Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases


Abstract
1. Introduction
2. Biosynthesis of Melatonin
3. Function of Melatonin
4. Action Mechanism of Melatonin
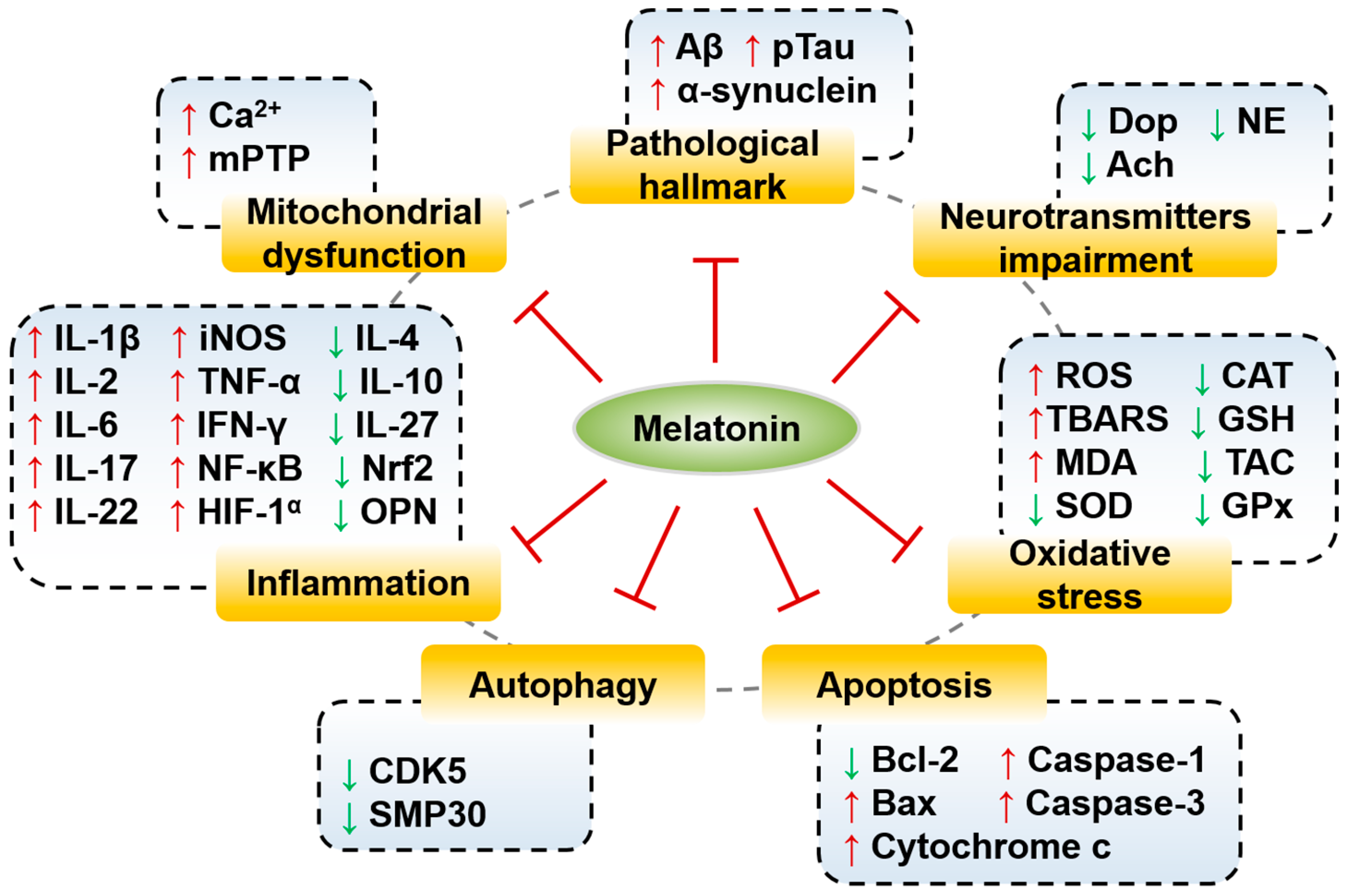
5. Effects and Molecular Mechanisms of Melatonin in Neurodegenerative Diseases
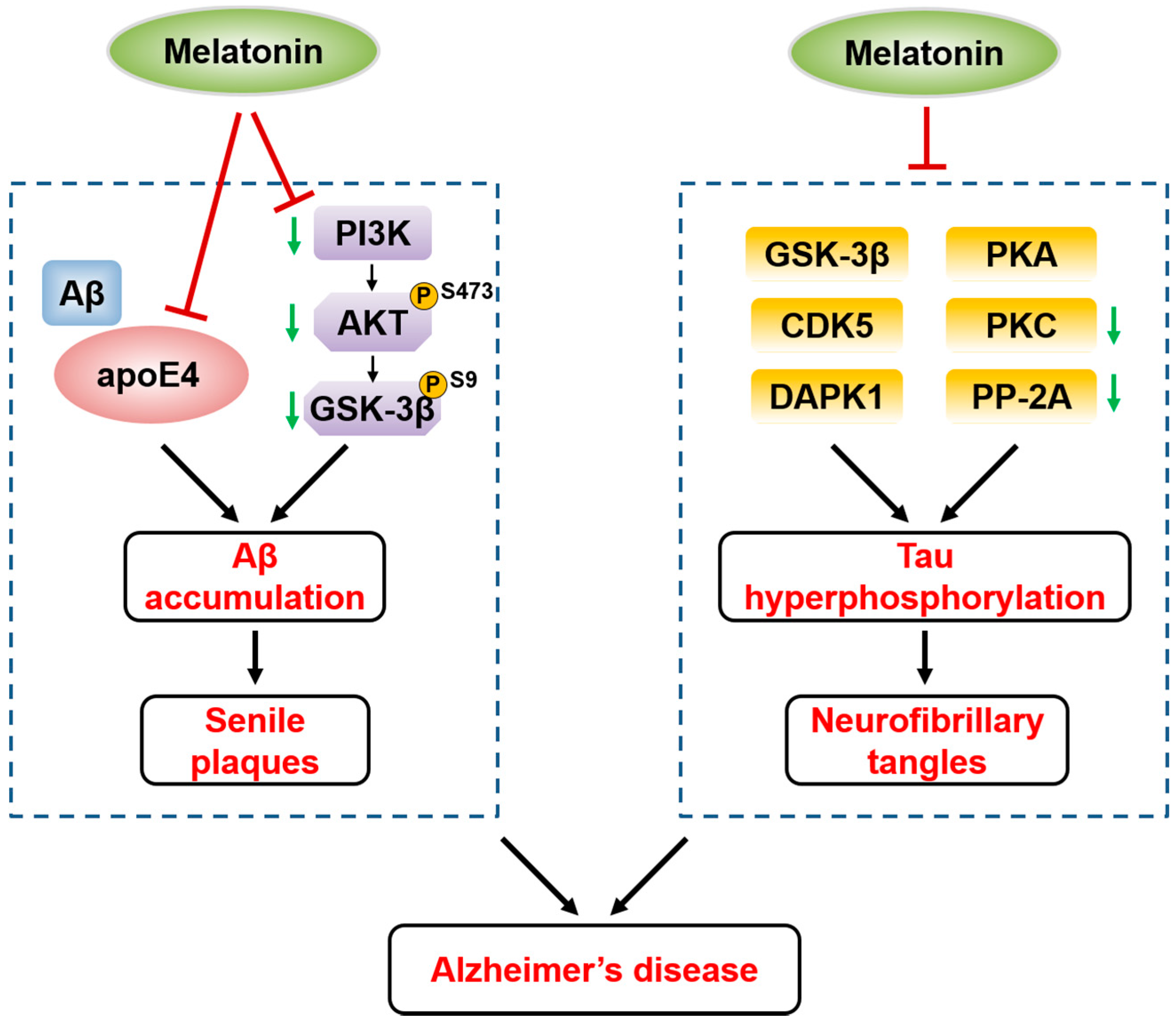
5.1. Melatonin and Alzheimer’s Disease
5.2. Melatonin and Parkinson’s Disease
5.3. Melatonin and Huntington’s Disease
5.4. Melatonin and Multiple Sclerosis
5.5. Melatonin and Amyotrophic Lateral Sclerosis
5.6. Melatonin and Vascular Dementia
6. Clinical Application of Melatonin in Neurodegenerative Diseases
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of melatonin, the pineal gland factor that lightens melanocytes1. J. Am. Chem. Soc. 1958, 80, 2587. [Google Scholar] [CrossRef]
- Claustrat, B.; Brun, J.; Chazot, G. The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 2005, 9, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Slats, D.; Claassen, J.A.; Verbeek, M.M.; Overeem, S. Reciprocal interactions between sleep, circadian rhythms and alzheimer’s disease: Focus on the role of hypocretin and melatonin. Ageing Res. Rev. 2013, 12, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Reiter, R.J. Melatonin feedback on clock genes: A theory involving the proteasome. J. Pineal Res. 2015, 58, 1–11. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a full service anti-cancer agent: Inhibition of initiation, progression and metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef]
- Cardinali, D.P. Melatonin: Clinical perspectives in neurodegeneration. Front. Endocrinol. 2019, 10, 480. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Liu, C.; Reiter, R.J. Covid-19: Melatonin as a potential adjuvant treatment. Life Sci. 2020, 250, 117583. [Google Scholar] [CrossRef]
- Talib, W.H. Melatonin and cancer hallmarks. Molecules 2018, 23, 518. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Gunata, M.; Parlakpinar, H.; Acet, H.A. Melatonin: A review of its potential functions and effects on neurological diseases. Revue Neurol. 2020, 176, 148–165. [Google Scholar] [CrossRef]
- Reiter, R.J. Pineal melatonin: Cell biology of its synthesis and of its physiological interactions. Endocr. Rev. 1991, 12, 151–180. [Google Scholar] [CrossRef] [PubMed]
- Stokkan, K.A.; Reiter, R.J. Melatonin rhythms in arctic urban residents. J. Pineal Res. 1994, 16, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Benot, S.; Molinero, P.; Soutto, M.; Goberna, R.; Guerrero, J.M. Circadian variations in the rat serum total antioxidant status: Correlation with melatonin levels. J. Pineal Res. 1998, 25, 1–4. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Cui, M.; Lin, H.; Zhao, L.; Wang, J.; Chen, S.; Shao, Z. Melatonin resists oxidative stress-induced apoptosis in nucleus pulposus cells. Life Sci. 2018, 199, 122–130. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Erren, T.C.; Fuentes-Broto, L.; Paredes, S.D. Light-mediated perturbations of circadian timing and cancer risk: A mechanistic analysis. Integr. Cancer Ther. 2009, 8, 354–360. [Google Scholar] [CrossRef]
- Gupta, M.; Gupta, Y.K.; Agarwal, S.; Aneja, S.; Kalaivani, M.; Kohli, K. Effects of add-on melatonin administration on antioxidant enzymes in children with epilepsy taking carbamazepine monotherapy: A randomized, double-blind, placebo-controlled trial. Epilepsia 2004, 45, 1636–1639. [Google Scholar] [CrossRef]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolín, I.; Herrera, F.; Martín, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Hirata, F.; Hayaishi, O.; Tokuyama, T.; Seno, S. In vitro and in vivo formation of two new metabolites of melatonin. J. Biol. Chem. 1974, 249, 1311–1313. [Google Scholar]
- Rosales-Corral, S.; Tan, D.X.; Reiter, R.J.; Valdivia-Velázquez, M.; Martínez-Barboza, G.; Acosta-Martínez, J.P.; Ortiz, G.G. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: A comparative, in vivo study versus vitamin c and e. J. Pineal Res. 2003, 35, 80–84. [Google Scholar] [CrossRef]
