Nucleotide Loading Modes of Human RNA Polymerase II as Deciphered by Molecular Simulations


Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. TFIIF Reconfiguration of the Transcription Bubble
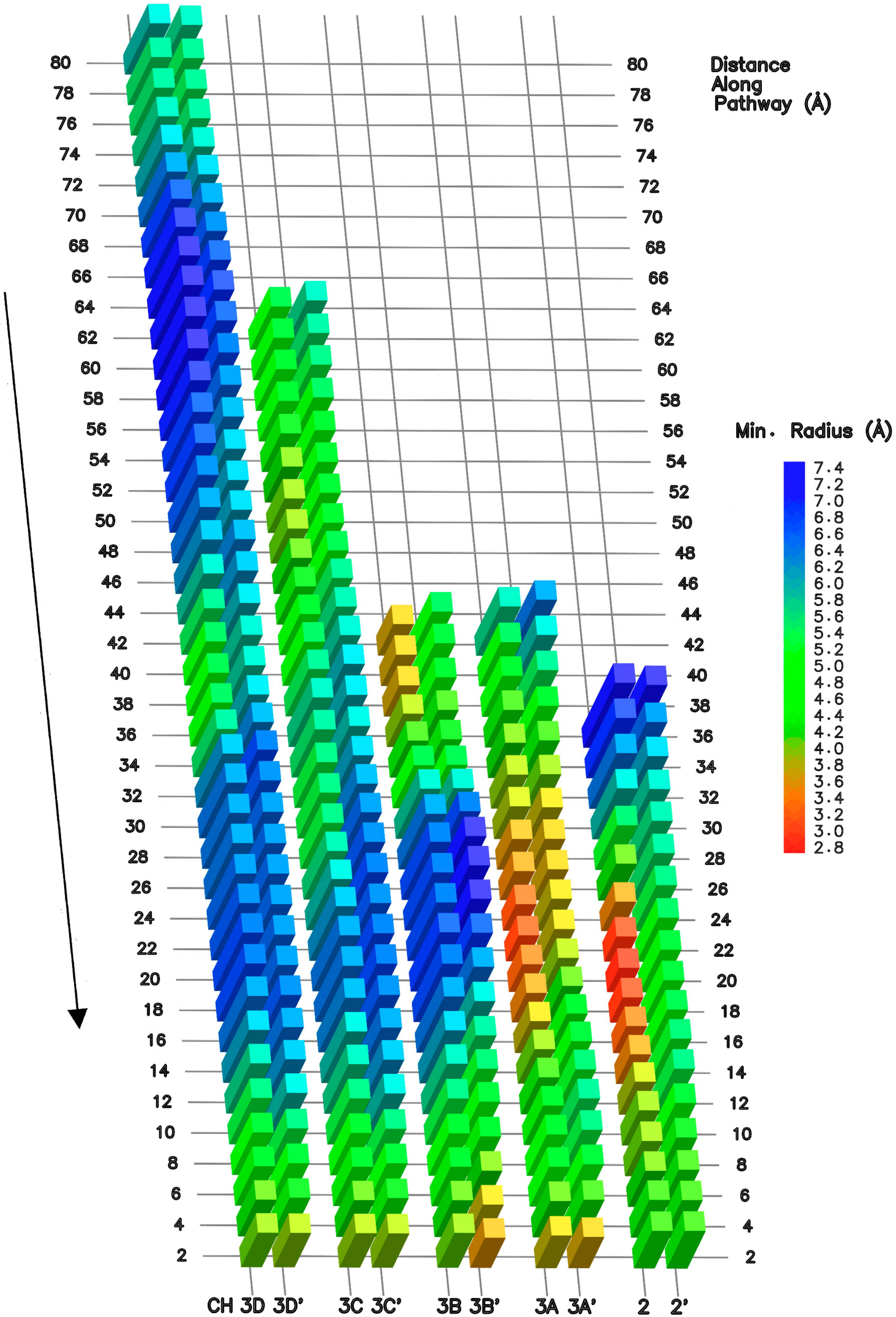
3.2. NTP Accessible Pathways
3.3. NTP Contacting Macro-Regions
3.4. NTP Loading Through CH2
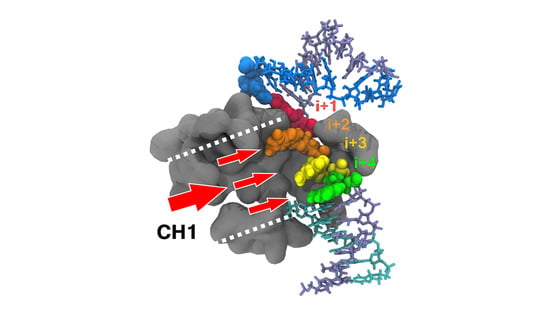
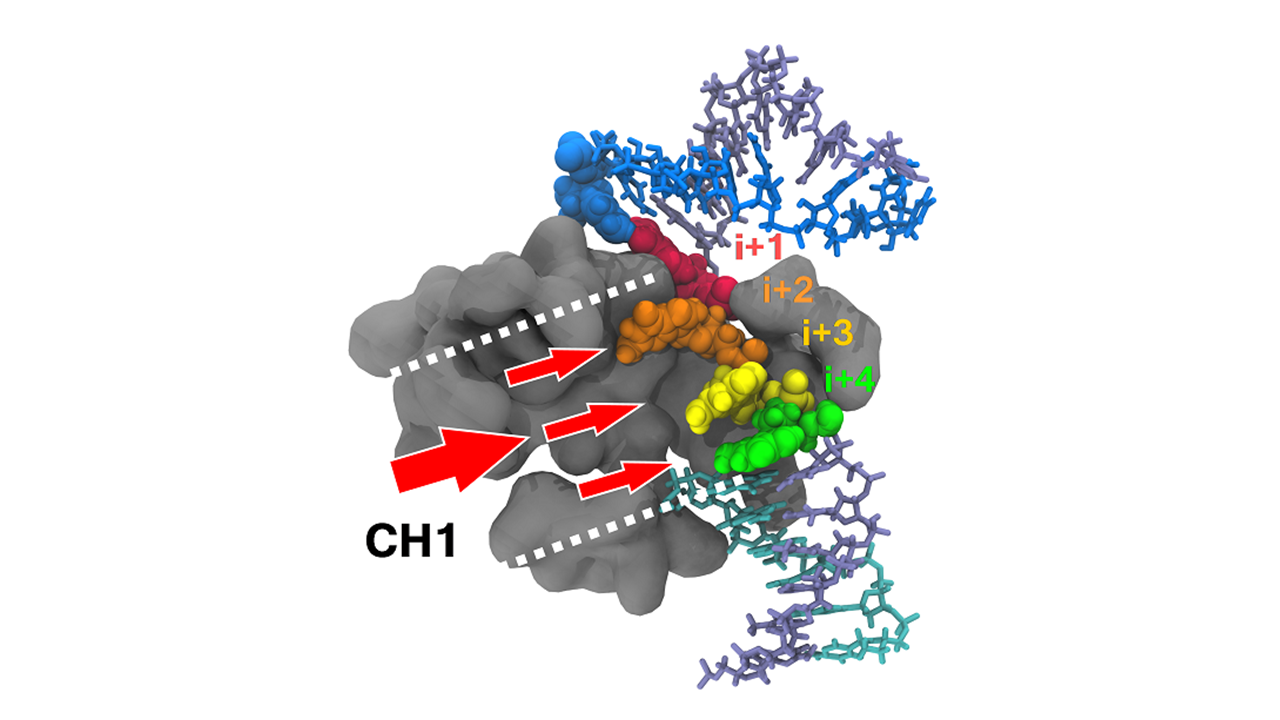
3.5. NTP Loading Through CH1
3.6. i + 2 NTP Isomerization, Downstream Scanning, and Transfer to the Active Site
3.7. Synergistic Coupling of Downstream Binding and Catalytic Isomerization
4. Discussion
- NTP selection time window. When NTPs were loaded via CH1, the enzyme performed two tasks at the same time. It selected the next nucleotide(s), up to the i + 4 register (Figure 7C,D), while incorporating the current NTP. This could be especially advantageous kinetically when NTP concentrations are low or stoichiometrically imbalanced.
- Coupling. In the context of several successful downstream NTP bindings, the induced refolding of the βD loop and stretching of the template DNA strongly isomerized the active site (Figure 8), thereby, potentially accelerating the incorporation of i + 1 NTP. As such, a coupling between catalysis and subsequent substrate availability could occur.
