Multi-Scale Understanding of NMDA Receptor Function in Schizophrenia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
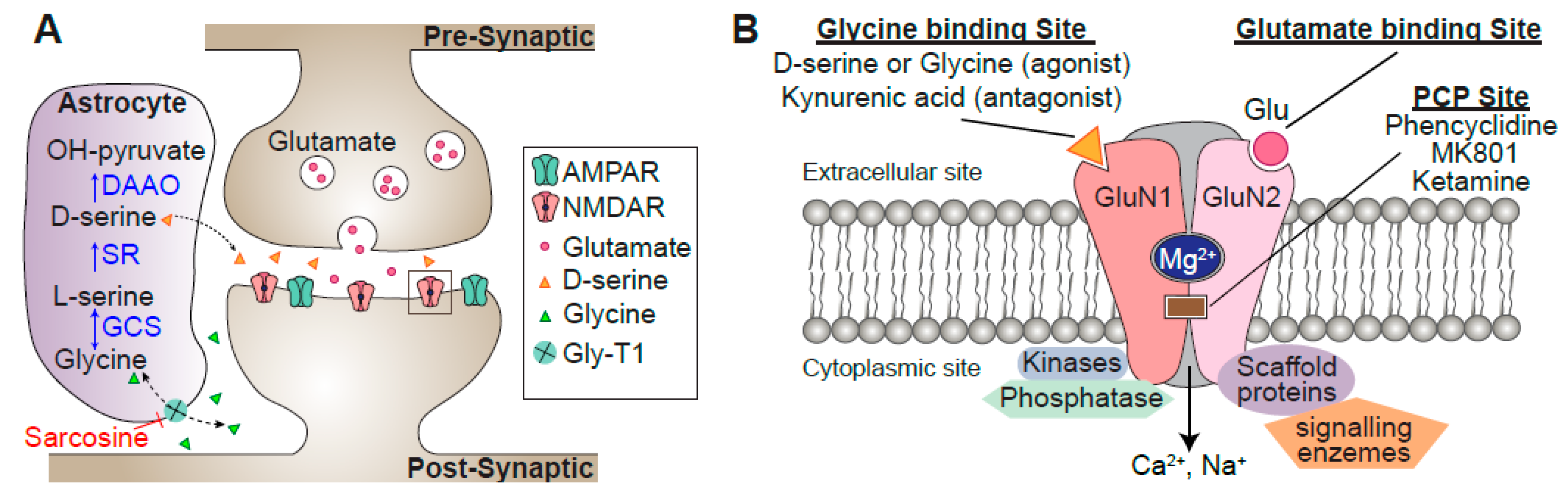
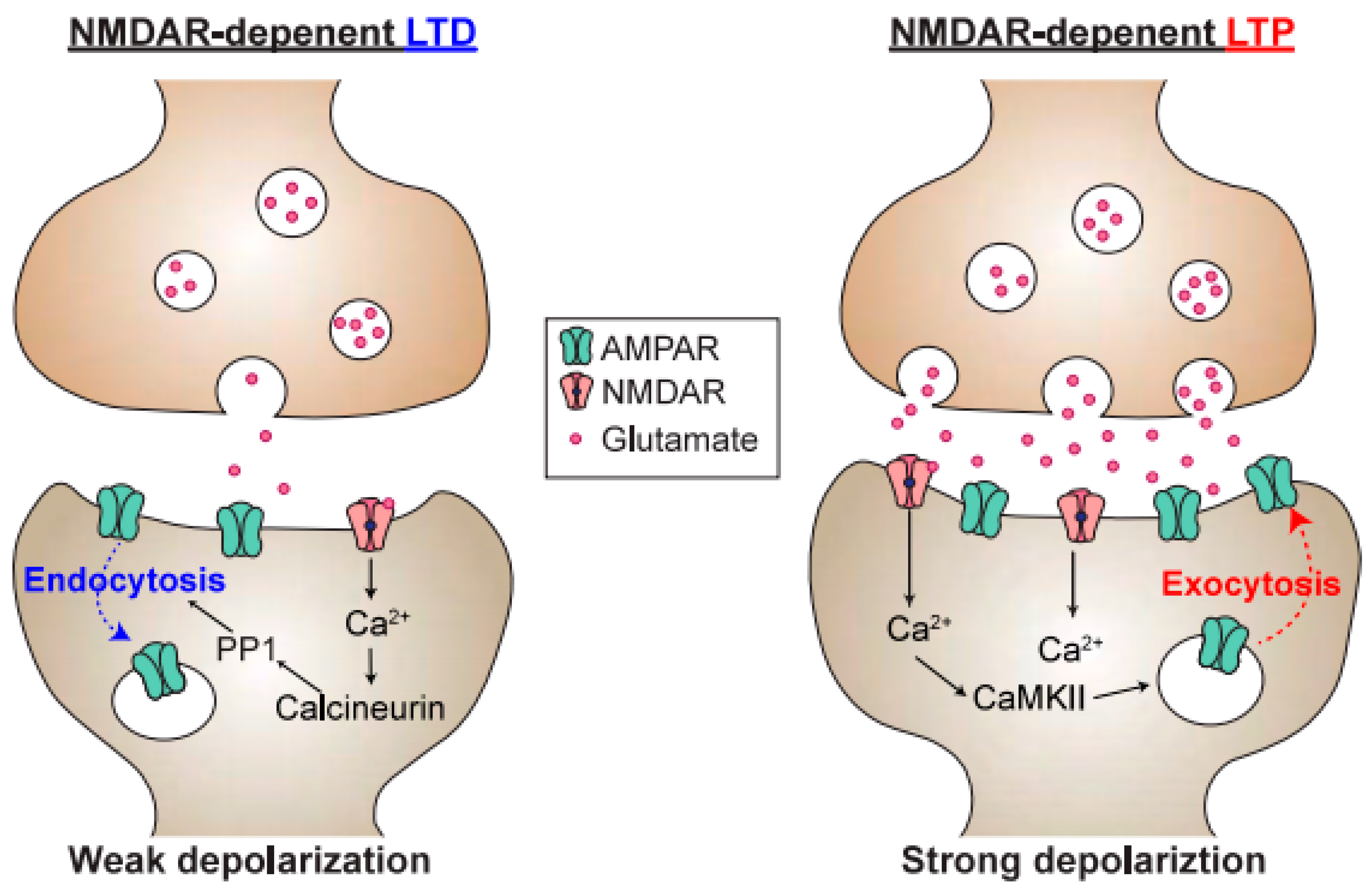
2. Functions of NMDAR Signaling
3. Evidence of NMDAR Dysregulation from Clinical Studies of Schizophrenia
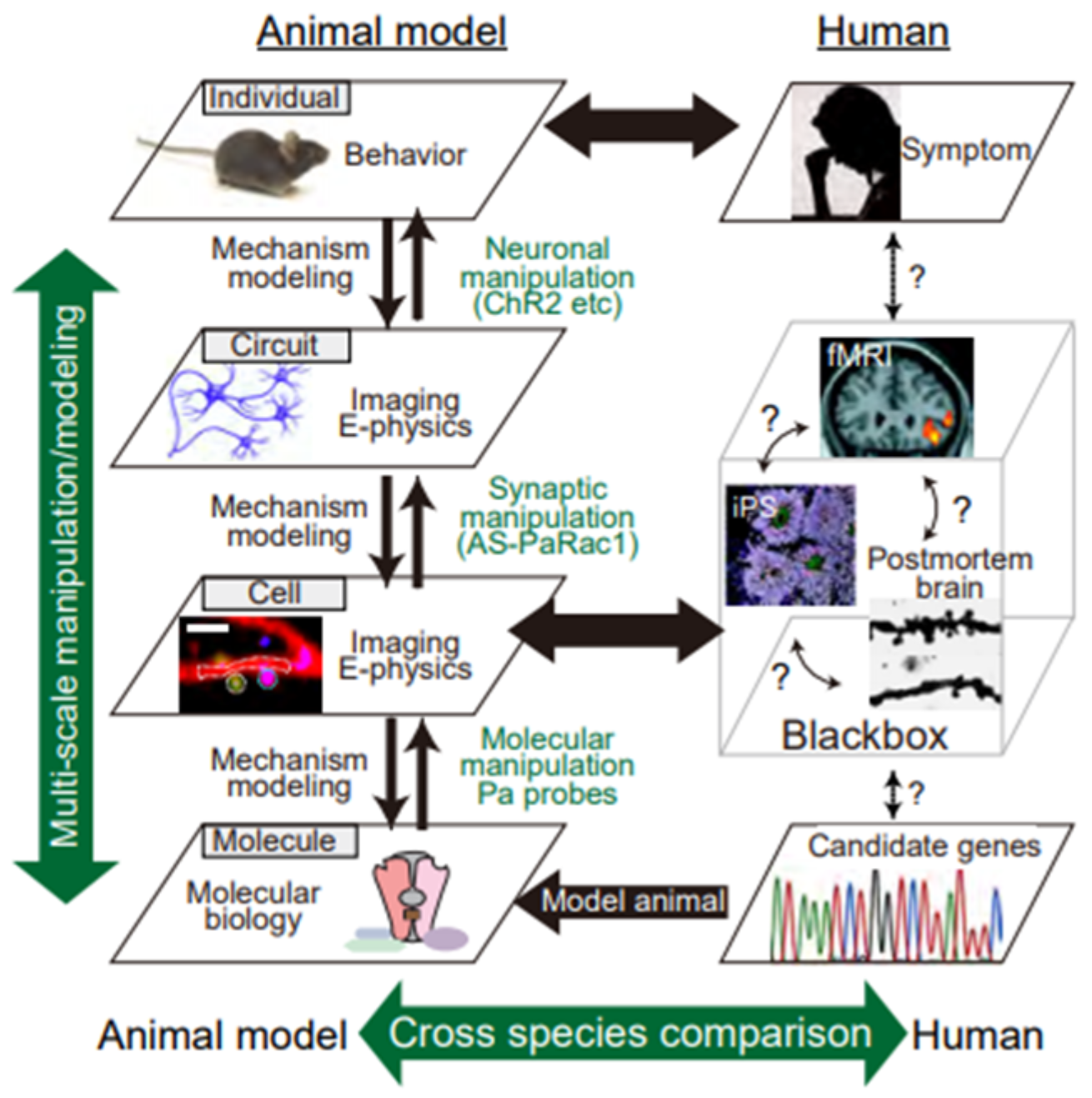
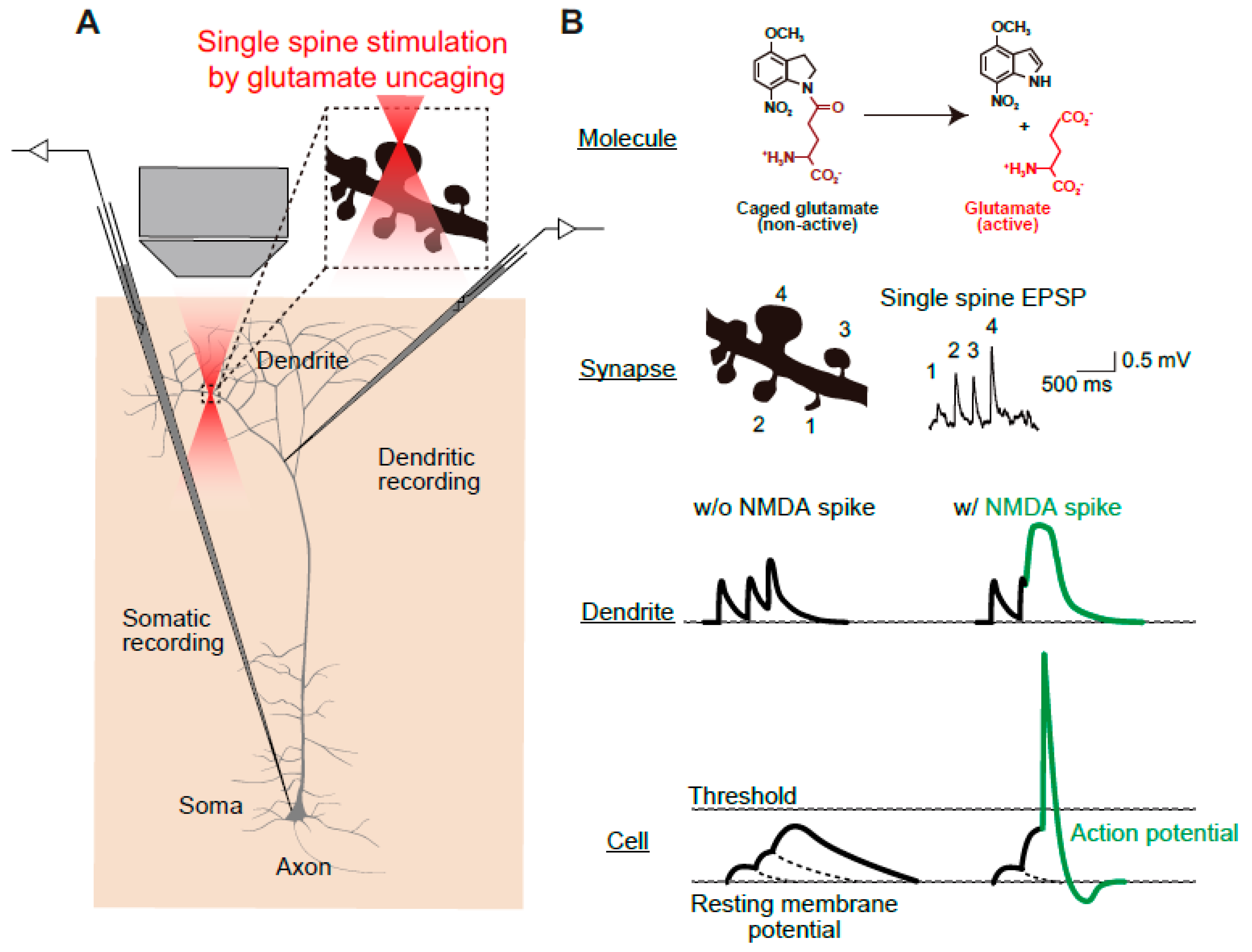
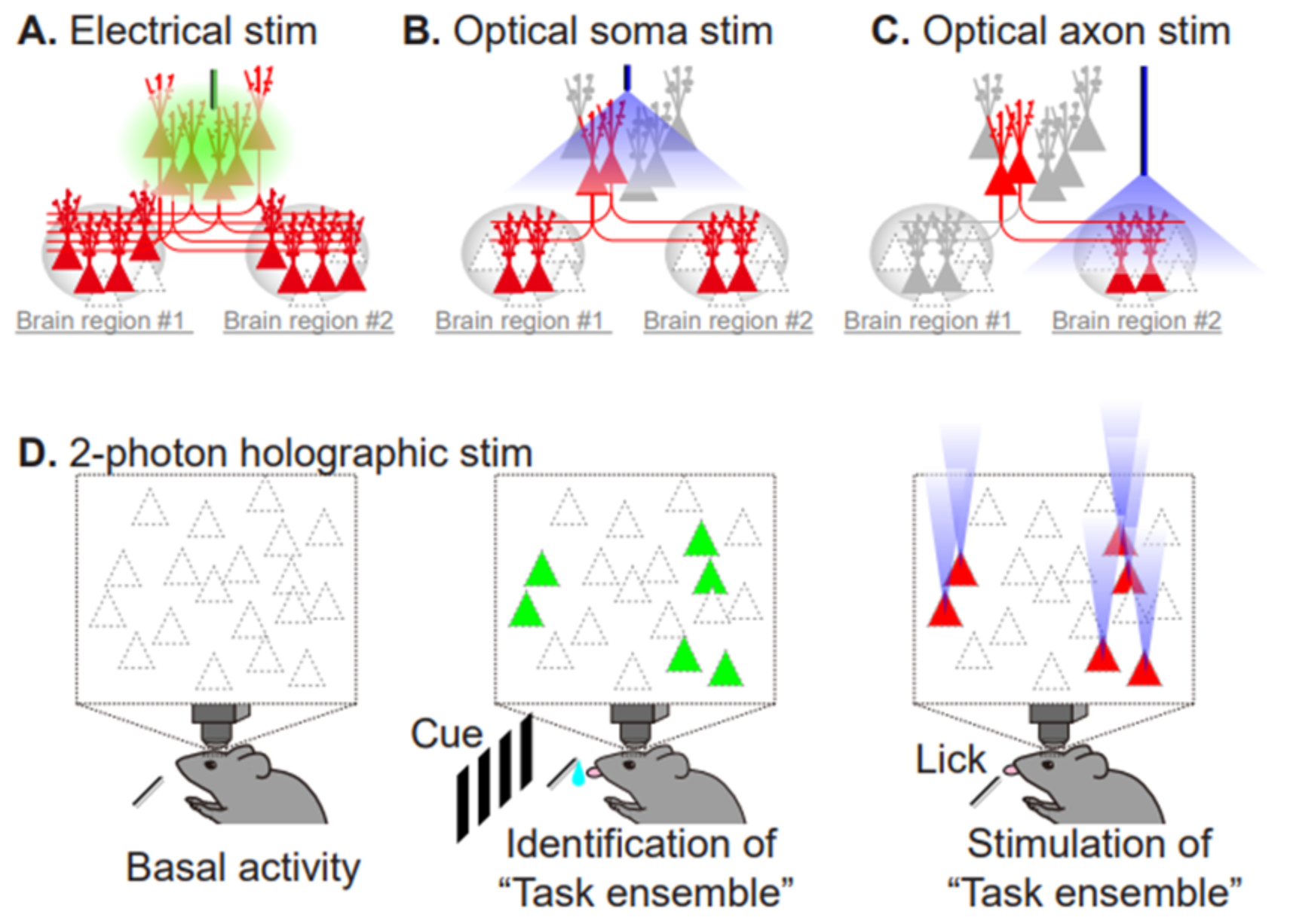
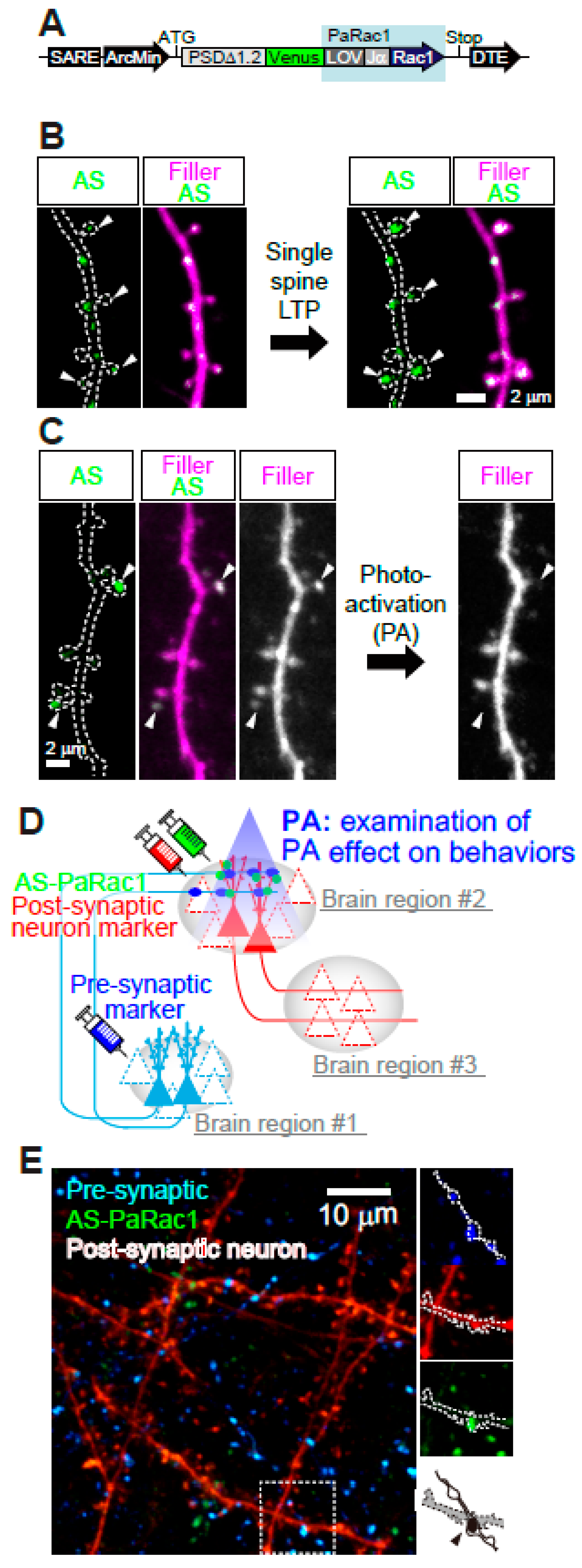
4. Utilization of Animal Models to Gain Insight into NMDAR-Dependent Synaptic Pathology at the Circuit Level
5. Closing Remarks/Conclusions
Funding
Conflicts of Interest
References
- Hayashi-Takagi, A. Synapse pathology and translational applications for schizophrenia. Neurosci. Res. 2017, 114, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dennison, C.A.; Legge, S.E.; Pardiñas, A.F.; Walters, J.T.R. Genome-wide association studies in schizophrenia: Recent advances, challenges and future perspective. Schizophr. Res. 2020, 217, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Smeland, O.B.; Frei, O.; Dale, A.M.; Andreassen, O.A. The polygenic architecture of schizophrenia—Rethinking pathogenesis and nosology. Nat. Rev. Neurol. 2020, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Hayashi-Takagi, A. Optical interrogation of multi-scale neuronal plasticity underlying behavioral learning. Curr. Opin. Neurobiol. 2021, 67, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Isaac, J.T.R.; Wang, Y.T. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 952–962. [Google Scholar] [CrossRef]
- Hashimoto, A.; Nishikawa, T.; Oka, T.; Takahashi, K. Endogenous d-Serine in Rat Brain: N-Methyl-d-Aspartate Receptor-Related Distribution and Aging. J. Neurochem. 1993, 60, 783–786. [Google Scholar] [CrossRef]