- Yokota, O.; Terada, S.; Ishizu, H.; Ishihara, T.; Ujike, H.; Nakashima, H.; Nakashima, Y.; Kugo, A.; Checler, F.; Kuroda, S. Cyclooxygenase-2 in the hippocampus is up-regulated in alzheimer’s disease but not in variant alzheimer’s disease with cotton wool plaques in humans. Neurosci. Lett. 2003, 343, 175–179. [Google Scholar] [CrossRef]
- Jang, M.H.; Jung, S.B.; Lee, M.H.; Kim, C.J.; Oh, Y.T.; Kang, I.; Kim, J.; Kim, E.H. Melatonin attenuates amyloid beta25-35-induced apoptosis in mouse microglial bv2 cells. Neurosci. Lett. 2005, 380, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, X.; Zhang, Y.; Shi, D.; Chen, W.; Fu, L.; Liu, L.; Xie, F.; Kang, T.; Huang, W.; et al. Simultaneous modulation of cox-2, p300, akt, and apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J. Pineal Res. 2012, 53, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Menéndez, J.; Hermida-Prado, F. Deciphering the molecular basis of melatonin protective effects on breast cells treated with doxorubicin: Twist1 a transcription factor involved in emt and metastasis, a novel target of melatonin. Cancers 2019, 11, 1011. [Google Scholar] [CrossRef] [PubMed]
- Ekmekcioglu, C. Melatonin receptors in humans: Biological role and clinical relevance. Biomed. Pharmacother. 2006, 60, 97–108. [Google Scholar] [CrossRef]
- Emet, M.; Ozcan, H.; Yayla, M.; Halici, Z.; Hacimuftuoglu, A. A review of melatonin, its receptors and drugs. Eurasian J. Med. 2016, 48, 135. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.; Zisapel, N.; Cardinali, D.P. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef]
- Comai, S.; Gobbi, G. Unveiling the role of melatonin mt2 receptors in sleep, anxiety and other neuropsychiatric diseases: A novel target in psychopharmacology. J. Psychiatry Neurosci. JPN 2014, 39, 6–21. [Google Scholar] [CrossRef]
- Nosjean, O.; Ferro, M.; Coge, F.; Beauverger, P.; Henlin, J.M.; Lefoulon, F.; Fauchere, J.L.; Delagrange, P.; Canet, E.; Boutin, J.A. Identification of the melatonin-binding site mt3 as the quinone reductase 2. J. Biol. Chem. 2000, 275, 31311–31317. [Google Scholar] [CrossRef]
- Carlberg, C. Gene regulation by melatonin. Ann. N. Y. Acad. Sci. 2000, 917, 387–396. [Google Scholar] [CrossRef]
- Garcia, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; Lopez, L.C.; Acuna-Castroviejo, D. Disruption of the nf-kappab/nlrp3 connection by melatonin requires retinoid-related orphan receptor-alpha and blocks the septic response in mice. FASEB J. 2015, 29, 3863–3875. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Freire, F. Melatonin effects on brain. Interaction with microtubule protein, inhibition of fast axoplasmic flow and induction of crystaloid and tubular formations in the hypothalamus. Mol. Cell. Endocrinol. 1975, 2, 317–330. [Google Scholar] [CrossRef]
- Melendez, J.; Maldonado, V.; Ortega, A. Effect of melatonin on beta-tubulin and map2 expression in nie-115 cells. Neurochem. Res. 1996, 21, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.; Escames, G.; Leon, J.; Coto, A.; Sbihi, Y.; Osuna, A.; Acuna-Castroviejo, D. Calreticulin-melatonin. An unexpected relationship. Eur. J. Biochem. 2003, 270, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Benitez-King, G.; Anton-Tay, F. Calmodulin mediates melatonin cytoskeletal effects. Experientia 1993, 49, 635–641. [Google Scholar] [CrossRef]
- Benitez-King, G.; Huerto-Delgadillo, L.; Anton-Tay, F. Melatonin modifies calmodulin cell levels in mdck and n1e-115 cell lines and inhibits phosphodiesterase activity in vitro. Brain Res. 1991, 557, 289–292. [Google Scholar] [CrossRef]
- Benitez-King, G.; Rios, A.; Martinez, A.; Anton-Tay, F. In vitro inhibition of ca2+/calmodulin-dependent kinase ii activity by melatonin. Biochim. Biophys. Acta 1996, 1290, 191–196. [Google Scholar] [CrossRef]
