A Virtual Screening Platform Identifies Chloroethylagelastatin A as a Potential Ribosomal Inhibitor

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials for Biological Testing (Cell Culture)
2.2. Protein Synthesis Assay
2.3. Statistical Methods for Analysis of Protein Synthesis Data
2.4. Molecular Modeling Methods—Evaluate Shape Fitting Algorithms
2.5. Evaluate De Novo Ligand Generation for AA Analogues
2.6. Molecular Dynamics Simulations
3. Results
3.1. Virtual Screening of AA and AA Analogues on Ribosomal A-Site Peptidyl Transferase Center
3.2. Ribosomal Docking Reveals that Therapeutically Active AA Analogues Possess Strong Ribosomal Binding Capacity
3.3. Structural Basis for Drug-Ribosome Interactions (3D-QSAR) Identifies Key Fingerprint Features (2D-Ligand Interaction Maps)
3.4. Modeling Shows Structural Underpinnings for Drug-Ribosome Interaction for AA Analogues
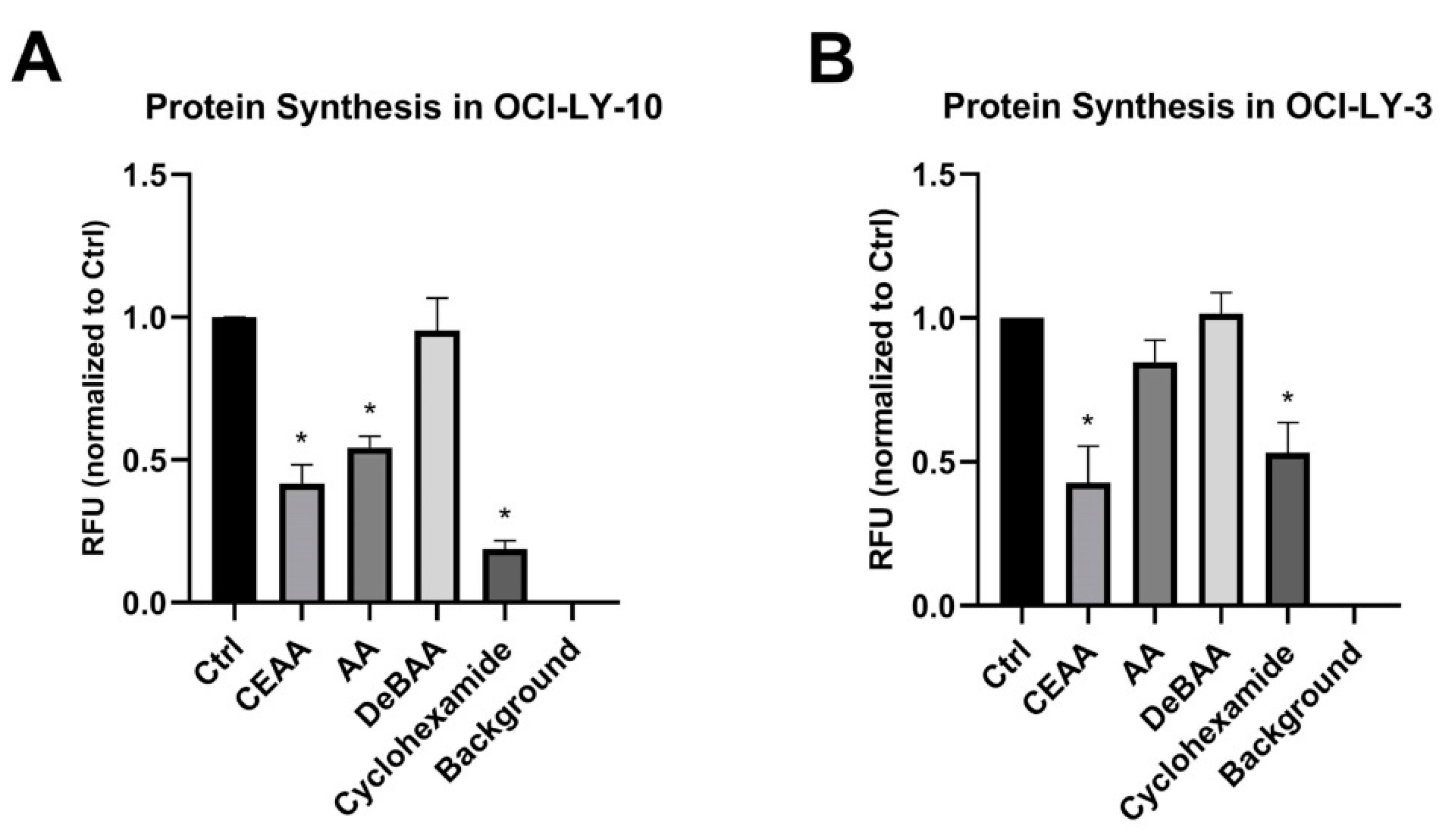
3.5. CEAA is an Inhibitor of Protein Synthesis
4. Discussion
4.1. Ribosomal Binding Capacity of CEAA Correlates with Inhibition of Protein Synthesis
4.2. Structural Modifications of AA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D’Ambrosio, M.; Guerriero, A.; Debitus, C.; Ribes, O.; Pusset, J.; Leroy, S.; Pietra, F. Agelastatin a, a new skeleton cytotoxic alkaloid of the oroidin family. Isolation from the axinellid sponge Agelas dendromorpha of the coral sea. J. Chem. Soc. Chem. Commun. 1993, 1305–1306. [Google Scholar] [CrossRef]
- Li, Z.; Kamon, T.; Personett, D.A.; Caulfield, T.; Copland, J.A.; Yoshimitsu, T.; Tun, H.W. Pharmacokinetics of Agelastatin A in the central nervous system. Med. Chem. Comm. 2012, 3, 233–237. [Google Scholar] [CrossRef]
- Li, Z.; Shigeoka, D.; Caulfield, T.R.; Kawachi, T.; Qiu, Y.; Kamon, T.; Arai, M.; Tun, H.W.; Yoshimitsu, T. An integrated approach to the discovery of potent agelastatin A analogues for brain tumors: Chemical synthesis and biological, physicochemical and CNS pharmacokinetic analyses. Med. Chem. Comm. 2013, 4, 1093–1098. [Google Scholar] [CrossRef]
- McClary, B.; Zinshteyn, B.; Meyer, M.; Jouanneau, M.; Pellegrino, S.; Yusupova, G.; Schuller, A.; Reyes, J.C.P.; Lu, J.; Guo, Z.; et al. Inhibition of Eukaryotic Translation by the Antitumor Natural Product Agelastatin A. Cell Chem. Biol. 2017, 24, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Paulus, A.; Akhtar, S.; Caulfield, T.R.; Samuel, K.; Yousaf, H.; Bashir, Y.; Paulus, S.M.; Tran, D.; Hudec, R.; Cogen, D.; et al. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib- or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016, 6, e492. [Google Scholar] [CrossRef]
- Kayode, O.; Wang, R.; Pendlebury, D.F.; Cohen, I.; Henin, R.D.; Hockla, A.; Soares, A.S.; Papo, N.; Caulfield, T.R.; Radisky, E.S. An Acrobatic Substrate Metamorphosis Reveals a Requirement for Substrate Conformational Dynamics in Trypsin Proteolysis. J. Biol. Chem. 2016, 291, 26304–26319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiesel, F.C.; Caulfield, T.R.; Moussaud-Lamodière, E.L.; Ogaki, K.; Dourado, D.F.A.R.; Flores, S.C.; Ross, O.A.; Springer, W. Structural and Functional Impact of Parkinson Disease-Associated Mutations in the E3 Ubiquitin Ligase Parkin. Hum. Mutat. 