- Transition. Transition between nucleotide addition rounds is likely to be stimulated in four ways. First, during induced isomerization, with NTPs bound to i + 2/i + 3/i + 4, an enhanced strain was applied onto the i + 1 NTP polyphosphate group (Figure 8E,F), which could result in improved PPi-MgB release. Pyrophosphate exit may not be rate limiting [81], therefore, only a modest stimulation would be expected. Second, the downstream NTP bindings stretched template DNA and shifted non-template DNA away (Figure 8A–C), which could bias translocation to the forward state. Third, the modification of the trigger loop dynamics via the NTP anchoring of TLc (Figure S10) could accelerate trigger loop closing. Fourth, because i + 2 NTP was, or was likely to be transferred to the active site concomitantly to translocation (Figure 7E–I and Figure S9), there is an increased probability that the ratchet was immediately incremented after translocation. However, employing the CH2 mechanism, the right NTP must first successfully be selected after vacation of the active site before translocation oscillation would be adjourned.
- Fidelity. When the next NTP binds to i + 2, first, it was isomerized in CH3P (Figure 7B), then detached from template DNA (Figure 7F), and transferred to the catalytic site (Figure 7I) before undergoing final catalytic confinement. Therefore, there is a double isomerization process at play, utilizing the energy of base pairs twice, and delayed in time. In addition, the re-enforced catalytic confinement occurring when multiple downstream positions were occupied (Figure 8) could reduce the probability of i + 1 misincorporation by reducing the possibility of altered Watson–Crick geometry stabilization in the active site.
- Pause escape. The same mechanism responsible for re-enforced catalytic isomerization on the i + 1 NTP that improves fidelity could also be involved in facilitating pause escape by stimulating realignment of the RNA 3′ end.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Fu, J.; Gnatt, A.L.; Bushnell, D.A.; Jensen, G.J.; Thompson, N.E.; Burgess, R.R.; David, P.R.; Kornberg, R.D. Yeast RNA polymerase II at 5 Å resolution. Cell 1999, 98, 799–810. [Google Scholar] [CrossRef][Green Version]
- Zhang, G.; Campbell, E.A.; Minakhin, L.; Richter, C.; Severinov, K.; Darst, S.A. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell 1999, 98, 811–824. [Google Scholar] [CrossRef]
- Sosunov, V.; Sosunova, E.; Mustaev, A.; Bass, I.; Nikiforov, V.; Goldfarb, A. Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase. EMBO J. 2003, 22, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Westover, K.D.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: Nucleotide selection by rotation in the RNA polymerase II active center. Cell 2004, 119, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bushnell, D.A.; Westover, K.D.; Kaplan, C.D.; Kornberg, R.D. Structural Basis of Transcription: Role of the Trigger Loop in Substrate Specificity and Catalysis. Cell 2006, 127, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Zenkin, N.; Yuzenkova, Y.; Severinov, K. Transcript-assisted transcriptional proofreading. Science 2006, 313, 518–520. [Google Scholar] [CrossRef]
- Zhang, Y.; Degen, D.; Ho, M.X.; Sineva, E.; Ebright, K.Y.; Ebright, Y.W.; Mekler, V.; Vahedian-Movahed, H.; Feng, Y.; Yin, R.; et al. GE23077 binds to the RNA polymerase ‘i’ and ‘i+1′ sites and prevents the binding of initiating nucleotides. eLife 2014, 3, e02450. [Google Scholar] [CrossRef]
- Yu, J.; Da, L.T.; Huang, X. Constructing kinetic models to elucidate structural dynamics of a complete RNA polymerase II elongation cycle. Phys. Biol. 2015, 12, 016004. [Google Scholar] [CrossRef]
