A Fluorescent Assay to Search for Inhibitors of HIV-1 Integrase Interactions with Human Ku70 Protein, and Its Application for Characterization of Oligonucleotide Inhibitors

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
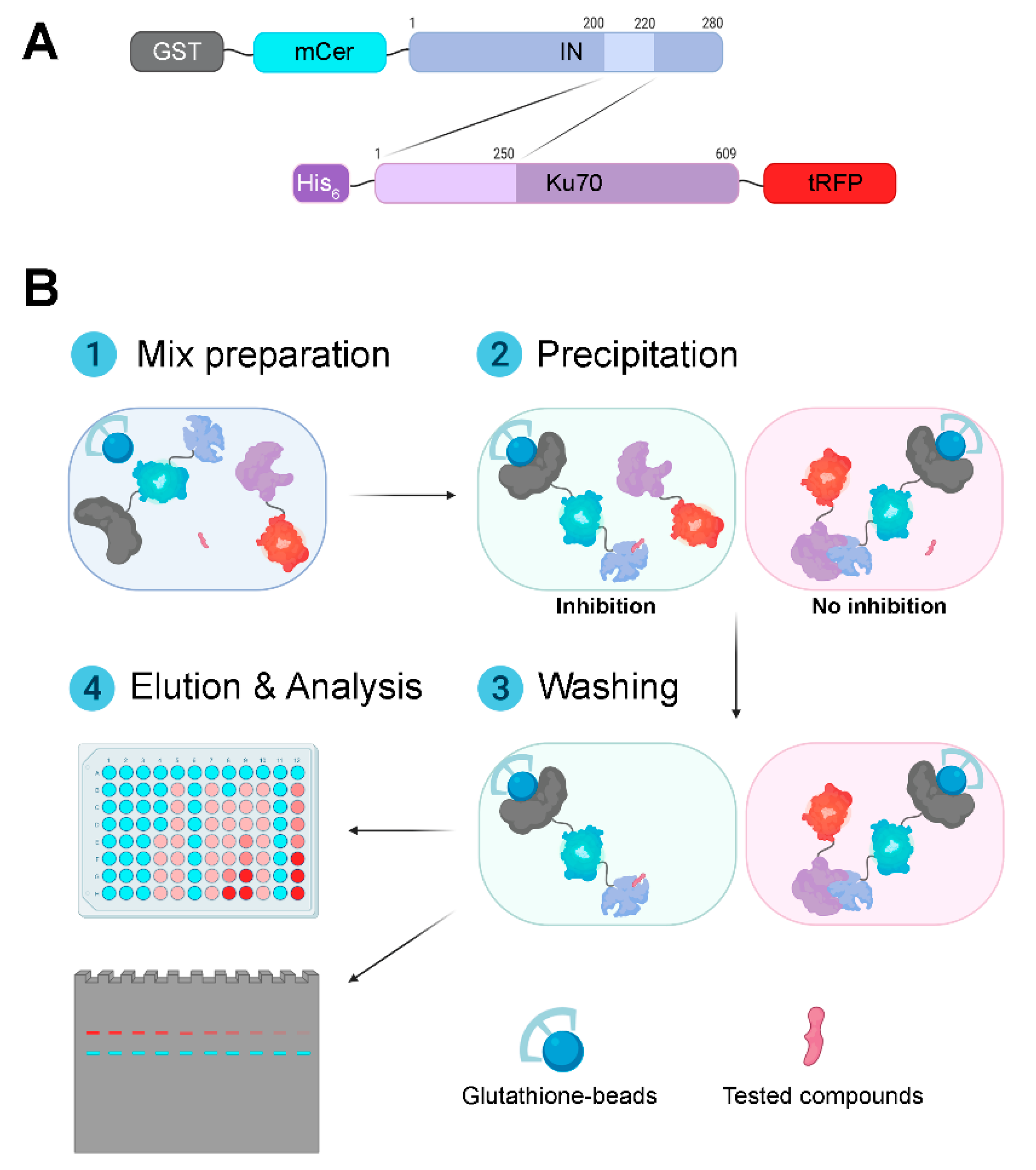
3.1. Design and Validation of a System for Search for Inhibitors of the Interaction between HIV-1 Integrase and Ku70
3.2. The Effect of the Oligonucleotide Inhibitor Structure on Binding of IN and Ku70
3.2.1. Nucleobases Rather than the Sugar-Phosphate Backbone of the Inhibitor Are Crucial for IN Binding with Ku70
3.2.2. The Inhibition Efficiency of the Eosin-Y-Conjugates Depends on the Length of Their Oligonucleotide Moiety
3.3. The Effect of the Oligonucleotide Moiety Structure on the Conjugates’ Inhibition Potential May Be Due to the Steric Shielding of the Integrase Surface Involved in the Complex Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef]
- Kuritzkes, D.R. Drug resistance in HIV-1. Curr. Opin. Virol. 2011, 1, 582–589. [Google Scholar] [CrossRef]
- Pennings, P.S. HIV drug resistance: Problems and perspectives. Infect. Dis. Rep. 2013, 5, 21–25. [Google Scholar] [CrossRef]
- Günthard, H.F.; Calvez, V.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Jacobsen, D.M.; Richman, D.D.; Francisco, S. Human Immunodeficiency Virus Drug Resistance: 2018 Recommendations of the International Antiviral Society-USA Panel and 8 International Antiviral Society-USA. HIV Drug Resist. Recomm. CID 2018, 2019, 177. [Google Scholar]
- Gupta, R.K.; Gregson, J.; Parkin, N.; Haile-Selassie, H.; Tanuri, A.; Andrade Forero, L.; Kaleebu, P.; Watera, C.; Aghokeng, A.; Mutenda, N.; et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: A systematic review and meta-regression analysis. Lancet Infect. Dis. 2018, 18, 346–355. [Google Scholar] [CrossRef]
- Taltynov, O.; Desimmie, B.A.; Demeulemeester, J.; Christ, F.; Debyser, Z. Cellular Cofactors of Lentiviral Integrase: From Target Validation to Drug Discovery. Mol. Biol. Int. 2012, 2012, 1–16. [Google Scholar] [CrossRef]
- Taltynov, O.; De Rijck, J.; Debyser, Z. Identification and Validation of HIV Cofactors. In Encyclopedia of AIDS; Springer: New York, NY, USA, 2018; pp. 1043–1047. [Google Scholar]
- Dürr, R.; Keppler, O.; Christ, F.; Crespan, E.; Garbelli, A.; Maga, G.; Dietrich, U. Targeting cellular cofactors in HIV therapy. Top. Med. Chem. 2015, 15, 183–222. [Google Scholar]
- Poeschla, E.M. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. 2008, 65, 1403–1424. [Google Scholar] [CrossRef]