- Mothet, J.-P.; Parent, A.T.; Wolosker, H.; Brady, R.O.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.-P.; Oliet, S.H. Synaptic and Extrasynaptic NMDA Receptors Are Gated by Different Endogenous Coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Bannerman, D.M.; Sprengel, R.; Sanderson, D.J.; McHugh, S.B.; Rawlins, J.N.P.; Monyer, H.; Seeburg, P.H. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 2014, 15, 181–192. [Google Scholar] [CrossRef]
- Major, G.; Larkum, M.E.; Schiller, J. Active Properties of Neocortical Pyramidal Neuron Dendrites. Annu. Rev. Neurosci. 2013, 36, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Larkum, M.E.; Nevian, T.; Sandler, M.; Polsky, A.; Schiller, J. Synaptic Integration in Tuft Dendrites of Layer 5 Pyramidal Neurons: A New Unifying Principle. Science 2009, 325, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.M.; Shai, A.S.; E Reeve, J.; Anderson, H.L.; Paulsen, O.; Larkum, M.E. NMDA spikes enhance action potential generation during sensory input. Nat. Neurosci. 2014, 17, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.; Schiller, Y.; Stuart, G.; Sakmann, B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J. Physiol. 1997, 505, 605–616. [Google Scholar] [CrossRef]
- Stuart, G.J.; Schiller, J.; Sakmann, B. Action potential initiation and propagation in rat neocortical pyramidal neurons. J. Physiol. 1997, 505, 617–632. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Ellis-Davies, G.C.R.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001, 4, 1086–1092. [Google Scholar] [CrossRef]
- Obi-Nagata, K.; Temma, Y.; Hayashi-Takagi, A. Synaptic functions and their disruption in schizophrenia: From clinical evidence to synaptic optogenetics in an animal model. Proc. Jpn. Acad. Ser. B 2019, 95, 179–197. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [CrossRef]