- Jenwitheesuk, A.; Nopparat, C.; Mukda, S.; Wongchitrat, P.; Govitrapong, P. Melatonin regulates aging and neurodegeneration through energy metabolism, epigenetics, autophagy and circadian rhythm pathways. Int. J. Mol. Sci. 2014, 15, 16848–16884. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Pilar Terron, M.; Flores, L.J.; Koppisepi, S. Medical implications of melatonin: Receptor-mediated and receptor-independent actions. Adv. Med. Sci. 2007, 52, 11–28. [Google Scholar]
- Reiter, R.J. The pineal gland and melatonin in relation to aging: A summary of the theories and of the data. Exp. Gerontol. 1995, 30, 199–212. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Poeggeler, B.; Menendez-Pelaez, A.; Chen, L.D.; Saarela, S. Melatonin as a free radical scavenger: Implications for aging and age-related diseases. Ann. N. Y. Acad. Sci. 1994, 719, 1–12. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Bartels, C.; Polking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Huther, G.; et al. Reduced oxidative damage in als by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Sainz, R.M.; Mayo, J.C.; Lopez-Burillo, S. Melatonin: Reducing the toxicity and increasing the efficacy of drugs. J. Pharm. Pharmacol. 2002, 54, 1299–1321. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.J.; Holtzman, D.M. Biomarker modeling of alzheimer’s disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Gotz, J. Amyloid-beta and tau complexity-towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Wu, Y.H.; Swaab, D.F. The human pineal gland and melatonin in aging and alzheimer’s disease. J. Pineal Res. 2005, 38, 145–152. [Google Scholar] [CrossRef]
- Wu, Y.H.; Feenstra, M.G.; Zhou, J.N.; Liu, R.Y.; Torano, J.S.; Van Kan, H.J.; Fischer, D.F.; Ravid, R.; Swaab, D.F. Molecular changes underlying reduced pineal melatonin levels in alzheimer disease: Alterations in preclinical and clinical stages. J. Clin. Endocrinol. Metab. 2003, 88, 5898–5906. [Google Scholar] [CrossRef]
- Zhou, J.N.; Liu, R.Y.; Kamphorst, W.; Hofman, M.A.; Swaab, D.F. Early neuropathological alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J. Pineal Res. 2003, 35, 125–130. [Google Scholar] [CrossRef]
- Mahlberg, R.; Kunz, D.; Sutej, I.; Kuhl, K.P.; Hellweg, R. Melatonin treatment of day-night rhythm disturbances and sundowning in alzheimer disease: An open-label pilot study using actigraphy. J. Clin. Psychopharmacol. 2004, 24, 456–459. [Google Scholar] [CrossRef]
- Brusco, L.I.; Marquez, M.; Cardinali, D.P. Melatonin treatment stabilizes chronobiologic and cognitive symptoms in alzheimer’s disease. Neuro Endocrinol. Lett. 2000, 21, 39–42. [Google Scholar] [PubMed]
- Zhang, T.; Chen, D.; Lee, T.H. Phosphorylation signaling in app processing in alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 209. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K. Melatonin affects the metabolism of the beta-amyloid precursor protein in different cell types. J. Pineal Res. 1999, 26, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Zhang, Y.C.; Chatterjie, N.; Grundke-Iqbal, I.; Iqbal, K.; Wang, J.Z. Effect of melatonin and melatonylvalpromide on beta-amyloid and neurofilaments in n2a cells. Neurochem. Res. 2008, 33, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Wang, Z.F.; Wang, Q.; Wang, Y.P.; Wang, J.Z. Melatonin attenuates beta-amyloid-induced inhibition of neurofilament expression. Acta Pharmacol. Sin. 2004, 25, 447–451. [Google Scholar]
- Chinchalongporn, V.; Shukla, M.; Govitrapong, P. Melatonin ameliorates abeta42 -induced alteration of betaapp-processing secretases via the melatonin receptor through the pin1/gsk3beta/nf-kappab pathway in sh-sy5y cells. J. Pineal Res. 2018, 64, e12470. [Google Scholar] [CrossRef]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of alzheimer beta-fibrillogenesis by melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef]