- Batada, N.N.; Westover, K.D.; Bushnell, D.A.; Levitt, M.; Kornberg, R.D. Diffusion of nucleoside triphosphates and role of the entry site to the RNA polymerase II active center. Proc. Natl. Acad. Sci. USA 2004, 101, 17361–17364. [Google Scholar] [CrossRef]
- Wang, B.; Sexton, R.E.; Feig, M. Kinetics of nucleotide entry into RNA polymerase active site provides mechanism for efficiency and fidelity. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 482–490. [Google Scholar] [CrossRef]
- Zhang, L.; Silva, D.A.; Pardo-Avila, F.; Wang, D.; Huang, X. Structural Model of RNA Polymerase II Elongation Complex with Complete Transcription Bubble Reveals NTP Entry Routes. PLoS Comput. Biol. 2015, 11, e1004354. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, F.; Cramer, P. Structural basis of transcription inhibition by α-amanitin and implications for RNA polymerase II translocation. Nat. Struct. Mol. Biol. 2008, 15, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Dangkulwanich, M.; Ishibashi, T.; Liu, S.; Kireeva, M.L.; Lubkowska, L.; Kashlev, M.; Bustamante, C.J. Complete dissection of transcription elongation reveals slow translocation of RNA polymerase II in a linear ratchet mechanism. eLife 2013, 2, e00971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pardo-Avila, F.; Unarta, I.C.; Cheung, P.P.H.; Wang, G.; Wang, D.; Huang, X. Elucidation of the Dynamics of Transcription Elongation by RNA Polymerase II using Kinetic Network Models. Acc. Chem. Res. 2016, 49, 687–694. [Google Scholar] [CrossRef]
- Foster, J.E.; Holmes, S.F.; Erie, D.A. Allosteric binding of nucleoside triphosphates to RNA polymerase regulates transcription elongation. Cell 2001, 106, 243–252. [Google Scholar] [CrossRef]
- Holmes, S.F.; Erie, D.A. Downstream DNA sequence effects on transcription elongation: Allosteric binding of nucleoside triphosphates facilitates translocation via a ratchet motion. J. Biol. Chem. 2003, 278, 35597–35608. [Google Scholar] [CrossRef]
- Gong, X.Q.; Zhang, C.; Feig, M.; Burton, Z.F. Dynamic error correction and regulation of downstream bubble opening by human RNA polymerase II. Mol. Cell 2005, 18, 461–470. [Google Scholar] [CrossRef]
- Xiong, Y.; Burton, Z.F. A tunable ratchet driving human RNA polymerase II translocation adjusted by accurately templated nucleoside triphosphates loaded at downstream sites and by elongation factors. J. Biol. Chem. 2007, 282, 36582–36592. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Erie, D.A. Templated nucleoside triphosphate binding to a noncatalytic site on RNA polymerase regulates transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 6079–6084. [Google Scholar] [CrossRef]
- Nedialkov, Y.A.; Gong, X.Q.; Hovde, S.L.; Yamaguchi, Y.; Handa, H.; Geiger, J.H.; Yan, H.; Burton, Z.F. NTP-driven translocation by human RNA polymerase II. J. Biol. Chem. 2003, 278, 18303–18312. [Google Scholar] [CrossRef]
- Zhang, C.; Burton, Z.F. Transcription factors IIF and IIS and nucleoside triphosphate substrates as dynamic probes of the human RNA polymerase II mechanism. J. Mol. Biol. 2004, 342, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zobeck, K.L.; Burton, Z.F. Human RNA Polymerase II Elongation in Slow Motion: Role of the TFIIF RAP74 α1 Helix in Nucleoside Triphosphate-Driven Translocation. Mol. Cell. Biol. 2005, 25, 3583–3595. [Google Scholar] [CrossRef] [PubMed]
- Toulokhonov, I.; Zhang, J.; Palangat, M.; Landick, R. A Central Role of the RNA Polymerase Trigger Loop in Active-Site Rearrangement during Transcriptional Pausing. Mol. Cell 2007, 27, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Hamelberg, D.; De Oliveira, C.A.F.; McCammon, J.A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 155102. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Genet. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.; Botello-Smith, W.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W. Characterization of the TIP4P-Ew water model: Vapor pressure and boiling point. J. Chem. Phys. 2005, 123, 194504. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of α/γ conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Otyepka, M.; Šponer, J.; Mládek, A.; Banáš, P.; Cheatham, T.E.; Jurečka, P. Refinement of the Cornell et al. Nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [Google Scholar] [CrossRef] [PubMed]