- Javitt, D.C. Glutamate and Schizophrenia: Phencyclidine, N-Methyl-d-Aspartate Receptors, and Dopamine-Glutamate Interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar] [CrossRef]
- Moghaddam, B. Reversal of Phencyclidine Effects by a Group II Metabotropic Glutamate Receptor Agonist in Rats. Science 1998, 281, 1349–1352. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Armangué, T.; Planagumà, J.; Radosevic, M.; Mannara, F.; Leypoldt, F.; Geis, C.; Lancaster, E.; Titulaer, M.J.; Rosenfeld, M.R.; et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: Mechanisms and models. Lancet Neurol. 2019, 18, 1045–1057. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; E Rossi, J.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Pearlman, D.; Najjar, S. Meta-analysis of the association between N-methyl-d-aspartate receptor antibodies and schizophrenia, schizoaffective disorder, bipolar disorder, and major depressive disorder. Schizophr. Res. 2014, 157, 249–258. [Google Scholar] [CrossRef]
- Dempster, K.; Jeon, P.; MacKinley, M.; Williamson, P.; Théberge, J.; Palaniyappan, L. Early treatment response in first episode psychosis: A 7-T magnetic resonance spectroscopic study of glutathione and glutamate. Mol. Psychiatry 2020, 25, 1640–1650. [Google Scholar] [CrossRef]
- Shah, P.; Plitman, E.; Iwata, Y.; Kim, J.; Nakajima, S.; Chan, N.; Brown, E.E.; Caravaggio, F.; Torres, E.; Hahn, M.; et al. Glutamatergic neurometabolites and cortical thickness in treatment-resistant schizophrenia: Implications for glutamate-mediated excitotoxicity. J. Psychiatr. Res. 2020, 124, 151–158. [Google Scholar] [CrossRef]