2015, 36, 774–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Hay, S.O.; Bannister, T.D.; Wang, H.; Cameron, M.D.; Caulfield, T.R.; Masson, A.; Bertrand, J.; Howard, E.A.; McGuire, M.P.; Crisafulli, U.; et al. Selective Targeting of Extracellular Insulin-Degrading Enzyme by Quasi-Irreversible Thiol-Modifying Inhibitors. ACS Chem. Biol. 2015, 10, 2716–2724. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Almeida, S.; Prudencio, M.; Caulfield, T.R.; Zhang, Y.J.; Tay, W.M.; Bauer, P.O.; Chew, J.; Sasaguri, H.; Jansen-West, K.R.; et al. Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum. Mol. Genet. 2014, 23, 1467–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caulfield, T.R.; Fiesel, F.C.; Moussaud-Lamodière, E.L.; Dourado, D.F.A.R.; Flores, S.C.; Springer, W. Phosphorylation by PINK1 Releases the UBL Domain and Initializes the Conformational Opening of the E3 Ubiquitin Ligase Parkin. PLoS Comput. Biol. 2014, 10, e1003935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivoli, M.; Caulfield, T.R.; Martinez-Mayorga, K.; Johnson, A.T.; Jiao, G.S.; Lindberg, I. Inhibition of prohormone convertases PC1/3 and PC2 by 2,5-dideoxystreptamine derivatives. Mol. Pharmacol. 2012, 81, 440–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caulfield, T.; Devkota, B. Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins 2012, 80, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.R.; Devkota, B.; Rollins, G.C. Examinations of tRNA Range of Motion Using Simulations of Cryo-EM Microscopy and X-Ray Data. J. Biophys. 2011, 2011, 219515. [Google Scholar] [CrossRef] [PubMed]
- Kuck, D.; Caulfield, T.; Lyko, F.; Medina-Franco, J.L. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010, 9, 3015–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, G.M.; Inakollu, V.S.; Sherman, W. Boosting Virtual Screening Enrichments with Data Fusion: Coalescing Hits from Two-Dimensional Fingerprints, Shape, and Docking. J. Chem. Inf. Model. 2013, 53, 1531–1542. [Google Scholar] [CrossRef]
- Pala, D.; Beuming, T.; Sherman, W.; Lodola, A.; Rivara, S.; Mor, M. Structure-Based Virtual Screening of MT2 Melatonin Receptor: Influence of Template Choice and Structural Refinement. J. Chem. Inf. Model. 2013, 53, 821–835. [Google Scholar] [CrossRef]
- Kalid, O.; Warshaviak, D.T.; Shechter, S.; Sherman, W.; Shacham, S. Consensus Induced Fit Docking (cIFD): Methodology, validation, and application to the discovery of novel Crm1 inhibitors. J. Comput. Aided Mol. Des. 2012, 26, 1217–1228. [Google Scholar] [CrossRef]
- Fu, J.; Si, P.; Zheng, M.; Chen, L.; Shen, X.; Tang, Y.; Li, W. Discovery of new non-steroidal FXR ligands via a virtual screening workflow based on Phase shape and induced fit docking. Bioorg. Med. Chem. Lett. 2012, 22, 6848–6853. [Google Scholar] [CrossRef]
- Sastry, G.M.; Dixon, S.L.; Sherman, W. Rapid Shape-Based Ligand Alignment and Virtual Screening Method Based on Atom/Feature-Pair Similarities and Volume Overlap Scoring. J. Chem. Inf. Model. 2011, 51, 2455–2466. [Google Scholar] [CrossRef]
- Schrodinger. Qikprop. v. 3.4; Schrödinger, LLC: New York, NY, USA, 2013. [Google Scholar]
