Single and Combined Methods to Specifically or Bulk-Purify RNA–Protein Complexes


Abstract
1. Opening Doors to the RNA-Binding Proteome
1.1. Why Study These Proteins? What New Exciting Insights Can Such Study Reveal?
1.2. The Study of the Ribonome: Where Are We Now?
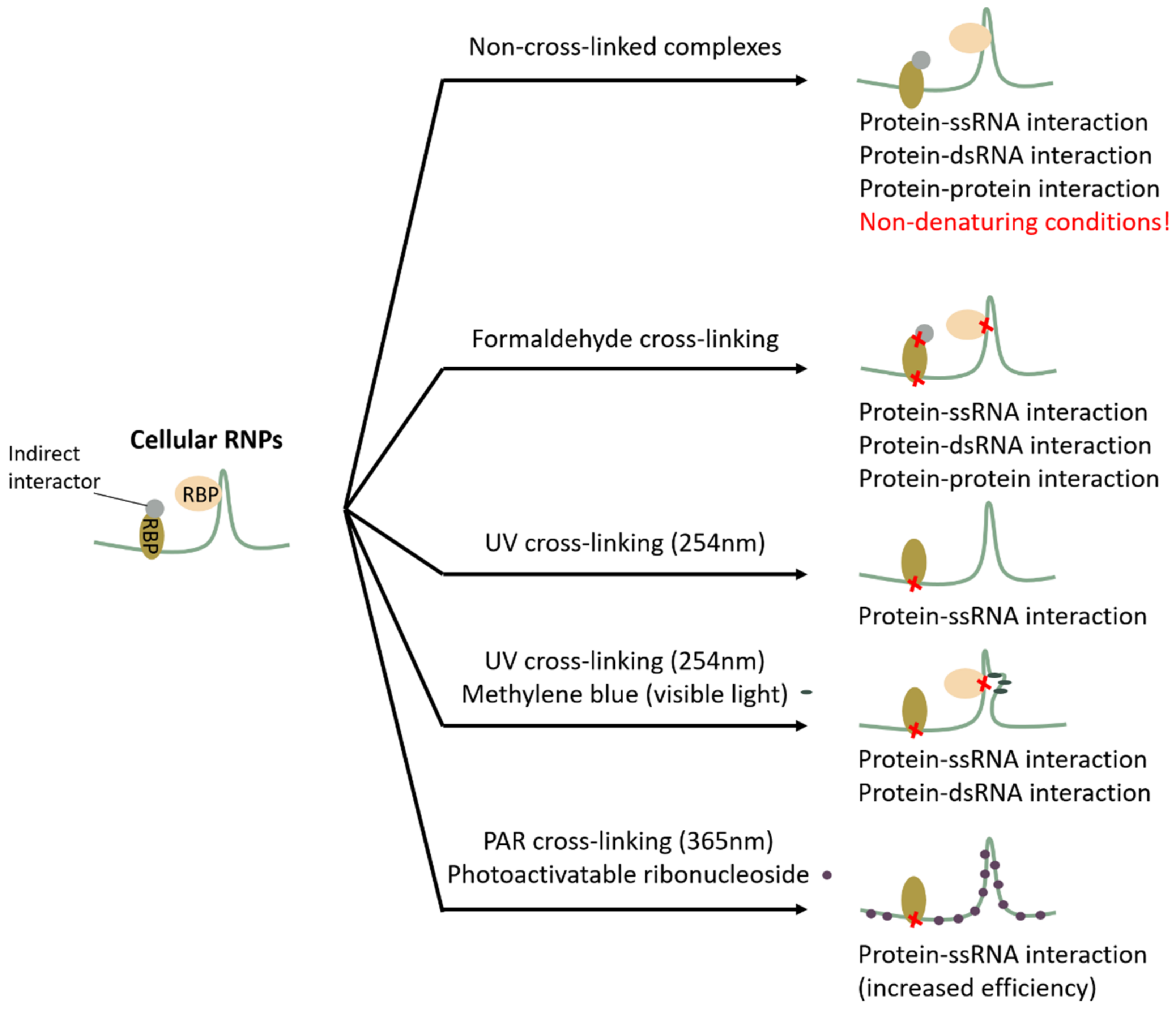
2. Cross-Linking Proteins to RNA or Not, and How?
2.1. Non-Cross-Linked Complexes: A Good Idea?
2.2. Formaldehyde as a Promiscuous Cross-Linker
2.3. UV Light as a Specific but Low-Efficiency Cross-Linker
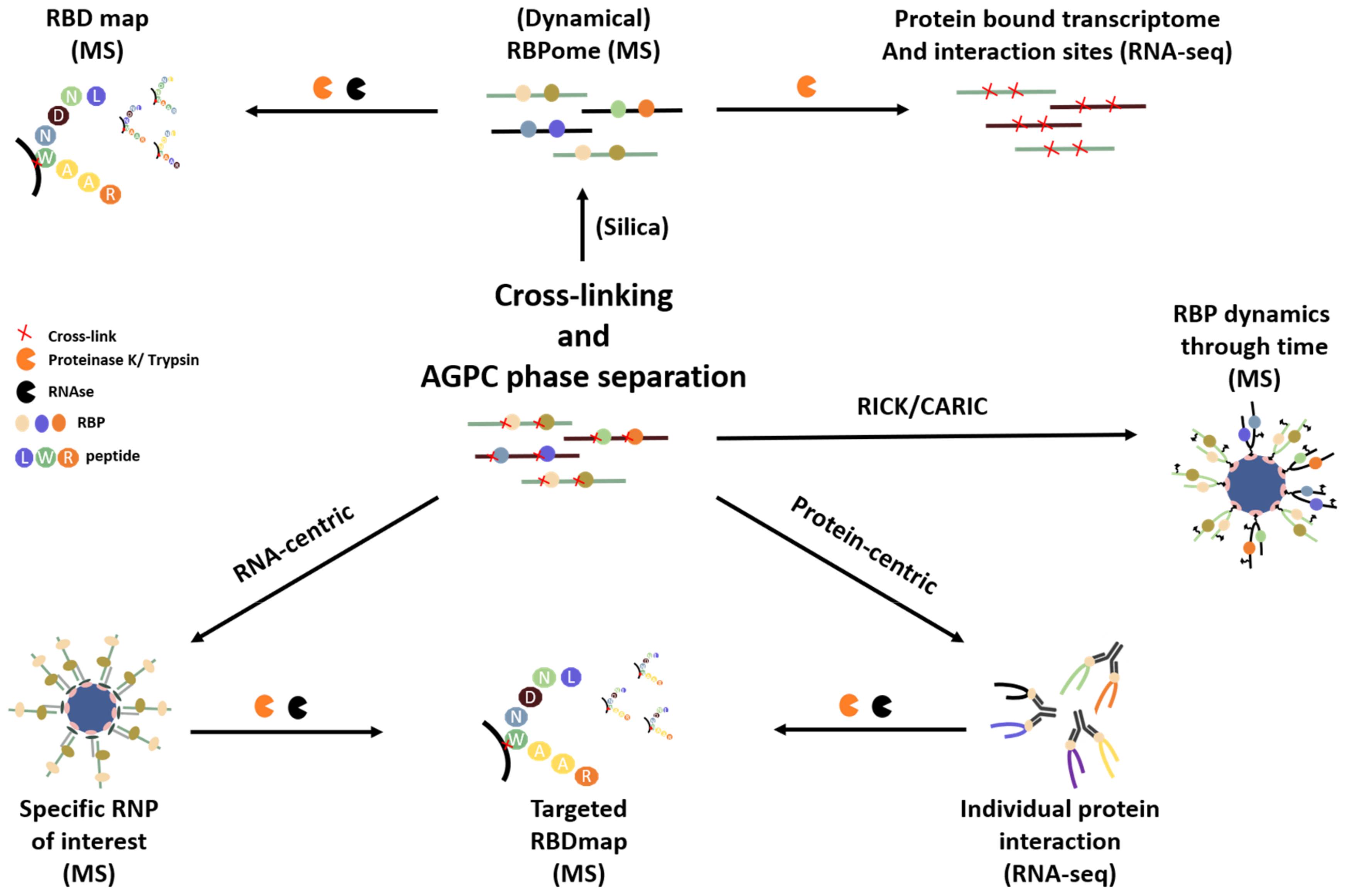
3. Overview of Available RNA- and RNP-Centric Purification Methods
3.1. Isolating the RNA-Bound Proteome to Study RNP Dynamics
3.1.1. Affinity-Based Separation
(Enhanced/Comparative) RNA Interactome Capture ((e/c) RIC)
RNA Interactome Using Click Chemistry (RICK) and Click Chemistry-Assisted RNA-Interactome Capture (CARIC)
3.1.2. Solid-Phase Separation
Complex Capture or 2C Method
Total RNA-Associated Protein Purification (TRAPP) and iTRAPP
Viral Cross-Linking and Solid Phase Purification (VIR-CLASP)
3.1.3. Phase Separation
Protein-Cross-Linked RNA eXtraction (XRNAX)
Orthogonal Organic Phase Separation (OOPS)
Phenol-Toluol Extraction (PTex)
3.2. Targeting a Specific RNA of Interest to Study Its Interacting Proteins
3.2.1. Modified/Tagged RNA Based Methods
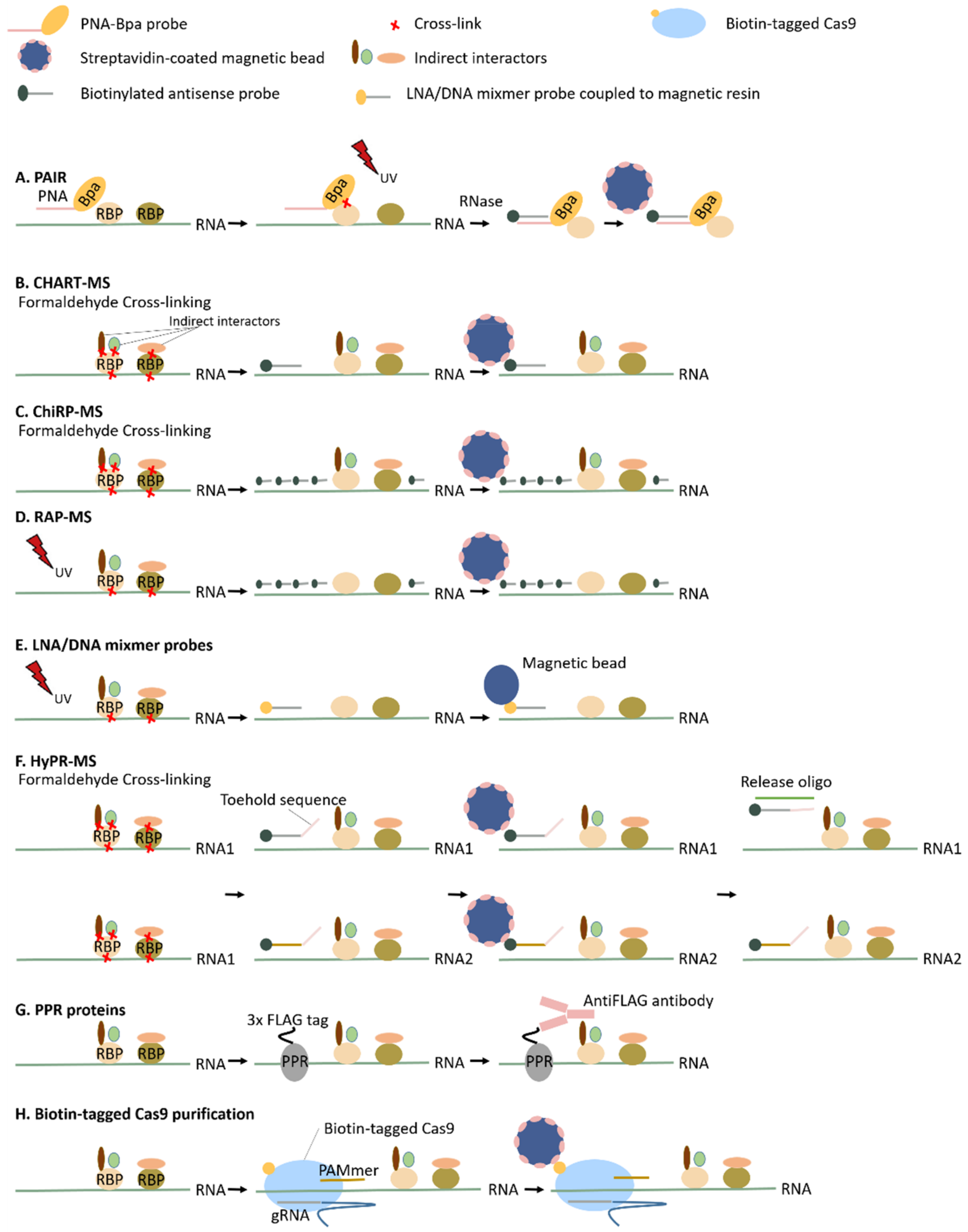
3.2.2. Antisense Probe-Based Methods
Peptide Nucleic Acid (PNA)-Assisted Identification of RBPs (PAIR)
Capture Hybridization Analysis of RNA Targets (CHART), Chromatin Isolation by RNA Purification (ChIRP) and RNA Antisense Purification (RAP)
Specific RNP Capture with Antisense LNA (Locked Nucleic Acid)/DNA Mixmers
Hybridization Purification of RNA–Protein Complexes Followed by Mass Spectrometry (HyPR-MS)
Expression of Proteins That Are Guided to or Recognize Specific RNA Molecules (PPR Proteins and Biotin-Tagged Cas9 Purification)
4. Overcoming Cost, Yield and Specificity Challenges by Combining Methods
5. Standardized Data Analysis and Visualization
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Mansfield, K.D.; Keene, J.D. The ribonome: A dominant force in co-ordinating gene expression. Biol. Cell 2009, 101, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Perez-Perri, J.I.; Rogell, B.; Schwarzl, T.; Stein, F.; Zhou, Y.; Rettel, M.; Brosig, A.; Hentze, M.W. Discovery of RNA-binding proteins and characterization of their dynamic responses by enhanced RNA interactome capture. Nat. Commun. 2018, 9, 4408. [Google Scholar] [CrossRef]
- Sysoev, V.O.; Fischer, B.; Frese, C.K.; Gupta, I.; Krijgsveld, J.; Hentze, M.W.; Castello, A.; Ephrussi, A. Global changes of the RNA-bound proteome during the maternal-to-zygotic transition in Drosophila. Nat. Commun. 2016, 7, 12128. [Google Scholar] [CrossRef]
- Garcia-Moreno, M.; Noerenberg, M.; Ni, S.; Järvelin, A.I.; González-Almela, E.; Lenz, C.E.; Bach-Pages, M.; Cox, V.; Avolio, R.; Davis, T.; et al. System-wide Profiling of RNA-Binding Proteins Uncovers Key Regulators of Virus Infection. Mol. Cell 2019, 74, 196–211.e11. [Google Scholar] [CrossRef]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef]
- Lukong, K.E.; Chang, K.W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef]
- Allerson, C.R.; Cazzola, M.; Rouault, T.A. Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia-cataract syndrome. J. Biol. Chem. 1999, 274, 26439–26447. [Google Scholar] [CrossRef]