- Wenneberg, C.; Glenthøj, B.Y.; Hjorthøj, C.R.; Zingenberg, F.J.B.; Glenthøj, L.B.; Rostrup, E.; Broberg, B.V.; Nordentoft, M. Cerebral glutamate and GABA levels in high-risk of psychosis states: A focused review and meta-analysis of 1H-MRS studies. Schizophr. Res. 2020, 215, 38–48. [Google Scholar] [CrossRef]
- Garey, L.J.; Ong, W.Y.; Patel, T.S.; Kanani, M.; Davis, A.; Mortimer, A.M.; E Barnes, T.R.; Hirsch, S.R. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 1998, 65, 446–453. [Google Scholar] [CrossRef]
- Glantz, L.A.; Lewis, D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef]
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. Jama Psychiatry 2014, 71, 1323–1331. [Google Scholar] [CrossRef]
- Kolluri, N.; Sun, Z.; Sampson, A.R.; A Lewis, D. Lamina-Specific Reductions in Dendritic Spine Density in the Prefrontal Cortex of Subjects with Schizophrenia. Am. J. Psychiatry 2005, 162, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Law, A.J.; Weickert, C.S.; Hyde, T.M.; Kleinman, J.E.; Harrison, P.J. Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: Molecular evidence for a pathology of dendritic spines. Am. J. Psychiatry 2004, 161, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Kass, K.E.; Thomas, E.A. Chronic haloperidol treatment results in a decrease in the expression of myelin/oligodendrocyte-related genes in the mouse brain. J. Neurosci. Res. 2007, 85, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Humphries, C.; Mortimer, A.; Hirsch, S.; De Belleroche, J.S. NMDA receptor mRNA correlation with antemortem cognitive impairment in schizophrenia. Neuroreport 1996, 7, 2051–2055. [Google Scholar] [CrossRef]