- Bavi, R.S.; Sambhare, S.B.; Sonawane, K.D. MD simulation studies to investigate iso-energetic conformational behaviour of modified nucleosides M2G AND M22G present in tRNA. Comput. Struct. Biotechnol. J. 2013, 5, e201302015. [Google Scholar] [CrossRef] [Green Version]
- Caulfield, T.; Medina-Franco, J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011, 176, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.R. Inter-ring rotation of apolipoprotein A-I protein monomers for the double-belt model using biased molecular dynamics. J. Mol. Graph. Model. 2011, 29, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz Jr, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Chem. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Reblova, K.; Lankas, F.; Razga, F.; Krasovska, M.V.; Koca, J.; Sponer, J. Structure, dynamics, and elasticity of free 16s rRNA helix 44 studied by molecular dynamics simulations. Biopolymers 2006, 82, 504–520. [Google Scholar] [CrossRef] [PubMed]
- Reblova, K.; Fadrna, E.; Sarzynska, J.; Kulinski, T.; Kulhanek, P.; Ennifar, E.; Koca, J.; Sponer, J. Conformations of flanking bases in HIV-1 RNA DIS kissing complexes studied by molecular dynamics. Biophys. J. 2007, 93, 3932–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheatham, T.E., III.; Young, M.A. Molecular dynamics simulation of nucleic acids: Successes, limitations and promise. Biopolymers 2001, 56, 232–256. [Google Scholar] [CrossRef]
- Brandman, O.; Hegde, R.S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016, 23, 7–15. [Google Scholar] [CrossRef]
- Jenner, L.; Melnikov, S.; de Loubresse, N.G.; Ben-Shem, A.; Iskakova, M.; Urzhumtsev, A.; Meskauskas, A.; Dinman, J.; Yusupova, G.; Yusupov, M. Crystal structure of the 80S yeast ribosome. Curr. Opin. Struct. Biol. 2012, 22, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Schmeing, T.M.; Voorhees, R.M.; Kelley, A.C.; Gao, Y.G.; Murphy, F.V.t.; Weir, J.R.; Ramakrishnan, V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science 2009, 326, 688–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochella, L.; Brunelle, J.L.; Green, R. Mutational analysis reveals two independent molecular requirements during transfer RNA selection on the ribosome. Nat. Struct. Mol. Biol. 2007, 14, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Selmer, M.; Dunham, C.M.; Murphy, F.V.T.; Weixlbaumer, A.; Petry, S.; Kelley, A.C.; Weir, J.R.; Ramakrishnan, V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 2006, 313, 1935–1942. [Google Scholar] [CrossRef] [Green Version]
- Ogle, J.M.; Ramakrishnan, V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005, 74, 129–177. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, S.C.; Kim, H.D.; Gonzalez, R.L., Jr.; Puglisi, J.D.; Chu, S. tRNA dynamics on the ribosome during translation. Proc. Natl. Acad. Sci. USA 2004, 101, 12893–12898. [Google Scholar] [CrossRef] [Green Version]
- Valle, M.; Zavialov, A.; Li, W.; Stagg, S.M.; Sengupta, J.; Nielsen, R.C.; Nissen, P.; Harvey, S.C.; Ehrenberg, M.; Frank, J. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat. Struct. Biol. 2003, 10, 899–906. [Google Scholar] [CrossRef]
- Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.H.; Noller, H.F. Crystal structure of the ribosome at 5.5 A resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef]
- Harms, J.; Schluenzen, F.; Zarivach, R.; Bashan, A.; Gat, S.; Agmon, I.; Bartels, H.; Franceschi, F.; Yonath, A. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell 2001, 107, 679–688. [Google Scholar] [CrossRef]
- Ibba, M.; Soll, D. Quality control mechanisms during translation. Science 1999, 286, 1893–1897. [Google Scholar] [CrossRef]
- Lomakin, I.B.; Stolboushkina, E.A.; Vaidya, A.T.; Zhao, C.; Garber, M.B.; Dmitriev, S.E.; Steitz, T.A. Crystal Structure of the Human Ribosome in Complex with DENR-MCT-1. Cell Rep. 2017, 20, 521–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaha, G.M.; Polikanov, Y.S.; Steitz, T.A. Elements of ribosomal drug resistance and specificity. Curr. Opin. Struct. Biol. 2012, 22, 750–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hu, Y.; Li, X.; Niu, L.; Teng, M. The crystal structure of the human nascent polypeptide-associated complex domain reveals a nucleic acid-binding region on the NACA subunit. Biochemistry 2010, 49, 2890–2896. [Google Scholar] [CrossRef] [PubMed]
- Doamekpor, S.K.; Lee, J.-W.; Hepowit, N.L.; Wu, C.; Charenton, C.; Leonard, M.; Bengtson, M.H.; Rajashankar, K.R.; Sachs, M.S.; Lima, C.D.; et al. Structure and function of the yeast listerin (Ltn1) conserved N-terminal domain in binding to stalled 60S ribosomal subunits. Proc. Natl. Acad. Sci. USA 2016, 113, E4151–E4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tun, H.W.; Yoshimitsu, T.; Shigeoka, D.; Kamon, T.; Li, Z.; Qiu, Y.; Caulfield, T.R. Substituted Imidazo[4‘,5’:4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazines and oxazolo[4’,5’:4,5]cyclopenta[1,2-e]pyrrolo[1,2-a]pyrazines for Treating Brain Cancer. U.S. Patent 9464093B2, 11 October 2016. [Google Scholar]
- Paulus, A.; Akhtar, S.; Yousaf, H.; Manna, A.; Paulus, S.M.; Bashir, Y.; Caulfield, T.R.; Kuranz-Blake, M.; Chitta, K.; Wang, X.; et al. Waldenstrom macroglobulinemia cells devoid of BTKC481S or CXCR4WHIM-like mutations acquire resistance to ibrutinib through upregulation of Bcl-2 and AKT resulting in vulnerability towards venetoclax or MK2206 treatment. Blood Cancer J. 2017, 7, e565. [Google Scholar] [CrossRef]
- Kayode, O.; Huang, Z.; Soares, A.S.; Caulfield, T.R.; Dong, Z.; Bode, A.M.; Radisky, E.S. Small molecule inhibitors of mesotrypsin from a structure-based docking screen. PLoS ONE 2017, 12, e0176694. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; D’Arcy, P.; Caulfield, T.R.; Paulus, A.; Chitta, K.; Mohanty, C.; Gullbo, J.; Chanan-Khan, A.; Linder, S. Synthesis and evaluation of derivatives of the proteasome deubiquitinase inhibitor b-AP15. Chem. Biol. Drug Des. 2015, 86, 1036–1048. [Google Scholar] [CrossRef] [Green Version]
- Caulfield, T.R.; Fiesel, F.C.; Springer, W. Activation of the E3 ubiquitin ligase Parkin. Biochem. Soc. Trans. 2015, 43, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Chitta, K.; Paulus, A.; Caulfield, T.R.; Akhtar, S.; Blake, M.K.; Ailawadhi, S.; Knight, J.; Heckman, M.G.; Pinkerton, A.; Chanan-Khan, A. Nimbolide targets BCL2 and induces apoptosis in preclinical models of Waldenstroms macroglobulinemia. Blood Cancer J. 2014, 4, e260. [Google Scholar] [CrossRef] [Green Version]