- Knoener, R.A.; Becker, J.T.; Scalf, M.; Sherer, N.M.; Smith, L.M. Elucidating the in vivo interactome of HIV-1 RNA by hybridization capture and mass spectrometry. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Phillips, S.L.; Garcia-Blanco, M.A.; Bradrick, S.S. Antisense-mediated affinity purification of dengue virus ribonucleoprotein complexes from infected cells. Methods 2015, 91, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ooi, Y.S.; Majzoub, K.; Flynn, R.A.; Mata, M.A.; Diep, J.; Li, J.K.; van Buuren, N.; Rumachik, N.; Johnson, A.G.; Puschnik, A.S.; et al. An RNA-centric dissection of host complexes controlling flavivirus infection. Nat. Microbiol. 2019, 4, 2369–2382. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nagy, P.D. Diverse roles of host RNA-binding proteins in RNA virus replication. RNA Biol. 2011, 8, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.U.; Paek, K.-H. Plant RNA binding proteins for control of RNA virus infection. Front. Physiol. 2013, 4, 1–5. [Google Scholar] [CrossRef]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP Identifies Nova-Regulated RNA Networks in the Brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Ule, J.; Jensen, K.; Mele, A.; Darnell, R.B. CLIP: A method for identifying protein–RNA interaction sites in living cells. Methods 2005, 37, 376–386. [Google Scholar] [CrossRef]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403.e19. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, R.M.L.; Smith, T.; Villanueva, E.; Marti-Solano, M.; Monti, M.; Pizzinga, M.; Mirea, D.M.; Ramakrishna, M.; Harvey, R.F.; Dezi, V.; et al. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 2019, 37, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Urdaneta, E.C.; Beckmann, B.M. Fast and unbiased purification of RNA-protein complexes after UV cross-linking. Methods 2020, 178, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, B.M. RNA interactome capture in yeast. Methods 2017, 118–119, 82–92. [Google Scholar] [CrossRef]
- Wheeler, E.C.; van Nostrand, E.L.; Yeo, G.W. Advances and challenges in the detection of transcriptome-wide protein-RNA interactions. Wiley Interdiscip. Rev. RNA 2018, 9. [Google Scholar] [CrossRef]
- Mili, S.; Steitz, J.A. Evidence for reassociation of RNA-binding proteins after cell lysis: Implications for the interpretation of immunoprecipitation analyses. RNA 2004, 10, 1692–1694. [Google Scholar] [CrossRef]
- Gilbert, C.; Svejstrup, J.Q. RNA Immunoprecipitation for Determining RNA-Protein Associations In Vivo. Curr. Protoc. Mol. Biol. 2006. [Google Scholar] [CrossRef]
- Patton, R.D.; Sanjeev, M.; Woodward, L.A.; Mabin, J.W.; Bundschuh, R.; Singh, G. Chemical crosslinking enhances RNA immunoprecipitation for efficient identification of binding sites of proteins that photo-crosslink poorly with RNA. RNA 2020. [Google Scholar] [CrossRef]
- BÄUMERT, H.G.; SKÖLD, S.-E.; KURLAND, C.G. RNA-Protein Neighbourhoods of the Ribosome Obtained by Crosslinking. Eur. J. Biochem. 1978, 89, 353–359. [Google Scholar] [CrossRef]
- Nygård, O.; Nika, H. Identification by RNA-protein cross-linking of ribosomal proteins located at the interface between the small and the large subunits of mammalian ribosomes. EMBO J. 1982, 1, 357–362. [Google Scholar] [CrossRef]
- Datta, B.; Weiner, A.M. Cross-linking of U2 snRNA using nitrogen mustard: Evidence for higher order structure. J. Biol. Chem. 1992, 267, 4497–4502. [Google Scholar] [PubMed]
- Sutherland, B.W.; Toews, J.; Kast, J. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J. Mass Spectrom. 2008, 43, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Meisenheimer, K.M.; Koch, T.H. Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 101–140. [Google Scholar] [CrossRef] [PubMed]
- Shetlar, M.D. Cross-Linking of Proteins to Nucleic Acids by Ultraviolet Light. In Photochemical and Photobiological Reviews; Smith, K.C., Ed.; Springer: Boston, MA, USA, 1980; Volume 5, pp. 105–197. [Google Scholar]