- O’Neill, G.C.; Sengupta, A.; Asghar, M.; Barratt, E.L.; Besle, J.; Schluppeck, D.; Francis, S.T.; Panchuelo, R.M.S. A probabilistic atlas of finger dominance in the primary somatosensory cortex. Neuroimage 2020, 217, 116880. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hülsmann, J.; Monoranu, C.-M.; Grünblatt, E.; Riederer, P. Neurochemical markers as potential indicators of postmortem tissue quality. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 150, pp. 119–127. [Google Scholar] [CrossRef]
- Bhaduri, A.; Andrews, M.G.; Leon, W.M.; Jung, D.; Shin, D.; Allen, D.; Jung, D.; Schmunk, G.; Haeussler, M.; Salma, J.; et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 2020, 578, 142–148. [Google Scholar] [CrossRef]
- Mohn, A.R.; Gainetdinov, R.; Caron, M.G.; Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef]
- Belforte, J.E.; Zsiros, V.; Sklar, E.R.; Jiang, Z.; Yu, G.; Li, Y.; Quinlan, E.M.; Nakazawa, K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2009, 13, 76–83. [Google Scholar] [CrossRef]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Boil. 2011, 21, 91–103. [Google Scholar] [CrossRef]
- Gerber, D.J.; Hall, D.; Miyakawa, T.; Demars, S.; Gogos, J.A.; Karayiorgou, M.; Tonegawa, S. Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin gamma subunit. Proc. Natl. Acad. Sci. USA 2003, 100, 8993–8998. [Google Scholar] [CrossRef]
- Yamada, K.; Gerber, D.J.; Iwayama, Y.; Ohnishi, T.; Ohba, H.; Toyota, T.; Aruga, J.; Minabe, Y.; Tonegawa, S.; Yoshikawa, T. Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 2815–2820. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chattarji, S.; Barbarosie, M.; Rondi-Reig, L.; Philpot, B.D.; Miyakawa, T.; Bear, M.F.; Tonegawa, S. Forebrain-Specific Calcineurin Knockout Selectively Impairs Bidirectional Synaptic Plasticity and Working/Episodic-like Memory. Cell 2001, 107, 617–629. [Google Scholar] [CrossRef]