- Krepl, M.; Zgarbová, M.; Stadlbauer, P.; Otyepka, M.; Banáš, P.; Koča, J.; Cheatham, T.E.; Jurečka, P.; Šponer, J. Reference simulations of noncanonical nucleic acids with different χ variants of the AMBER FORCE Field: Quadruplex DNA, quadruplex RNA, and Z-DNA. J. Chem. Theory Comput. 2012, 8, 2506–2520. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Luque, F.J.; Šponer, J.; Cheatham, T.E.; Otyepka, M.; Jurečka, P. Toward improved description of DNA backbone: Revisiting epsilon and zeta torsion force field parameters. J. Chem. Theory Comput. 2013, 9, 2339–2354. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E.; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar-Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef]
- Panteva, M.T.; Giambaşu, G.M.; York, D.M. Force Field for Mg2+, Mn2+, Zn2+, and Cd2+ Ions That Have Balanced Interactions with Nucleic Acids. J. Phys. Chem. B 2015, 119, 15460–15470. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Taking into account the ion-induced dipole interaction in the nonbonded model of ions. J. Chem. Theory Comput. 2014, 10, 289–297. [Google Scholar] [CrossRef]
- Li, P.; Song, L.F.; Merz, K.M. Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 2015, 11, 1645–1657. [Google Scholar] [CrossRef]
- Panteva, M.T.; Giambaşu, G.M.; York, D.M. Comparison of structural, thermodynamic, kinetic and mass transport properties of Mg2+ ion models commonly used in biomolecular simulations. J. Comput. Chem. 2015, 36, 970–982. [Google Scholar] [CrossRef]
- Meagher, K.L.; Redman, L.T.; Carlson, H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003, 24, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Demir, Ö.; Amaro, R.E. Elements of nucleotide specificity in the trypanosoma brucei mitochondrial RNA editing enzyme RET2. J. Chem. Inf. Model. 2012, 52, 1308–1318. [Google Scholar] [CrossRef]
- He, Y.; Yan, C.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Nature 2016, 533, 359–365. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, D.; Weiss, D.R.; Bushnell, D.A.; Kornberg, R.D.; Levitt, M. RNA polymerase II trigger loop residues stabilize and position the incoming nucleotide triphosphate in transcription. Proc. Natl. Acad. Sci. USA 2010, 107, 15745–15750. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating molecular dynamic simulation on graphics processing units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef]
- Eastman, P.; Pande, V.S. Constant constraint matrix approximation: A robust, parallelizable constraint method for molecular simulations. J. Chem. Theory Comput. 2010, 6, 434–437. [Google Scholar] [CrossRef]
- Eastman, P.; Pande, V.S. Efficient nonbonded interactions for molecular dynamics on a graphics processing unit. J. Comput. Chem. 2010, 31, 1268–1272. [Google Scholar] [CrossRef]
- Eastman, P.; Pande, V.S. OpenMM: A Hardware-Independent Framework for Molecular Simulations. Comput. Sci. Eng. 2010, 12, 34–39. [Google Scholar] [CrossRef]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.P.; Shukla, D.; et al. OpenMM 4: A reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput. 2013, 9, 461–469. [Google Scholar] [CrossRef]
- Varshney, A.; Brooks, F.P.; Wright, W. V Computing smooth molecular surfaces. IEEE Comput. Graph. Appl. 1994, 14, 19–25. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinforma. 1995, 23, 566–579. [Google Scholar] [CrossRef]
