorolysis in the Initiation-to-Elongation Transition by E. coli RNA Polymerase. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Komissarova, N.; Kashlev, M. Functional topography of nascent RNA in elongation intermediates of RNA polymerase. Proc. Natl. Acad. Sci. USA 1998, 95, 14699–14704. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, E.; Lee, J.; Ozerova, M.; Semenova, E.; Datsenko, K.; Wanner, B.L.; Severinov, K.; Borukhov, S. Analysis of promoter targets for Escherichia coli transcription elongation factor GreA in vivo and in vitro. J. Bacteriol. 2007, 189, 8772–8785. [Google Scholar] [CrossRef]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
- Schurr, J.M. The one-dimensional diffusion coefficient of proteins absorbed on DNA. Hydrodynamic considerations. Biophys. Chem. 1979, 9, 413–414. [Google Scholar] [CrossRef]
- Sela, I.; Lukatsky, D.B. DNA sequence correlations shape nonspecific transcription factor-DNA binding affinity. Biophys. J. 2011, 101, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Afek, A.; Schipper, J.L.; Horton, J.; Gordân, R.; Lukatsky, D.B. Protein-DNA binding in the absence of specific base-pair recognition. Proc. Natl. Acad. Sci. USA 2014, 111, 17140–17145. [Google Scholar] [CrossRef]
- Imashimizu, M.; Lukatsky, D.B. Transcription pausing: Biological significance of thermal fluctuations biased by repetitive genomic sequences. Transcription 2018, 9, 196–203. [Google Scholar] [CrossRef]
- Imashimizu, M.; Kireeva, M.L.; Lubkowska, L.; Gotte, D.; Parks, A.R.; Strathern, J.N.; Kashlev, M. Intrinsic Translocation Barrier as an Initial Step in Pausing by RNA Polymerase II. J. Mol. Biol. 2013, 425, 697–712. [Google Scholar] [CrossRef]
- Imashimizu, M.; Oshima, T.; Lubkowska, L.; Kashlev, M. Direct assessment of transcription fidelity by high-resolution RNA sequencing. Nucleic Acids Res. 2013, 41, 9090–9104. [Google Scholar] [CrossRef]
- Komissarova, N.; Becker, J.; Solter, S.; Kireeva, M.; Kashlev, M. Shortening of RNA:DNA hybrid in the elongation complex of RNA polymerase is a prerequisite for transcription termination. Mol. Cell 2002, 10, 1151–1162. [Google Scholar] [CrossRef]
- Vahed, M.; Ishihara, J.I.; Takahashi, H. DIpartite: A tool for detecting bipartite motifs by considering base interdependencies. PLoS ONE 2019, 14, e0220207. [Google Scholar] [CrossRef] [PubMed]
- Hook-Barnard, I.G.; Hinton, D.M. The promoter spacer influences transcription initiation via sigma70 region 1.1 of Escherichia coli RNA polymerase. Proc. Natl. Acad. Sci. USA 2009, 106, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Takanami, M.; Ohtsuka, E.; Taniyama, Y.; Marumoto, R.; Sato, H.; Ikehara, M. Essential structure of E. coli promoter: Effect of spacer length between the two consensus sequences on promoter function. Nucleic Acids Res. 1983, 11, 5855–5864. [Google Scholar] [CrossRef] [PubMed]
- Feklistov, A.; Darst, S.A. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase σ subunit. Cell 2011, 147, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Yamazaki, Y.; Tanaka, K.; Ishihama, A. The whole set of constitutive promoters recognized by RNA polymerase RpoD holoenzyme of Escherichia coli. PLoS ONE 2014, 9, e90447. [Google Scholar] [CrossRef]
- Huang, Y.; Weng, X.; Russu, I.M. Structural energetics of the adenine tract from an intrinsic transcription terminator. J. Mol. Biol. 2010, 397, 677–688. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gueron, M.; Kochoyan, M.; Leroy, J.L. A single mode of DNA base-pair opening drives imino proton exchange. Nature 1987, 328, 89–92. [Google Scholar] [CrossRef]
- Kapanidis, A.N.; Margeat, E.; Ho, S.O.; Kortkhonjia, E.; Weiss, S.; Ebright, R.H. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science 2006, 314, 1144–1147. [Google Scholar] [CrossRef]
- Winkelman, J.T.; Chandrangsu, P.; Ross, W.; Gourse, R.L. Open complex scrunching before nucleotide addition accounts for the unusual transcription start site of E. coli ribosomal RNA promoters. Proc. Natl. Acad. Sci. USA 2016, 113, E1787–E1795. [Google Scholar] [CrossRef]
- Imashimizu, M.; Shimamoto, N.; Oshima, T.; Kashlev, M. Transcription elongation. Heterogeneous tracking of RNA polymerase and its biological implications. Transcription 2014, 5, e28285. [Google Scholar] [CrossRef]
- Ruff, E.F.; Record, M.T.; Artsimovitch, I. Initial events in bacterial transcription initiation. Biomolecules 2015, 5, 1035–1062. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Steitz, T.A. Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol. Cell 2015, 58, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Bellissent-Funel, M.C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef] [PubMed]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imashimizu, M.; Tokunaga, Y.; Afek, A.; Takahashi, H.; Shimamoto, N.; Lukatsky, D.B. Control of Transcription Initiation by Biased Thermal Fluctuations on Repetitive Genomic Sequences. Biomolecules 2020, 10, 1299. https://doi.org/10.3390/biom10091299
Imashimizu M, Tokunaga Y, Afek A, Takahashi H, Shimamoto N, Lukatsky DB. Control of Transcription Initiation by Biased Thermal Fluctuations on Repetitive Genomic Sequences. Biomolecules. 2020; 10(9):1299. https://doi.org/10.3390/biom10091299
Chicago/Turabian StyleImashimizu, Masahiko, Yuji Tokunaga, Ariel Afek, Hiroki Takahashi, Nobuo Shimamoto, and David B. Lukatsky. 2020. "Control of Transcription Initiation by Biased Thermal Fluctuations on Repetitive Genomic Sequences" Biomolecules 10, no. 9: 1299. https://doi.org/10.3390/biom10091299
APA StyleImashimizu, M., Tokunaga, Y., Afek, A., Takahashi, H., Shimamoto, N., & Lukatsky, D. B. (2020). Control of Transcription Initiation by Biased Thermal Fluctuations on Repetitive Genomic Sequences. Biomolecules, 10(9), 1299. https://doi.org/10.3390/biom10091299