- Miyakawa, T.; Leiter, L.M.; Gerber, D.J.; Gainetdinov, R.; Sotnikova, T.D.; Zeng, H.; Caron, M.G.; Tonegawa, S. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc. Natl. Acad. Sci. USA 2003, 100, 8987–8992. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, H.; Hayashi-Takagi, A.; Nagaoka, A.; Negishi, M.; Ucar, H.; Yagishita, S.; Ishii, K.; Toyoizumi, T.; Fox, K.; Kasai, H. Calcineurin knockout mice show a selective loss of small spines. Neurosci. Lett. 2018, 671, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, M.L.; Alhassan, J.; Newman, J.T.; Richard, M.; Gu, H.; Kelly, R.M.; Sampson, A.R.; Fish, K.N.; Penzes, P.; Wills, Z.P.; et al. Selective Loss of Smaller Spines in Schizophrenia. Am. J. Psychiatry 2017, 174, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Brandon, N.J.; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 2011, 12, 707–722. [Google Scholar] [CrossRef]
- Teng, S.; A Thomson, P.; McCarthy, S.; Kramer, M.; Muller, S.; Lihm, J.; Morris, S.; Soares, D.C.; Hennah, W.; Harris, S.; et al. Rare disruptive variants in the DISC1 Interactome and Regulome: Association with cognitive ability and schizophrenia. Mol. Psychiatry 2017, 23, 1270–1277. [Google Scholar] [CrossRef]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.M.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 2010, 13, 327–332. [Google Scholar] [CrossRef]
- Millar, J.K. DISC1 and PDE4B Are Interacting Genetic Factors in Schizophrenia That Regulate cAMP Signaling. Science 2005, 310, 1187–1191. [Google Scholar] [CrossRef]
- Kuroda, K.; Yamada, S.; Tanaka, M.; Iizuka, M.; Yano, H.; Mori, D.; Tsuboi, D.; Nishioka, T.; Namba, T.; Iizuka, Y.; et al. Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum. Mol. Genet. 2011, 20, 4666–4683. [Google Scholar] [CrossRef]