- Poeggeler, B.; Miravalle, L.; Zagorski, M.G.; Wisniewski, T.; Chyan, Y.J.; Zhang, Y.; Shao, H.; Bryant-Thomas, T.; Vidal, R.; Frangione, B.; et al. Melatonin reverses the profibrillogenic activity of apolipoprotein e4 on the alzheimer amyloid abeta peptide. Biochemistry 2001, 40, 14995–15001. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, J.T. Long-term melatonin or 17beta-estradiol supplementation alleviates oxidative stress in ovariectomized adult rats. Free Radic. Biol. Med. 2005, 39, 195–204. [Google Scholar] [CrossRef]
- Hoppe, J.B.; Frozza, R.L.; Horn, A.P.; Comiran, R.A.; Bernardi, A.; Campos, M.M.; Battastini, A.M.; Salbego, C. Amyloid-beta neurotoxicity in organotypic culture is attenuated by melatonin: Involvement of gsk-3beta, tau and neuroinflammation. J. Pineal Res. 2010, 48, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via pi3/akt/gsk3β pathway in the mouse hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. Ltp inhibits ltd in the hippocampus via regulation of gsk3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Whitcomb, D.J.; Olsen, K.M.; Kerrigan, T.L.; Lo, S.C.; Bru-Mercier, G.; Dickinson, B.; Scullion, S.; Sheng, M.; Collingridge, G.; et al. Aβ(1-42) inhibition of ltp is mediated by a signaling pathway involving caspase-3, akt1 and gsk-3β. Nat. Neurosci. 2011, 14, 545–547. [Google Scholar] [CrossRef]
- Lee, K.Y.; Koh, S.H.; Noh, M.Y.; Kim, S.H.; Lee, Y.J. Phosphatidylinositol-3-kinase activation blocks amyloid beta-induced neurotoxicity. Toxicology 2008, 243, 43–50. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Stoothoff, W.H.; de Calignon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends NeuroSci. 2009, 32, 150–159. [Google Scholar] [CrossRef]
- Li, S.P.; Deng, Y.Q.; Wang, X.C.; Wang, Y.P.; Wang, J.Z. Melatonin protects sh-sy5y neuroblastoma cells from calyculin a-induced neurofilament impairment and neurotoxicity. J. Pineal Res. 2004, 36, 186–191. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Fu, Z.; Li, Y.; Feng, J.; Luo, J.; Zhang, Q.; Wang, Q.; Tian, Q. Melatonin ameliorates alzheimer-like pathological changes and spatial memory retention impairment induced by calyculin a. J. Psychopharmacol. 2011, 25, 1118–1125. [Google Scholar] [CrossRef]
- Ling, Z.Q.; Tian, Q.; Wang, L.; Fu, Z.Q.; Wang, X.C.; Wang, Q.; Wang, J.Z. Constant illumination induces alzheimer-like damages with endoplasmic reticulum involvement and the protection of melatonin. J. Alzheimers Dis. 2009, 16, 287–300. [Google Scholar] [CrossRef]
- Shi, C.; Zeng, J.; Li, Z.; Chen, Q.; Hang, W.; Xia, L.; Wu, Y.; Chen, J.; Shi, A. Melatonin mitigates kainic acid-induced neuronal tau hyperphosphorylation and memory deficits through alleviating er stress. Front. Mol. Neurosci. 2018, 11, 5. [Google Scholar] [CrossRef]
- Wang, D.L.; Ling, Z.Q.; Cao, F.Y.; Zhu, L.Q.; Wang, J.Z. Melatonin attenuates isoproterenol-induced protein kinase a overactivation and tau hyperphosphorylation in rat brain. J. Pineal Res. 2004, 37, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Q.; Xu, G.G.; Duan, P.; Zhang, Q.; Wang, J.Z. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol. Sin. 2005, 26, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Gomez-Isla, T.; Puig, B.; Freixes, M.; Ribé, E.; Dalfó, E.; Avila, J. Current advances on different kinases involved in tau phosphorylation, and implications in alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2005, 2, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.X.; Hu, J.; Liu, D.; Hong, X.P.; Wu, Y.Y.; Zhu, L.Q.; Wang, J.Z. Disease-modified glycogen synthase kinase-3β intervention by melatonin arrests the pathology and memory deficits in an alzheimer’s animal model. Neurobiol. Aging 2013, 34, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhu, L.; Shi, H.; Zheng, H.; Tian, Q.; Wang, Q.; Liu, R.; Wang, J.Z. Inhibition of melatonin biosynthesis induces neurofilament hyperphosphorylation with activation of cyclin-dependent kinase 5. Neurochem. Res. 2007, 32, 1329–1335. [Google Scholar] [CrossRef]