- Liu, Z.R.; Wilkie, A.M.; Clemens, M.J.; Smith, C.W.J. Detection of double-stranded RNA-protein interactions by methylene blue- mediated photo-crosslinking. RNA 1996, 2, 611–621. [Google Scholar] [PubMed]
- Wower, I.; Wower, J.; Meinke, M.; Brimacombe, R. The use of 2-iminothiolane as an RNA-protein cross-linking agent in Escherichia coli ribosomes, and the localisation on 23S RNA of sites cross-linked to proteins L4, L6, L21, L23, L27 and L29. Nucleic Acids Res. 1981, 9, 4285–4302. [Google Scholar] [CrossRef]
- Urlaub, H.; Kruft, V.; Bischof, O.; Müller, E.C.; Wittmann-Liebold, B. Protein-rRNA binding features and their structural and functional implications in ribosomes as determined by cross-linking studies. EMBO J. 1995, 14, 4578–4588. [Google Scholar] [CrossRef]
- Zaman, U.; Richter, F.M.; Hofele, R.; Kramer, K.; Sachsenberg, T.; Kohlbacher, O.; Lenz, C.; Urlaub, H. Dithiothreitol (DTT) acts as a specific, UV-inducible cross-linker in elucidation of protein-RNA interactions. Mol. Cell. Proteom. 2015, 14, 3196–3210. [Google Scholar] [CrossRef]
- Zhang, Z.; Boonen, K.; Ferrari, P.; Schoofs, L.; Janssens, E.; Noort, V.; Rolland, F.; Geuten, K. UV crosslinked mRNA-binding proteins captured from leaf mesophyll protoplasts. Plant Methods 2016, 12, 1–12. [Google Scholar] [CrossRef]
- Darnell, R.B. HITS-CLIP: Panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. PAR-CliP-A method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. 2010, 2034. [Google Scholar] [CrossRef]
- Kramer, K.; Sachsenberg, T.; Beckmann, B.M.; Qamar, S.; Boon, K.L.; Hentze, M.W.; Kohlbacher, O.; Urlaub, H. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 2014, 11, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Shchepachev, V.; Bresson, S.; Spanos, C.; Petfalski, E.; Fischer, L.; Rappsilber, J.; Tollervey, D. Defining the RNA interactome by total RNA-associated protein purification. Mol. Syst. Biol. 2019, 15, e8689. [Google Scholar] [CrossRef]
- Beckstead, A.A.; Zhang, Y.; de Vries, M.S.; Kohler, B. Life in the light: Nucleic acid photoproperties as a legacy of chemical evolution. Phys. Chem. Chem. Phys. 2016, 18, 24228–24238. [Google Scholar] [CrossRef]
- Zou, X.; Dai, X.; Liu, K.; Zhao, H.; Song, D.; Su, H. Photophysical and photochemical properties of 4-thiouracil: Time-resolved ir spectroscopy and DFT studies. J. Phys. Chem. B 2014, 118, 5864–5872. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.C.; Yi, H.; Eichelbaum, K.; Föhr, S.; Fischer, B.; You, K.T.; Castello, A.; Krijgsveld, J.; Hentze, M.W.; Kim, V.N. The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Castello, A.; Fischer, B.; Leicht, S.; Föehr, S.; Frese, C.K.; Ragan, C.; Kurscheid, S.; Pagler, E.; Yang, H.; et al. The Cardiomyocyte RNA-Binding Proteome: Links to Intermediary Metabolism and Heart Disease. Cell Rep. 2016, 16, 1456–1469. [Google Scholar] [CrossRef]
- Marondedze, C.; Thomas, L.; Serrano, N.L.; Lilley, K.S.; Gehring, C. The RNA-binding protein repertoire of Arabidopsis thaliana. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Marondedze, C.; Thomas, L.; Gehring, C.; Lilley, K.S. Changes in the Arabidopsis RNA-binding proteome reveal novel stress response mechanisms. BMC Plant Biol. 2019, 19, 139. [Google Scholar] [CrossRef]
- Lueong, S.; Merce, C.; Fischer, B.; Hoheisel, J.D.; Erben, E.D. Gene expression regulatory networks in Trypanosoma brucei: Insights into the role of the mRNA-binding proteome. Mol. Microbiol. 2016, 100, 457–471. [Google Scholar] [CrossRef]
- De Pablos, L.M.; Ferreira, T.R.; Dowle, A.A.; Forrester, S.; Parry, E.; Newling, K.; Walrad, P.B. The mRNA-bound Proteome of Leishmania mexicana: Novel genetic insight into an ancient parasite. Mol. Cell Proteom. 2019, 18, 1271–1284. [Google Scholar] [CrossRef]