- Christ, F.; Debyser, Z. The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology 2013, 435, 102–109. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van Der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268.e12. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Mallareddy, J.R.; Lu, D.; Kolluru, S. Recent advances in the discovery of small-molecule inhibitors of HIV-1 integrase. Future Sci. OA 2018, 4, FSO338. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ao, Z.; Wang, B.; Jayappa, K.D.; Yao, X. Host protein Ku70 binds and protects HIV-1 integrase from proteasomal degradation and is required for HIV replication. J. Biol. Chem. 2011, 286, 17722–17735. [Google Scholar] [CrossRef]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Zalevsky, A.O.; Agapkina, J.Y.; Sizov, A.I.; Zatsepin, T.S.; Gottikh, M.B. Characterization of HIV-1 integrase interaction with human Ku70 protein and initial implications for drug targeting. Sci. Rep. 2017, 7, 5649. [Google Scholar] [CrossRef]
- Knyazhanskaya, E.; Anisenko, A.; Shadrina, O.; Kalinina, A.; Zatsepin, T.; Zalevsky, A.; Mazurov, D.; Gottikh, M. NHEJ pathway is involved in post-integrational DNA repair due to Ku70 binding to HIV-1 integrase. Retrovirology 2019, 16, 30. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Sunand Kumar, G.N. Protein-Protein Interaction Detection: Methods and Analysis. Int. J. Proteom. 2014, 2014, 147648. [Google Scholar] [CrossRef]
- Eglen, R.M.; Reisine, T.; Roby, P.; Rouleau, N.; Illy, C.; Bossé, R.; Bielefeld, M. The Use of AlphaScreen Technology in HTS: Current Status. Curr. Chem. Genom. 2008, 1, 2–10. [Google Scholar] [CrossRef]
- Ossiboff, R.J.; Zhou, Y.; Lightfoot, P.J.; Prasad, B.V.V.; Parker, J.S.L. Conformational Changes in the Capsid of a Calicivirus upon Interaction with Its Functional Receptor. J. Virol. 2010, 84, 5550–5564. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Zhao, Q. Utilizing ELISA to monitor protein-protein interaction. In Protein-Protein Interactions: Methods and Applications, 2nd ed.; Springer: New York, NY, USA, 2015; pp. 341–352. ISBN 9781493924257. [Google Scholar]
- Pinto, M.G.V.; Baiker, A. LuMPIS: Luciferase-based MBP-Pull-down protein interaction screening system. Methods Mol. Biol. 2012, 815, 265–275. [Google Scholar] [PubMed]
- Bacart, J.; Corbel, C.; Jockers, R.; Bach, S.; Couturier, C. The BRET technology and its application to screening assays. Biotechnol. J. 2008, 3, 311–324. [Google Scholar] [CrossRef]
- Al-Mugotir, M.; Kolar, C.; Vance, K.; Kelly, D.L.; Natarajan, A.; Borgstahl, G.E.O. A simple fluorescent assay for the discovery of protein-protein interaction inhibitors. Anal. Biochem. 2019, 569, 46–52. [Google Scholar] [CrossRef]