- Rivera-Bermudez, M.A.; Gerdin, M.J.; Earnest, D.J.; Dubocovich, M.L. Regulation of basal rhythmicity in protein kinase c activity by melatonin in immortalized rat suprachiasmatic nucleus cells. Neurosci. Lett. 2003, 346, 37–40. [Google Scholar] [CrossRef]
- Li, X.C.; Wang, Z.F.; Zhang, J.X.; Wang, Q.; Wang, J.Z. Effect of melatonin on calyculin a-induced tau hyperphosphorylation. Eur. J. Pharmacol. 2005, 510, 25–30. [Google Scholar] [CrossRef]
- Zhu, L.Q.; Wang, S.H.; Ling, Z.Q.; Wang, D.L.; Wang, J.Z. Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phosphorylation in rat. J. Pineal Res. 2004, 37, 71–77. [Google Scholar] [CrossRef]
- Alvira, D.; Tajes, M.; Verdaguer, E.; Acuña-Castroviejo, D.; Folch, J.; Camins, A.; Pallas, M. Inhibition of the cdk5/p25 fragment formation may explain the antiapoptotic effects of melatonin in an experimental model of parkinson’s disease. J. Pineal Res. 2006, 40, 251–258. [Google Scholar] [CrossRef]
- Chen, D.; Mei, Y.; Kim, N.; Lan, G.; Gan, C.L.; Fan, F.; Zhang, T.; Xia, Y.; Wang, L.; Lin, C.; et al. Melatonin directly binds and inhibits death-associated protein kinase 1 function in alzheimer’s disease. J. Pineal Res. 2020, 69, e12665. [Google Scholar] [CrossRef]
- Kim, N.; Chen, D.; Zhou, X.Z.; Lee, T.H. Death-associated protein kinase 1 phosphorylation in neuronal cell death and neurodegenerative disease. Int. J. Mol. Sci. 2019, 20, 3131. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhou, X.Z.; Lee, T.H. Death-associated protein kinase 1 as a promising drug target in cancer and alzheimer’s disease. Recent Pat. Anti Cancer Drug Discov. 2019, 14, 144–157. [Google Scholar] [CrossRef]
- Chen, D.; Wang, L.; Lee, T.H. Post-translational modifications of the peptidyl-prolyl isomerase pin1. Front. Cell Dev. Biol. 2020, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, Y.; Chen, D.; Lee, T.H. Peptidyl-prolyl cis/trans isomerase pin1 and alzheimer’s disease. Front. Cell Dev. Biol. 2020, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Cookson, M.R. The biochemistry of parkinson’s disease. Annu. Rev. Biochem. 2005, 74, 29–52. [Google Scholar] [CrossRef]
- Sidhu, A.; Wersinger, C.; Moussa, C.E.; Vernier, P. The role of alpha-synuclein in both neuroprotection and neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1035, 250–270. [Google Scholar] [CrossRef]
- Miller, D.W.; Hague, S.M.; Clarimon, J.; Baptista, M.; Gwinn-Hardy, K.; Cookson, M.R.; Singleton, A.B. Alpha-synuclein in blood and brain from familial parkinson disease with snca locus triplication. Neurology 2004, 62, 1835–1838. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-synuclein in filamentous inclusions of lewy bodies from parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Singhal, N.K.; Srivastava, G.; Patel, D.K.; Jain, S.K.; Singh, M.P. Melatonin or silymarin reduces maneb- and paraquat-induced parkinson’s disease phenotype in the mouse. J. Pineal Res. 2011, 50, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Patki, G.; Lau, Y.S. Melatonin protects against neurobehavioral and mitochondrial deficits in a chronic mouse model of parkinson’s disease. Pharmacol. Biochem. Behav. 2011, 99, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30pro mutation in the gene encoding alpha-synuclein in parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Brito-Armas, J.M.; Baekelandt, V.; Castro-Hernández, J.R.; González-Hernández, T.; Rodríguez, M.; Castro, R. Melatonin prevents dopaminergic cell loss induced by lentiviral vectors expressing a30p mutant alpha-synuclein. Histol. Histopathol. 2013, 28, 999–1006. [Google Scholar]
- Chang, C.F.; Huang, H.J.; Lee, H.C.; Hung, K.C.; Wu, R.T.; Lin, A.M. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J. Pineal Res. 2012, 52, 312–321. [Google Scholar] [CrossRef]