- Bunnik, E.M.; Batugedara, G.; Saraf, A.; Prudhomme, J.; Florens, L.; le Roch, K.G. The mRNA-bound proteome of the human malaria parasite Plasmodium falciparum. Genome Biol. 2016, 17, 147. [Google Scholar] [CrossRef]
- Matia-González, A.M.; Laing, E.E.; Gerber, A.P. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 2015, 22, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.F.; Jain, S.; She, M.; Parker, R. Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 2013, 20, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, B.M.; Horos, R.; Fischer, B.; Castello, A.; Eichelbaum, K.; Alleaume, A.M.; Schwarzl, T.; Curk, T.; Foehr, S.; Huber, W.; et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015, 6, 10127. [Google Scholar] [CrossRef] [PubMed]
- Wessels, H.H.; Imami, K.; Baltz, A.G.; Kolinski, M.; Beldovskaya, A.; Selbach, M.; Small, S.; Ohler, U.; Landthaler, M. The mRNA-bound proteome of the early fly embryo. Genome Res. 2016, 26, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Despic, V.; Dejung, M.; Gu, M.; Krishnan, J.; Zhang, J.; Herzel, L.; Straube, K.; Gerstein, M.B.; Butter, F.; Neugebauer, K.M. Dynamic RNA-protein interactions underlie the zebrafish maternal-to-zygotic transition. Genome Res. 2017, 27, 1184–1194. [Google Scholar] [CrossRef]
- Köster, T.; Reichel, M.; Staiger, D. CLIP and RNA interactome studies to unravel genome-wide RNA-protein interactions in vivo in Arabidopsis thaliana. Methods 2020, 178, 63–71. [Google Scholar] [CrossRef]
- Reichel, M.; Liao, Y.; Rettel, M.; Ragan, C.; Evers, M.; Alleaume, A.M.; Horos, R.; Hentze, M.W.; Preiss, T.; Millar, A.A. In planta determination of the mRNA-binding proteome of arabidopsis etiolated seedlings. Plant Cell 2016, 28, 2435–2452. [Google Scholar] [CrossRef]
- Bach-Pages, M.; Homma, F.; Kourelis, J.; Kaschani, F.; Mohammed, S.; Kaiser, M.; van der Hoorn, R.A.L.; Castello, A.; Preston, G.M. Discovering the RNA-binding proteome of plant leaves with an improved RNA interactome capture method. Biomolecules 2020, 10, 661. [Google Scholar] [CrossRef]
- Urdaneta, E.C.; Vieira-Vieira, C.H.; Hick, T.; Wessels, H.H.; Figini, D.; Moschall, R.; Medenbach, J.; Ohler, U.; Granneman, S.; Selbach, M.; et al. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 2019, 10, 990. [Google Scholar] [CrossRef]
- Marchese, D.; de Groot, N.S.; Lorenzo Gotor, N.; Livi, C.M.; Tartaglia, G.G. Advances in the characterization of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 2016, 7, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Horos, R.; Strein, C.; Fischer, B.; Eichelbaum, K.; Steinmetz, L.M.; Krijgsveld, J.; Hentze, M.W. Comprehensive identification of RNA-binding proteins by RNA interactome capture. Methods Mol. Biol. 2016, 1358, 131–139. [Google Scholar] [PubMed]
- Huang, R.; Han, M.; Meng, L.; Chen, X. Capture and Identification of RNA-binding Proteins by Using Click Chemistry-assisted RNA-interactome Capture (CARIC) Strategy. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Guo, X.; Yin, M.; Tariq, M.; Lai, Y.; Kanwal, S.; Zhou, J.; Li, N.; Lv, Y.; Pulido-Quetglas, C.; et al. Capturing the interactome of newly transcribed RNA. Nat. Methods 2018, 15, 213–220. [Google Scholar] [CrossRef]
- Asencio, C.; Chatterjee, A.; Hentze, M.W. Silica-based solid-phase extraction of cross-linked nucleic acid–bound proteins. Life Sci. Alliance 2018, 1, e201800088. [Google Scholar] [CrossRef]