- Prikazchikova, T.A.; Volkov, E.M.; Zubin, E.M.; Romanova, E.A.; Gottikh, M.B. Inhibition of HIV-1 integrase by modified oligonucleotides: Optimization of the inhibitor structure. Mol. Biol. 2007, 41, 118–125. [Google Scholar] [CrossRef]
- Agapkina, J.; Zatsepin, T.; Knyazhanskaya, E.; Mouscadet, J.F.; Gottikh, M. Structure-activity relationship studies of HIV-1 integrase oligonucleotide inhibitors. ACS Med. Chem. Lett. 2011, 2, 532–537. [Google Scholar] [CrossRef][Green Version]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Zatsepin, T.S.; Gottikh, M.B. Human Ku70 protein binds hairpin RNA and double stranded DNA through two different sites. Biochimie 2017, 132, 85–93. [Google Scholar] [CrossRef]
- Knyazhanskaya, E.S.; Smolov, M.A.; Kondrashina, O.V.; Gottikh, M.B. Relative Comparison of Catalytic Characteristics of Human Foamy Virus and HIV-1 Integrases. Acta Nat. 2009, 1, 78–80. [Google Scholar] [CrossRef]
- Li, S.; Olson, W.K.; Lu, X.-J. Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Web Serv. Issue Publ. Online 2019, 47, W26–W34. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinforma. Appl. NOTE 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.A.; Guallar, V. pyRMSD: A Python package for efficient pairwise RMSD matrix calculation and handling. Bioinformatics 2013, 29, 2363–2364. [Google Scholar] [CrossRef] [PubMed]
- Team, T. Pandas development Pandas-dev/pandas: Pandas. Zenodo 2020, 21, 1–9. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 99–104. [Google Scholar] [CrossRef]
- Saeed, I.A.; Ashraf, S.S. Denaturation studies reveal significant differences between GFP and blue fluorescent protein. Int. J. Biol. Macromol. 2009, 45, 236–241. [Google Scholar] [CrossRef]
- Shcherbo, D.; Merzlyak, E.M.; Chepurnykh, T.V.; Fradkov, A.F.; Ermakova, G.V.; Solovieva, E.A.; Lukyanov, K.A.; Bogdanova, E.A.; Zaraisky, A.G.; Lukyanov, S.; et al. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods 2007, 4, 741–746. [Google Scholar] [CrossRef]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: A web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, 441–445. [Google Scholar] [CrossRef]
- Kalliokoski, T.; Kramer, C.; Vulpetti, A.; Gedeck, P. Comparability of Mixed IC50 Data–A Statistical Analysis. PLoS ONE 2013, 8, e61007. [Google Scholar] [CrossRef]
- Guy, A.T.; Piggot, T.J.; Khalid, S. Single-stranded DNA within nanopores: Conformational dynamics and implications for sequencing; A molecular dynamics simulation study. Biophys. J. 2012, 103, 1028–1036. [Google Scholar] [CrossRef]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein–protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Benoıˆt De Chassey, B.; Ne Meyniel-Schicklin, L.; Aublin-Gex, A.; André, P.; Lotteau, V. New horizons for antiviral drug discovery from virus-host protein interaction networks. Curr. Opin. Virol. 