- Lin, A.M.; Fang, S.F.; Chao, P.L.; Yang, C.H. Melatonin attenuates arsenite-induced apoptosis in rat brain: Involvement of mitochondrial and endoplasmic reticulum pathways and aggregation of alpha-synuclein. J. Pineal Res. 2007, 43, 163–171. [Google Scholar] [CrossRef]
- Su, L.Y.; Li, H.; Lv, L.; Feng, Y.M.; Li, G.D.; Luo, R.; Zhou, H.J.; Lei, X.G.; Ma, L.; Li, J.L.; et al. Melatonin attenuates mptp-induced neurotoxicity via preventing cdk5-mediated autophagy and snca/α-synuclein aggregation. Autophagy 2015, 11, 1745–1759. [Google Scholar] [CrossRef]
- Sae-Ung, K.; Uéda, K.; Govitrapong, P.; Phansuwan-Pujito, P. Melatonin reduces the expression of alpha-synuclein in the dopamine containing neuronal regions of amphetamine-treated postnatal rats. J. Pineal Res. 2012, 52, 128–137. [Google Scholar] [CrossRef]
- Zampol, M.A.; Barros, M.H. Melatonin improves survival and respiratory activity of yeast cells challenged by alpha-synuclein and menadione. Yeast 2018, 35, 281–290. [Google Scholar] [CrossRef]
- Adi, N.; Mash, D.C.; Ali, Y.; Singer, C.; Shehadeh, L.; Papapetropoulos, S. Melatonin mt1 and mt2 receptor expression in parkinson’s disease. Med. Sci. Monit. 2010, 16, BR61–BR67. [Google Scholar]
- Willis, G.L. Parkinson’s disease as a neuroendocrine disorder of circadian function: Dopamine-melatonin imbalance and the visual system in the genesis and progression of the degenerative process. Rev. Neurosci. 2008, 19, 245–316. [Google Scholar] [CrossRef] [PubMed]
- Alexiuk, N.A.; Vriend, J.P. Melatonin reduces dopamine content in the neurointermediate lobe of male syrian hamsters. Brain Res. Bull. 1993, 32, 433–436. [Google Scholar] [CrossRef]
- Willis, G.L. The role of ml-23 and other melatonin analogues in the treatment and management of parkinson’s disease. Drug News Perspect. 2005, 18, 437–444. [Google Scholar] [CrossRef]
- Willis, G.L. Intraocular microinjections repair experimental parkinson’s disease. Brain Res. 2008, 1217, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- van der Burg, J.M.; Björkqvist, M.; Brundin, P. Beyond the brain: Widespread pathology in huntington’s disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
- Sawa, A.; Wiegand, G.W.; Cooper, J.; Margolis, R.L.; Sharp, A.H.; Lawler, J.F.J.; Greenamyre, J.T.; Snyder, S.H.; Ross, C.A. Increased apoptosis of huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat. Med. 1999, 5, 1194–1198. [Google Scholar] [CrossRef]
- Kalliolia, E.; Silajdžić, E.; Nambron, R.; Hill, N.R.; Doshi, A.; Frost, C.; Watt, H.; Hindmarsh, P.; Björkqvist, M.; Warner, T.T. Plasma melatonin is reduced in huntington’s disease. Mov. Disord. 2014, 29, 1511–1515. [Google Scholar] [CrossRef]
- Xue, F.; Shi, C.; Chen, Q.; Hang, W.; Xia, L.; Wu, Y.; Tao, S.Z.; Zhou, J.; Shi, A.; Chen, J. Melatonin mediates protective effects against kainic acid-induced neuronal death through safeguarding er stress and mitochondrial disturbance. Front. Mol. Neurosci. 2017, 10, 49. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Qi, W.; Kim, S.J.; El-Sokkary, G.H. Melatonin protects hippocampal neurons in vivo against kainic acid-induced damage in mice. J. Neurosci. Res. 1998, 54, 382–389. [Google Scholar] [CrossRef]
- Manev, H.; Uz, T.; Kharlamov, A.; Cagnoli, C.M.; Franceschini, D.; Giusti, P. In vivo protection against kainate-induced apoptosis by the pineal hormone melatonin: Effect of exogenous melatonin and circadian rhythm. Restor. Neurol. Neurosci. 1996, 9, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Túnez, I.; Montilla, P.; Del Carmen Muñoz, M.; Feijóo, M.; Salcedo, M. Protective effect of melatonin on 3-nitropropionic acid-induced oxidative stress in synaptosomes in an animal model of huntington’s disease. J. Pineal Res. 2004, 37, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.; Lee, S.M.; Koh, S.E.; Joo, W.S.; Maeng, S.; Im, H.I.; Kim, Y.S. Melatonin protects against neuronal damage induced by 3-nitropropionic acid in rat striatum. Brain Res. 2005, 1046, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Lin, E.; Liu, B.; Ma, Y.; OuYang, L.; Li, Y.; Chen, S.; Zhang, J.; Lei, W. Melatonin reduces projection neuronal injury induced by 3-nitropropionic acid in the rat striatum. Neuro Degener. Dis. 2014, 14, 139–150. [Google Scholar] [CrossRef]