- Kim, B.; Arcos, S.; Rothamel, K.; Jian, J.; Rose, K.L.; McDonald, W.H.; Bian, Y.; Reasoner, S.; Barrows, N.J.; Bradrick, S.; et al. Discovery of Widespread Host Protein Interactions with the Pre-replicated Genome of CHIKV Using VIR-CLASP. Mol. Cell 2020, 78, 624–640.e7. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Boom, R.; Sol, C.J.; Salimans, M.M.; Jansen, C.L.; Wertheim-van Dillen, P.M.; van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [CrossRef]
- Mendes, M.L.; Fischer, L.; Chen, Z.A.; Barbon, M.; O’Reilly, F.J.; Giese, S.H.; Bohlke-Schneider, M.; Belsom, A.; Dau, T.; Combe, C.W.; et al. An integrated workflow for crosslinking mass spectrometry. Mol. Syst. Biol. 2019, 15, e8994. [Google Scholar] [CrossRef]
- Javorovic, M.; Pohla, H.; Frankenberger, B.; Wölfel, T.; Schendel, D.J. RNA transfer by electroporation into mature dendritic cells leading to reactivation of effector-memory cytotoxic T lymphocytes: A quantitative analysis. Mol. Ther. 2005, 12, 734–743. [Google Scholar] [CrossRef]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S. el; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug. Deliv. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Faoro, C.; Ataide, S.F. Ribonomic approaches to study the RNA-binding proteome. FEBS Lett. 2014, 588, 3649–3664. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, J.; Kilk, K.; Peritz, T.; Kannanayakal, T.; Miyashiro, K.Y.; Eiríksdóttir, E.; Jochems, J.; Langel, Û.; Eberwine, J. In vivo identification of ribonucleoprotein-RNA interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 1557–1562. [Google Scholar] [CrossRef]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Rogell, B.; Fischer, B.; Rettel, M.; Krijgsveld, J.; Castello, A.; Hentze, M.W. Specific RNP capture with antisense LNA/DNA mixmers. RNA 2017, 23, 1290–1302. [Google Scholar] [CrossRef]
- Spiniello, M.; Knoener, R.; Steinbrink, M.; Yang, B.; Cesnik, A.; Buxton, K.; Scalf, M.; Jarrard, D.; Smith, L. HyPR-MS for Multiplexed Discovery of MALAT1, NEAT1, and NORAD IncRNA Protein Interactomes. J. Proteome Res. 2018, 17, 3022–3038. [Google Scholar] [CrossRef]
- McDermott, J.J.; Watkins, K.P.; Williams-Carrier, R.; Barkan, A. Ribonucleoprotein capture by in vivo expression of a designer pentatricopeptide repeat protein in arabidopsis. Plant Cell 2019, 31, 1723–1733. [Google Scholar] [CrossRef]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef]
- Beach, D.L.; Keene, J.D. Ribotrap: Targeted purification of RNA-specific RNPs from cell lysates through immunoaffinity precipitation to identify regulatory proteins and RNAs. Methods Mol. Biol. 2008, 419, 69–91. [Google Scholar]
- Simmonds, H.M.K.J. TRAP-Tagging: A Novel Method for the Identification and Purification of RNA-Protein Complexes. US Patent US2006105341, 7 April 2005. [Google Scholar]
- Tsai, B.P.; Wang, X.; Huang, L.; Waterman, M.L. Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol. Cell. Proteom. 2011, 10, 1–15. [Google Scholar]
- Ramanathan, M.; Majzoub, K.; Rao, D.S.; Neela, P.H.; Zarnegar, B.J.; Mondal, S.; Roth, J.G.; Gai, H.; Kovalski, J.R.; Siprashvili, Z.; et al. RNA–protein interaction detection in living cells. Nat. Methods 2018, 15, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Leppek, K.; Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2014, 42, e13. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. MS2-TRAP: Tagging RNA to identify associated miRNAs. Methods 2012, 58, 81–87. [Google Scholar]
- Lee, H.Y.; Haurwitz, R.E.; Apffel, A.; Zhou, K.; Smart, B.; Wenger, C.D.; Laderman, S.; Bruhn, L.; Doudna, J.A. RNA-protein analysis using a conditional CRISPR nuclease. Proc. Natl. Acad. Sci. USA 2013, 110, 5416–5421. [Google Scholar] [CrossRef]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Ho, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and nontoxic antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 2000, 97, 5633–5638. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Kennedy-Darling, J.; Shortreed, M.R.; Scalf, M.; Gasch, A.P.; Smith, L.M. Multiplexed sequence-specific capture of chromatin and mass spectrometric discovery of associated proteins. Anal. Chem. 2017, 89, 7841–7846. [Google Scholar] [CrossRef]