- Stone, J.E. Efficient Library for Parallel Ray Tracing and Animation. Master’s Thesis, Missouri University of Science and Technology, Rolla, MO, USA, 1998. [Google Scholar]
- Markwick, P.R.L.; McCammon, J.A. Studying functional dynamics in bio-molecules using accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2011, 13, 20053–20065. [Google Scholar] [CrossRef]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; McCammon, J.A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Kappel, K.; Miao, Y.; McCammon, J.A. Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 2015, 48, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.G.; Chen, J.S.; Miao, Y.; Jinek, M.; Doudna, J.A.; McCammon, J.A.; Palermo, G. Deciphering Off-Target Effects in CRISPR-Cas9 through Accelerated Molecular Dynamics. ACS Cent. Sci. 2019, 5, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P.; Bushnell, D.A.; Fu, J.; Gnatt, A.L.; Maier-Davis, B.; Thompson, N.E.; Burgess, R.R.; Edwards, A.M.; David, P.R.; Kornberg, R.D. Architecture of RNA polymerase II and implications for the transcription mechanism. Science 2000, 288, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Kettenberger, H.; Armache, K.J.; Cramer, P. Architecture of the RNA polymerase II-TFIIS complex and implications for mRNA cleavage. Cell 2003, 114, 347–357. [Google Scholar] [CrossRef]
- Murakami, K.S.; Shin, Y.; Turnbough, C.L.; Molodtsov, V. X-ray crystal structure of a reiterative transcription complex reveals an atypical RNA extension pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 8211–8216. [Google Scholar] [CrossRef] [PubMed]
- Molodtsov, V.; Sineva, E.; Zhang, L.; Huang, X.; Cashel, M.; Ades, S.E.; Murakami, K.S. Allosteric Effector ppGpp Potentiates the Inhibition of Transcript Initiation by DksA. Mol. Cell 2018, 69, 828.e5–839.e5. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Steitz, T.A. Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol. Cell 2015, 58, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, M.N.; Mogni, M.; Millet, O.; Bell, S.D.; Abrescia, N.G.A. Structural and functional analyses of the interaction of archaeal RNA polymerase with DNA. Nucleic Acids Res. 2012, 40, 9941–9952. [Google Scholar] [CrossRef] [PubMed]
- Miropolskaya, N.; Artsimovitch, I.; Klimašauskas, S.; Nikiforov, V.; Kulbachinskiy, A. Allosteric control of catalysis by the F loop of RNA polymerase. Proc. Natl. Acad. Sci. USA 2009, 106, 18942–18947. [Google Scholar] [CrossRef]
- Miropolskaya, N.; Esyunina, D.; Klimašauskas, S.; Nikiforov, V.; Artsimovitch, I.; Kulbachinskiy, A. Interplay between the trigger loop and the F loop during RNA polymerase catalysis. Nucleic Acids Res. 2014, 42, 544–552. [Google Scholar] [CrossRef]
- Liu, X.; Farnung, L.; Wigge, C.; Cramer, P. Cryo-EM structure of a mammalian RNA polymerase II elongation complex inhibited by α-amanitin. J. Biol. Chem. 2018, 293, 7189–7194. [Google Scholar] [CrossRef]
- Walmacq, C.; Kireeva, M.L.; Irvin, J.; Nedialkov, Y.; Lubkowska, L.; Malagon, F.; Strathern, J.N.; Kashlev, M. Rpb9 subunit controls transcription fidelity by delaying NTP sequestration in RNA polymerase II. J. Biol. Chem. 2009, 284, 19601–19612. [Google Scholar] [CrossRef]
- Kettenberger, H.; Armache, K.J.; Cramer, P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol. Cell 2004, 16, 955–965. [Google Scholar] [CrossRef]
- Vassylyev, D.G.; Vassylyeva, M.N.; Zhang, J.; Palangat, M.; Artsimovitch, I.; Landick, R. Structural basis for substrate loading in bacterial RNA polymerase. Nature 2007, 448, 163–168. [Google Scholar] [CrossRef]
- Cramer, P.; Bushnell, D.A.; Kornberg, R.D. Structural Basis of Transcription: RNA Polymerase II at 2.8 Angstrom Resolution. Science 2001, 292, 1863–1876. [Google Scholar] [CrossRef]