- Koike, H.; Arguello, P.A.; Kvajo, M.; Karayiorgou, M.; Gogos, J.A. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3693–3697. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.S.; Zhang, F.; Bamberg, E.; Nagel, G.; Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 2005, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Boyden, E.S. Multiple-Color Optical Activation, Silencing, and Desynchronization of Neural Activity, with Single-Spike Temporal Resolution. PLoS ONE 2007, 2, e299. [Google Scholar] [CrossRef] [PubMed]
- Shirai, F.; Hayashi-Takagi, A.; Msc, F.S. Optogenetics: Applications in psychiatric research. Psychiatry Clin. Neurosci. 2017, 71, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Tye, K.M.; Mirzabekov, J.J.; Warden, M.R.; Ferenczi, E.A.; Tsai, H.-C.; Finkelstein, J.; Kim, S.-Y.; Adhikari, A.; Thompson, K.R.; Andalman, A.S.; et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 2012, 493, 537–541. [Google Scholar] [CrossRef]
- Witten, I.B.; Lin, S.-C.; Brodsky, M.; Prakash, R.; Diester, I.; Anikeeva, P.; Gradinaru, V.; Ramakrishnan, C.; Deisseroth, K. Cholinergic Interneurons Control Local Circuit Activity and Cocaine Conditioning. Science 2010, 330, 1677–1681. [Google Scholar] [CrossRef]
- Namburi, P.; Beyeler, A.; Yorozu, S.; Calhoon, G.G.; Halbert, S.A.; Wichmann, R.; Holden, S.S.; Mertens, K.L.; Anahtar, M.; Felix-Ortiz, A.C.; et al. A circuit mechanism for differentiating positive and negative associations. Nature 2015, 520, 675–678. [Google Scholar] [CrossRef]
- Ahmari, S.E.; Spellman, T.; Douglass, N.L.; Kheirbek, M.A.; Simpson, H.B.; Deisseroth, K.; Gordon, J.A.; Hen, R. Repeated Cortico-Striatal Stimulation Generates Persistent OCD-Like Behavior. Science 2013, 340, 1234–1239. [Google Scholar] [CrossRef]