- Kennedy-Darling, J.; Holden, M.T.; Shortreed, M.R.; Smith, L.M. Multiplexed Programmable Release of Captured DNA. ChemBioChem 2014, 15, 2353–2356. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Rojas, M.; Fujii, S.; Yap, A.; Chong, Y.S.; Bond, C.S.; Small, I. A Combinatorial Amino Acid Code for RNA Recognition by Pentatricopeptide Repeat Proteins. PLoS Genet. 2012, 8, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.D.; McCann, K.L.; Wang, Y.; Fonseca, D.R.; Shukla, T.; Alexander, J.C.; Qiu, C.; Wickens, M.; Lo, T.W.; Tanaka Hall, T.M.; et al. Engineering a conserved RNA regulatory protein repurposes its biological function in vivo. Elife 2019, 8, e43788. [Google Scholar] [CrossRef] [PubMed]
- Campbell, Z.T.; Valley, C.T.; Wickens, M. A protein-RNA specificity code enables targeted activation of an endogenous human transcript.(RESOURCE)(Report). Nat. Struct. Mol. Biol. 2014, 21, 732. [Google Scholar] [CrossRef]
- Colas des Francs-Small, C.; Vincis Pereira Sanglard, L.; Small, I. Targeted cleavage of nad6 mRNA induced by a modified pentatricopeptide repeat protein in plant mitochondria. Commun. Biol. 2018, 1, 166. [Google Scholar] [CrossRef]
- Coquille, S.; Filipovska, A.; Chia, T.; Rajappa, L.; Lingford, J.P.; Razif, M.F.M.; Thore, S.; Rackham, O. An artificial PPR scaffold for programmable RNA recognition. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Kindgren, P.; Yap, A.; Bond, C.S.; Small, I. Predictable alteration of sequence recognition by RNA editing factors from Arabidopsis. Plant Cell 2015, 27, 403. [Google Scholar] [CrossRef]
- Miranda, R.G.; Mcdermott, J.J.; Barkan, A. RNA-binding specificity landscapes of designer pentatricopeptide repeat proteins elucidate principles of PPR-RNA interactions. Nucleic Acids Res. 2018, 46, 2613–2623. [Google Scholar] [CrossRef]
- Yan, J.; Yao, Y.; Hong, S.; Yang, Y.; Shen, C.; Zhang, Q.; Zhang, D.; Zou, T.; Yin, P. Delineation of pentatricopeptide repeat codes for target RNA prediction. Nucleic Acids Res. 2019, 47, 3728–3738. [Google Scholar] [CrossRef]
- Miranda, R.G.; Rojas, M.; Montgomery, M.P.; Gribbin, K.P.; Barkan, A. RNA-binding specificity landscape of the pentatricopeptide repeat protein PPR10. RNA 2017, 23, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Zhang, D.; Guan, Z.; Liu, Y.; Yang, Z.; Yang, Y.; Wang, X.; Wang, Q.; Zhang, Q.X.; Fan, S.; et al. Structural basis for specific single-stranded RNA recognition by designer pentatricopeptide repeat proteins. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Matia-González, A.M.; Iadevaia, V.; Gerber, A.P. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 2017, 118–119, 93–100. [Google Scholar]
- Viktorovskaya, O.V.; Greco, T.M.; Cristea, I.M.; Thompson, S.R. Identification of RNA Binding Proteins Associated with Dengue Virus RNA in Infected Cells Reveals Temporally Distinct Host Factor Requirements. PLoS Negl. Trop. Dis. 2016, 10, e0004921. [Google Scholar] [CrossRef] [PubMed]
- De Troyer, L.; Zhao, P.; Pastor, T.; Baietti, M.F.; Barra, J.; Vendramin, R.; Dok, R.; Lechat, B.; Najm, P.; Van Haver, D.; et al. Stress-induced lncRNA LASTR fosters cancer cell fitness by regulating the activity of the U4/U6 recycling factor SART3. Nucleic Acids Res. 2020, 48, 2502–2517. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Mullari, M.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Specifying RNA-Binding Regions in Proteins by Peptide Cross-Linking and Affinity Purification. J. Proteome Res. 2017, 16, 2762–2772. [Google Scholar] [CrossRef]