2012, 2, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Pinskaya, M.; Romanova, E.; Volkov, E.; Deprez, E.; Leh, H.; Brochon, J.C.; Mouscadet, J.F.; Gottikh, M. HIV-1 integrase complexes with DNA dissociate in the presence of short oligonucleotides conjugated to acridine. Biochemistry 2004, 43, 8735–8743. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.H.; Krucinski, J.; Miercke, L.J.W.; Finer-Moore, J.S.; Tang, A.H.; Leavitt, A.D.; Stroud, R.M. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: A model for viral DNA binding. Proc. Natl. Acad. Sci. USA 2000, 97, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Lutzke, R.A.P.; Vink, C.; Plasterk, R.H.A. Characterization of the Minimal DNA-Binding Domain of the HIV Integrase Protein. Nucleic Acids Res. 1994, 22, 4125–4131. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}

| ODN-E | Sequence, 5′-3′ | IC50, nM |
|---|---|---|
| 11-OM-E | GGUUUUUGUGU-EosinY | 135 ± 20 |
| 11-D-E | GGTTTTTGTGT-EosinY | 150 ± 30 |
| 11-DX-E-1 | GGTTTTTxGTxGTx-EosinY | 160 ± 55 |
| 11-DX-E-2 | GGTxTxTxTTGTGT-EosinY | 150 ± 40 |
| 11-DS-E | GsGsTsTsTsTsTsGsTsGsTs-EosinY | 40 ± 10 |
| Hex-11-D | HEX-GGTTTTTGTGT | 170 ± 45 |
| Hex-11-ddR | HEX-(ddR)10T | No inhibition up to 3200 nM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galkin, S.; Rozina, A.; Zalevsky, A.; Gottikh, M.; Anisenko, A. A Fluorescent Assay to Search for Inhibitors of HIV-1 Integrase Interactions with Human Ku70 Protein, and Its Application for Characterization of Oligonucleotide Inhibitors. Biomolecules 2020, 10, 1236. https://doi.org/10.3390/biom10091236
Galkin S, Rozina A, Zalevsky A, Gottikh M, Anisenko A. A Fluorescent Assay to Search for Inhibitors of HIV-1 Integrase Interactions with Human Ku70 Protein, and Its Application for Characterization of Oligonucleotide Inhibitors. Biomolecules. 2020; 10(9):1236. https://doi.org/10.3390/biom10091236
Chicago/Turabian StyleGalkin, Simon, Anna Rozina, Arthur Zalevsky, Marina Gottikh, and Andrey Anisenko. 2020. "A Fluorescent Assay to Search for Inhibitors of HIV-1 Integrase Interactions with Human Ku70 Protein, and Its Application for Characterization of Oligonucleotide Inhibitors" Biomolecules 10, no. 9: 1236. https://doi.org/10.3390/biom10091236
APA StyleGalkin, S., Rozina, A., Zalevsky, A., Gottikh, M., & Anisenko, A. (2020). A Fluorescent Assay to Search for Inhibitors of HIV-1 Integrase Interactions with Human Ku70 Protein, and Its Application for Characterization of Oligonucleotide Inhibitors. Biomolecules, 10(9), 1236. https://doi.org/10.3390/biom10091236