- Tye, K.M.; Prakash, R.; Kim, S.-Y.; Fenno, L.E.; Grosenick, L.; Zarabi, H.; Thompson, K.R.; Gradinaru, V.; Ramakrishnan, C.; Deisseroth, K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 2011, 471, 358–362. [Google Scholar] [CrossRef]
- Carrillo-Reid, L.; Han, S.; Yang, W.; Akrouh, A.; Yuste, R. Controlling Visually Guided Behavior by Holographic Recalling of Cortical Ensembles. Cell 2019, 178, 447–457.e5. [Google Scholar] [CrossRef]
- Hoshiba, Y.; Wada, T.; Hayashi-Takagi, A. Synaptic Ensemble Underlying the Selection and Consolidation of Neuronal Circuits during Learning. Front. Neural Circuits 2017, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Takagi, A.; Yagishita, S.; Nakamura, M.; Shirai, F.; Wu, Y.I.; Loshbaugh, A.L.; Kuhlman, B.; Hahn, K.M.; Kasai, H. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 2015, 525, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.E.; Milner, P.M. The legacy of Donald O. Hebb: More than the Hebb Synapse. Nat. Rev. Neurosci. 2003, 4, 1013–1019. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyun, J.S.; Inoue, T.; Hayashi-Takagi, A. Multi-Scale Understanding of NMDA Receptor Function in Schizophrenia. Biomolecules 2020, 10, 1172. https://doi.org/10.3390/biom10081172
Hyun JS, Inoue T, Hayashi-Takagi A. Multi-Scale Understanding of NMDA Receptor Function in Schizophrenia. Biomolecules. 2020; 10(8):1172. https://doi.org/10.3390/biom10081172
Chicago/Turabian StyleHyun, Jo Soo, Takafumi Inoue, and Akiko Hayashi-Takagi. 2020. "Multi-Scale Understanding of NMDA Receptor Function in Schizophrenia" Biomolecules 10, no. 8: 1172. https://doi.org/10.3390/biom10081172
APA StyleHyun, J. S., Inoue, T., & Hayashi-Takagi, A. (2020). Multi-Scale Understanding of NMDA Receptor Function in Schizophrenia. Biomolecules, 10(8), 1172. https://doi.org/10.3390/biom10081172