- Silverman, I.M.; Li, F.; Alexander, A.; Goff, L.; Trapnell, C.; Rinn, J.L.; Gregory, B.D. RNase-mediated protein footprint sequencing reveals protein-binding sites throughout the human transcriptome. Genome Biol. 2014, 15, 1–16. [Google Scholar] [CrossRef]
- Lundgren, D.H.; Hwang, S.I.; Wu, L.; Han, D.K. Role of spectral counting in quantitative proteomics. Expert Rev. Proteom. 2010, 7, 39–53. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | RNA Target | Advantage | Disadvantage | Cell Type | Cell Number |
|---|---|---|---|---|---|
| Affinity-Based Separation | |||||
| RIC [3,61] | Poly(A) tailed RNA | Isolates only mRNA complexes (if subset is of interest) Widely used protocol | Isolates only mRNA complexes Low signal-to-noise ratios Additionally, co-purification of non-cross-linked (free) RNA Can purify off-target RNA containing poly(A) stretches within its sequence | All poly(A) tailed containing organisms | 7500 cm2 HeLa cells [3] 107 cells [39] 108 cells [63] |
| e/cRIC [7,9] | Poly(A) tailed RNA | Isolates only mRNA complexes (if subset is of interest) Better signal-to-noise ratios than RIC | Isolates only mRNA complexes Additionally, co-purification of free RNA Can purify off-target RNA containing poly(A) stretches within its sequence | All poly(A) tailed containing organisms | 1–1.3 × 108 cells [7] 3 × 15 cm dishes at 80% confluence [9] |
| CARIC [64] | Newly transcribed RNA | All RNA types RNP monitoring through time | Use of nucleoside analogs Potential co-purification of naturally biotinylated proteins Additionally, co-purification of free RNA | Limited to cell cultures receptive for nucleoside analogs | 4 × 107 cells |
| RICK [65] | Newly transcribed RNA | All RNA types RNP monitoring through time | Use of nucleoside analogs Potential co-purification of naturally biotinylated proteins Additionally, co-purification of free RNA | Limited to cell cultures receptive for nucleoside analogs | Not specified |
| Solid Phase Separation | |||||
| 2C [66] | All RNPs | Fast and cost-effective method | Contamination of both free protein and free RNA Dependent on the scale of the silica columns A nucleotide size limitation can occur inherent to silica matrices | All cell types and tissue | Not specified |
| (PAR)-TRAPP [43] | All RNPs | Cost-effective method Scalable protocol | DNA is co-eluted Additionally, co-purification of free RNA A nucleotide size limitation can occur inherent to silica matrices | All cell types and tissue | 750 mL of media containing cells at an OD600 of 0.5 |
| VIR-CLASP [67] | Pre-replicated viral RNPs | Study of early-stage viral infection Theoretically adaptable to every type of in vitro transcribed RNA molecule Cost-effective method | The current field of application is a highly interesting but small niche SPRI beads can have size-selective artefacts | Limited to cell cultures receptive for nucleoside analogs | 15 cm2 of cells |
| Organic Phase Separation | |||||
| XRNAX [21] | All RNPs | All RNA types Little free RNA Cost-effective method Easily scalable Good starting point for more specific techniques | Glycoproteins and RNA–protein adducts cannot be distinguished Technically challenging Crude fraction | All cell types and tissue | 1 × 108 cells |
| OOPS [22] | All RNPs | All RNA types Cost-effective method Easily scalable | Technically challenging Cannot be used as a starting point for more specific techniques | All cell types and tissue | 28.2 cm2 of 90% confluence |