- Mochel, F.; Haller, R.G. Energy deficit in huntington disease: Why it matters. J. Clin. Investig. 2011, 121, 493–499. [Google Scholar] [CrossRef]
- Wang, X.; Sirianni, A.; Pei, Z.; Cormier, K.; Smith, K.; Jiang, J.; Zhou, S.; Wang, H.; Zhao, R.; Yano, H.; et al. The melatonin mt1 receptor axis modulates mutant huntingtin-mediated toxicity. J. Neurosci. 2011, 31, 14496–14507. [Google Scholar] [CrossRef]
- Zhang, L.; Li, F.; Su, X.; Li, Y.; Wang, Y.; Fang, R.; Guo, Y.; Jin, T.; Shan, H.; Zhao, X.; et al. Melatonin prevents lung injury by regulating apelin 13 to improve mitochondrial dysfunction. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Ruiz, A.; Matute, C.; Alberdi, E. Intracellular ca2+ release through ryanodine receptors contributes to ampa receptor-mediated mitochondrial dysfunction and er stress in oligodendrocytes. Cell Death Dis. 2010, 1, e54. [Google Scholar] [CrossRef]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in huntington’s disease. Cell Death Dis. 2011, 2, e228. [Google Scholar] [CrossRef]
- Leung, A.W.; Halestrap, A.P. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2008, 1777, 946–952. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Sayeed, I.; Siemen, D.; Wolf, G.; Horn, T.F. Direct inhibition of the mitochondrial permeability transition pore: A possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci. Ther. 2009, 15, 345–357. [Google Scholar] [CrossRef]
- Mohseni, M.; Mihandoost, E.; Shirazi, A.; Sepehrizadeh, Z.; Bazzaz, J.T.; Ghazi-khansari, M. Melatonin may play a role in modulation of bax and bcl-2 expression levels to protect rat peripheral blood lymphocytes from gamma irradiation-induced apoptosis. Mutat. Res. 2012, 738–739, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Radogna, F.; Cristofanon, S.; Paternoster, L.; D’Alessio, M.; De Nicola, M.; Cerella, C.; Dicato, M.; Diederich, M.; Ghibelli, L. Melatonin antagonizes the intrinsic pathway of apoptosis via mitochondrial targeting of bcl-2. J. Pineal Res. 2008, 44, 316–325. [Google Scholar] [CrossRef]
- Radogna, F.; Albertini, M.C.; De Nicola, M.; Diederich, M.; Bejarano, I.; Ghibelli, L. Melatonin promotes bax sequestration to mitochondria reducing cell susceptibility to apoptosis via the lipoxygenase metabolite 5-hydroxyeicosatetraenoic acid. Mitochondrion 2015, 21, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple sclerosis: Pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017, 19, 1. [Google Scholar]
- Kamm, C.P.; Uitdehaag, B.M.; Polman, C.H. Multiple sclerosis: Current knowledge and future outlook. Eur. Neurol. 2014, 72, 132–141. [Google Scholar] [CrossRef]
- Wootla, B.; Eriguchi, M.; Rodriguez, M. Is multiple sclerosis an autoimmune disease? Autoimmune Dis. 2012, 2012, 969657. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Akpinar, Z.; Tokgöz, S.; Gökbel, H.; Okudan, N.; Uğuz, F.; Yilmaz, G. The association of nocturnal serum melatonin levels with major depression in patients with acute multiple sclerosis. Psychiatry Res. 2008, 161, 253–257. [Google Scholar] [CrossRef]
- Farhadi, N.; Oryan, S.; Nabiuni, M. Serum levels of melatonin and cytokines in multiple sclerosis. Biomed. J. 2014, 37, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, A.; Moraes, A.S.; Farias, A.; Damasceno, B.P.; dos Santos, L.M.; Cendes, F. Disruption of melatonin circadian rhythm production is related to multiple sclerosis severity: A preliminary study. J. Neurol. Sci. 2015, 353, 166–168. [Google Scholar] [CrossRef]