- Ruprich-Robert, G.; Thuriaux, P. Non-canonical DNA transcription enzymes and the conservation of two-barrel RNA polymerases. Nucleic Acids Res. 2010, 38, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Weinzierl, R.O.J. Nanomechanical constraints acting on the catalytic site of cellular RNA polymerases. Biochem. Soc. Trans. 2010, 38, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Domecq, C.; Kireeva, M.; Archambault, J.; Kashlev, M.; Coulombe, B.; Burton, Z.F. Site-directed mutagenesis, purification and assay of Saccharomyces cerevisiae RNA polymerase II. Protein Expr. Purif. 2010, 69, 83–90. [Google Scholar] [CrossRef][Green Version]
- Hein, P.P.; Landick, R. The bridge helix coordinates movements of modules in RNA polymerase. BMC Biol. 2010, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Seibold, S.A.; Singh, B.N.; Zhang, C.; Kireeva, M.; Domecq, C.; Bouchard, A.; Nazione, A.M.; Feig, M.; Cukier, R.I.; Coulombe, B.; et al. Conformational coupling, bridge helix dynamics and active site dehydration in catalysis by RNA polymerase. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, Y.; Xu, L.; Xiao, R.; Lu, X.; Chen, L.; Chong, J.; Li, H.; He, C.; Fu, X.D.; et al. Molecular basis for 5-carboxycytosine recognition by RNA polymerase II elongation complex. Nature 2015, 523, 621–625. [Google Scholar] [CrossRef]
- Chang, J.W.; Wu, Y.M.; Chen, Z.Y.; Huang, S.H.; Wang, C.H.; Wu, P.L.; Weng, Y.P.; You, C.; Piehler, J.; Chang, W.H. Hybrid electron microscopy-FRET imaging localizes the dynamical C-terminus of Tfg2 in RNA polymerase II-TFIIF with nanometer precision. J. Struct. Biol. 2013, 184, 52–62. [Google Scholar] [CrossRef]
- Schweikhard, V.; Meng, C.; Murakami, K.; Kaplan, C.D.; Kornberg, R.D.; Block, S.M. Transcription factors TFIIF and TFIIS promote transcript elongation by RNA polymerase II by synergistic and independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 6642–6647. [Google Scholar] [CrossRef]
- Da, L.T.; Wang, D.; Huang, X. Dynamics of pyrophosphate ion release and its coupled trigger loop motion from closed to open state in RNA polymerase II. J. Am. Chem. Soc. 2012, 134, 2399–2406. [Google Scholar] [CrossRef]
- Chakraborty, A.; Wang, D.; Ebright, Y.W.; Korlann, Y.; Kortkhonjia, E.; Kim, T.; Chowdhury, S.; Wigneshweraraj, S.; Irschik, H.; Jansen, R.; et al. Opening and Closing of the Bacterial RNA Polymerase Clamp. Science 2012, 337, 591–595. [Google Scholar] [CrossRef]
- Schulz, S.; Gietl, A.; Smollett, K.; Tinnefeld, P.; Werner, F.; Grohmann, D. TFE and Spt4/5 open and close the RNA polymerase clamp during the transcription cycle. Proc. Natl. Acad. Sci. USA 2016, 113, E1816–E1825. [Google Scholar] [CrossRef] [PubMed]
- Duchi, D.; Mazumder, A.; Malinen, A.M.; Ebright, R.H.; Kapanidis, A.N. The RNA polymerase clamp interconverts dynamically among three states and is stabilized in a partly closed state by ppGpp. Nucleic Acids Res. 2018, 46, 7284–7295. [Google Scholar] [CrossRef] [PubMed]
- Geiger, S.R.; Lorenzen, K.; Schreieck, A.; Hanecker, P.; Kostrewa, D.; Heck, A.J.R.; Cramer, P. RNA Polymerase I Contains a TFIIF-Related DNA-Binding Subcomplex. Mol. Cell 2010, 39, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.I.; Baudin, F.; Von Scheven, G.; Müller, C.W. RNA polymerase III-specific general transcription factor IIIC contains a heterodimer resembling TFIIF Rap30/Rap74. Nucleic Acids Res. 2013, 41, 9183–9196. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Lin, Y.-C.; Chen, H.-T. The TFIIF-Like Rpc37/53 Dimer Lies at the Center of a Protein Network To Connect TFIIIC, Bdp1, and the RNA Polymerase III Active Center. Mol. Cell. Biol. 2011, 31, 2715–2728. [Google Scholar] [CrossRef]