| PTex [23] | All RNP >30 bp | All RNA types Little free RNA Cost-effective method Easily scalable Good starting point for more specific techniques | Glycoproteins and RNA–protein adducts cannot be distinguished Technically challenging 25–30% recovery | All cell types and tissue | 2 × 106 cells |
| Method | Advantage | Disadvantage | Cell Number |
|---|---|---|---|
| Antisense Probe-Based Methods | |||
| PAIR [74] | Direct interactors RBPs of a specific region Different transcript isoforms Denaturing purification conditions | Difficult to study whole interactome of a specific RNA Potential co-purification of naturally biotinylated proteins Costs of CPP-PNA-Bpa probes | 2 × 106–1 × 107 cells |
| CHART-MS [75] | Different transcript isoforms Denaturing purification conditions | Direct and indirect interactors RNase H assay to determine accessibility of the probes Potential co-purification of naturally biotinylated proteins | 8 × 107 cells |
| ChiRP-MS [10] | No prior knowledge of the RNA structure and accessibility required Capture of RNA with impaired integrity Denaturing purification conditions | Direct and indirect interactors Large number of probes may result in higher background contamination Potential co-purification of naturally biotinylated proteins Costs of large number of probes | 1–5 × 108 cells depending on the cell type |
| RAP-MS [11] | Direct interactors No prior knowledge of the RNA structure and accessibility required Long probes may result in high specificity Capture of RNA with impaired integrity Denaturing purification conditions | Large number of probes may result in higher background contamination Potential co-purification of naturally biotinylated proteins Costs of large number of probes Cost of synthesizing long probes | 2 × 107 cells |
| LNA/DNA mixmers [76] | Direct interactors Different transcript isoforms Increased hybridization specificity Denaturing purification conditions | Cost of LNA-containing probes Use of only one probe results in RNA integrity to be critical 20% recovery Less optimal for low-copy-number transcripts | 2000 cm2 of HeLa cells at 70% confluence |
| HyPR-MS [77] | Cost- and labor-effective protocol Reduced sample and background variance Different transcript isoforms Denaturing purification conditions | Direct and indirect interactors Potential co-purification of naturally biotinylated proteins | 1 × 108 cells |
| PPR proteins [78] | Different transcript isoforms | Direct and indirect interactors Efficacy only proven in chloroplasts and mitochondria Restriction to use denaturing purification conditions | Chloroplasts isolated from 40 g tissue |
| Biotin-tagged Cas9 purification [79] | Different transcript isoforms | Direct and indirect interactors Restriction to use denaturing purification conditions Potential co-purification of naturally biotinylated proteins | 5x106 cells |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Ende, R.; Balzarini, S.; Geuten, K. Single and Combined Methods to Specifically or Bulk-Purify RNA–Protein Complexes. Biomolecules 2020, 10, 1160. https://doi.org/10.3390/biom10081160
Van Ende R, Balzarini S, Geuten K. Single and Combined Methods to Specifically or Bulk-Purify RNA–Protein Complexes. Biomolecules. 2020; 10(8):1160. https://doi.org/10.3390/biom10081160
Chicago/Turabian StyleVan Ende, Roosje, Sam Balzarini, and Koen Geuten. 2020. "Single and Combined Methods to Specifically or Bulk-Purify RNA–Protein Complexes" Biomolecules 10, no. 8: 1160. https://doi.org/10.3390/biom10081160
APA StyleVan Ende, R., Balzarini, S., & Geuten, K. (2020). Single and Combined Methods to Specifically or Bulk-Purify RNA–Protein Complexes. Biomolecules, 10(8), 1160. https://doi.org/10.3390/biom10081160