- Sindhikara, D.; Spronk, S.A.; Day, T.; Borrelli, K.; Cheney, D.L.; Posy, S.L. Improving Accuracy, Diversity, and Speed with Prime Macrocycle Conformational Sampling. J. Chem. Inf. Model. 2017, 57, 1881–1894. [Google Scholar] [CrossRef]
- Bhachoo, J.; Beuming, T. Investigating Protein–Peptide Interactions Using the Schrödinger Computational Suite. In Modeling Peptide-Protein Interactions: Methods and Protocols; Schueler-Furman, O., London, N., Eds.; Springer New York: New York, NY, USA, 2017; pp. 235–254. [Google Scholar]
- Lorber, D.M.; Shoichet, B.K. Flexible ligand docking using conformational ensembles. Protein Sci. 1998, 7, 938–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain A J. Neurol. 2017, 140, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Caulfield, T.; Xu, Y.F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Caulfield, T. Advances in the computational development of DNA methyltransferase inhibitors. Drug Discov. Today 2011, 16, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Caulfield, T.; Coban, M.; Tek, A.; Flores, S.C. Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of -1 Frameshifting by tRNA(Ser3). Biomolecules 2019, 9, 745. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Predicted CSM Affinity (-log10(KD|Ki)) | Docking score (ΔG) | Conformation 1 Predicted MM-GBSA Affinity (ΔG) | Conformation 2 Predicted MM-GBSA Affinity (ΔG) |
|---|---|---|---|---|---|
| CEAA |  | −11.1 | −6.8 | −50.81 | −32.52 |
| AA |  | −10.8 | −4.59 | −48.12 | −21.33 |
| CAA |  | −10.9 | −6.65 | −32.92 | −16.3 |
| DCEAA |  | −10.5 | −6.13 | −15.32 | −15.42 |
| PAA |  | −10.4 | −5.62 | −25.95 | 77.85 |
| EAA |  | −10.5 | −4.24 | 1293.69 | 3472.33 |
| DeBAA |  | −10.1 | −4.16 | 933.01 | 49.55 |
| DeBEAA |  | −9.4 | −4.38 | 1279.36 | 2815.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caulfield, T.R.; Hayes, K.E.; Qiu, Y.; Coban, M.; Seok Oh, J.; Lane, A.L.; Yoshimitsu, T.; Hazlehurst, L.; Copland, J.A.; Tun, H.W. A Virtual Screening Platform Identifies Chloroethylagelastatin A as a Potential Ribosomal Inhibitor. Biomolecules 2020, 10, 1407. https://doi.org/10.3390/biom10101407
Caulfield TR, Hayes KE, Qiu Y, Coban M, Seok Oh J, Lane AL, Yoshimitsu T, Hazlehurst L, Copland JA, Tun HW. A Virtual Screening Platform Identifies Chloroethylagelastatin A as a Potential Ribosomal Inhibitor. Biomolecules. 2020; 10(10):1407. https://doi.org/10.3390/biom10101407
Chicago/Turabian StyleCaulfield, Thomas R., Karen E. Hayes, Yushi Qiu, Mathew Coban, Joon Seok Oh, Amy L. Lane, Takehiko Yoshimitsu, Lori Hazlehurst, John A. Copland, and Han W. Tun. 2020. "A Virtual Screening Platform Identifies Chloroethylagelastatin A as a Potential Ribosomal Inhibitor" Biomolecules 10, no. 10: 1407. https://doi.org/10.3390/biom10101407
APA StyleCaulfield, T. R., Hayes, K. E., Qiu, Y., Coban, M., Seok Oh, J., Lane, A. L., Yoshimitsu, T., Hazlehurst, L., Copland, J. A., & Tun, H. W. (2020). A Virtual Screening Platform Identifies Chloroethylagelastatin A as a Potential Ribosomal Inhibitor. Biomolecules, 10(10), 1407. https://doi.org/10.3390/biom10101407