- Kubicek, C.E.; Chisholm, R.D.; Takayama, S.; Hawley, D.K. RNA Polymerase II Mutations Conferring Defects in Poly(A) Site Cleavage and Termination in Saccharomyces cerevisiae. G3 Genes Genomes Genet. 2013, 3, 167–180. [Google Scholar] [CrossRef]
- Mullen Davis, M.A.; Guo, J.; Price, D.H.; Luse, D.S. Functional interactions of the RNA polymerase II-interacting Proteins Gdown1 and TFIIF. J. Biol. Chem. 2014, 289, 11143–11152. [Google Scholar] [CrossRef]
- DeLaney, E.; Luse, D.S. Gdown1 associates efficiently with RNA polymerase II after promoter clearance and displaces TFIIF during transcript elongation. PLoS ONE 2016, 11, e0163649. [Google Scholar] [CrossRef]
- Jishage, M.; Yu, X.; Shi, Y.; Ganesan, S.J.; Chen, W.Y.; Sali, A.; Chait, B.T.; Asturias, F.J.; Roeder, R.G. Architecture of Pol II(G) and molecular mechanism of transcription regulation by Gdown1. Nat. Struct. Mol. Biol. 2018, 25, 859–867. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulation ID | Protein Structure | Transitive NTP Restart 1 | Observations |
|---|---|---|---|
| aMD_A1–6 | PDB#5IY9 | Initial structure. | |
| aMD_B1–4 | aMD_A1 168.5 ns | aMD_A6 233 ns | CH1 loading state sampled in aMD_A6 is aligned into aMD_A1 displaying a fully stabilized RNAPII-TFIIF complex. |
| aMD_B’1–2 | aMD_A1 168.5 ns | aMD_A6 233 ns | Same as above but the trigger loop is in closed conformation (templated from PDB#2E2H). |
| aMD_C1–4 | aMD_B2 89.75 ns | aMD_B2 comprises an NTP loaded through CH1, RNAPII-TFIIF complex is stable. | |
| aMD_D1–15 | aMD_A1 181 ns (D1 to D5), aMD_B2 72.5 ns (D6 to D10) and aMD_B3 108 ns (D11 to D15) | aMD_C1 6.5 ns | CH1 preinsertion state sampled in aMD_C1 is aligned into structures with three different open trigger loop conformations. |
| aMD_E1–6 | aMD_D12 9 ns | An NTP is bound to the i + 2 register. The nucleic pair is kept attached with restraints. | |
| aMD_F1–6 | aMD_E4 56 ns | The i + 2 bound NTP is isomerized. | |
| aMD_F’1–6 | aMD_E4 56 ns | Same as above, but the i + 4 DNA pair is restrained. | |
| aMD_G1–2 | aMD_A1 87 ns | aMD_E4 56 ns | The i + 2 isomerized NTP state is aligned into a structure displaying a bent bridge helix. |
| aMD_H1–5 | aMD_G1 11.4 ns | NTP forward transfer intermediate towards the active site. | |
| aMD_I1–3 | aMD_H2 55.8 ns | Forward transfer intermediate. | |
| aMD_J1–7 | aMD_I3 21.4 ns | Forward transfer intermediate. | |
| aMD_K1–2 | aMD_A1 110 ns | aMD_A5 359 ns | CH2 loading state sampled in aMD_A5 is aligned into aMD_A1 displaying a stabilized RNAPII-TFIIF complex. |
| aMD_L1–6 | aMD_K2 34.5 ns | NTP loaded through CH2 corridor. | |
| aMD_M1–3 | aMD_D12 9 ns (M1), aMD_D7 12 ns (M2, M3) | aMD_M1–3 inputs are completed with i + 2, i + 2/+ 3 and i + 2/+ 3/+ 4 NTP bound registers. | |
| aMD_N1–4 | aMD_B’1 12 ns (N1), aMD_B4 117 ns (N2), aMD_A1 107 ns (N3), aMD_A1 182 ns (N4) | Hybrid accelerated/steered MD method to investigate the loading through the different CH1 entry paths. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Génin, N.E.J.; Weinzierl, R.O.J. Nucleotide Loading Modes of Human RNA Polymerase II as Deciphered by Molecular Simulations. Biomolecules 2020, 10, 1289. https://doi.org/10.3390/biom10091289
Génin NEJ, Weinzierl ROJ. Nucleotide Loading Modes of Human RNA Polymerase II as Deciphered by Molecular Simulations. Biomolecules. 2020; 10(9):1289. https://doi.org/10.3390/biom10091289
Chicago/Turabian StyleGénin, Nicolas E. J., and Robert O. J. Weinzierl. 2020. "Nucleotide Loading Modes of Human RNA Polymerase II as Deciphered by Molecular Simulations" Biomolecules 10, no. 9: 1289. https://doi.org/10.3390/biom10091289
APA StyleGénin, N. E. J., & Weinzierl, R. O. J. (2020). Nucleotide Loading Modes of Human RNA Polymerase II as Deciphered by Molecular Simulations. Biomolecules, 10(9), 1289. https://doi.org/10.3390/biom10091289