- Melamud, L.; Golan, D.; Luboshitzky, R.; Lavi, I.; Miller, A. Melatonin dysregulation, sleep disturbances and fatigue in multiple sclerosis. J. Neurol. Sci. 2012, 314, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Abo Taleb, H.A.; Alghamdi, B.S. Neuroprotective effects of melatonin during demyelination and remyelination stages in a mouse model of multiple sclerosis. J. Mol. Neurosci. MN 2020, 70, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Vakilzadeh, G.; Khodagholi, F.; Ghadiri, T.; Ghaemi, A.; Noorbakhsh, F.; Sharifzadeh, M.; Gorji, A. The effect of melatonin on behavioral, molecular, and histopathological changes in cuprizone model of demyelination. Mol. Neurobiol. 2016, 53, 4675–4684. [Google Scholar] [CrossRef]
- Kashani, I.R.; Rajabi, Z.; Akbari, M.; Hassanzadeh, G.; Mohseni, A.; Eramsadati, M.K.; Rafiee, K.; Beyer, C.; Kipp, M.; Zendedel, A. Protective effects of melatonin against mitochondrial injury in a mouse model of multiple sclerosis. Exp. Brain Res. 2014, 232, 2835–2846. [Google Scholar] [CrossRef]
- Mascanfroni, I.D.; Yeste, A.; Vieira, S.M.; Burns, E.J.; Patel, B.; Sloma, I.; Wu, Y.; Mayo, L.; Ben-Hamo, R.; Efroni, S.; et al. Il-27 acts on dcs to suppress the t cell response and autoimmunity by inducing expression of the immunoregulatory molecule cd39. Nat. Immunol. 2013, 14, 1054–1063. [Google Scholar] [CrossRef]
- Álvarez-Sánchez, N.; Cruz-Chamorro, I.; Díaz-Sánchez, M.; Sarmiento-Soto, H.; Medrano-Campillo, P.; Martínez-López, A.; Lardone, P.J.; Guerrero, J.M.; Carrillo-Vico, A. Melatonin reduces inflammatory response in peripheral t helper lymphocytes from relapsing-remitting multiple sclerosis patients. J. Pineal Res. 2017, 63, e12442. [Google Scholar] [CrossRef]
- Farez, M.F.; Mascanfroni, I.D.; Méndez-Huergo, S.P.; Yeste, A.; Murugaiyan, G.; Garo, L.P.; Balbuena Aguirre, M.E.; Patel, B.; Ysrraelit, M.C.; Zhu, C.; et al. Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell 2015, 162, 1338–1352. [Google Scholar] [CrossRef]
- von Lewinski, F.; Keller, B.U. Ca2+, mitochondria and selective motoneuron vulnerability: Implications for als. Trends Neurosci. 2005, 28, 494–500. [Google Scholar] [CrossRef]
- Zhang, Y.; Cook, A.; Kim, J.; Baranov, S.V.; Jiang, J.; Smith, K.; Cormier, K.; Bennett, E.; Browser, R.P.; Day, A.L.; et al. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits mt1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 55, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Rogério, F.; Teixeira, S.A.; de Rezende, A.C.; de Sá, R.C.; de Souza Queiroz, L.; De Nucci, G.; Muscará, M.N.; Langone, F. Superoxide dismutase isoforms 1 and 2 in lumbar spinal cord of neonatal rats after sciatic nerve transection and melatonin treatment. Brain Res. Dev. Brain Res. 2005, 154, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Donnenfeld, H.; Sasaki, S.; Nakano, I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1984, 43, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, G.A.; Clark, A.W.; Rooke, K.; Pramatarova, A.; Krizus, A.; Suchowersky, O.; Julien, J.P.; Figlewicz, D. Sod1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 128–131. [Google Scholar] [CrossRef]
- Julien, J.P.; Beaulieu, J.M. Cytoskeletal abnormalities in amyotrophic lateral sclerosis: Beneficial or detrimental effects? J. Neurol. Sci. 2000, 180, 7–14. [Google Scholar] [CrossRef]
- Estévez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, M.M.; Barbeito, L.; Beckman, J.S. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 1999, 286, 2498–2500. [Google Scholar]
- Crow, J.P.; Sampson, J.B.; Zhuang, Y.; Thompson, J.A.; Beckman, J.S. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 1997, 69, 1936–1944. [Google Scholar] [CrossRef]
- Schiavon, A.P.; Soares, L.M.; Bonato, J.M.; Milani, H.; Guimarães, F.S.; Weffort de Oliveira, R.M. Protective effects of cannabidiol against hippocampal cell death and cognitive impairment induced by bilateral common carotid artery occlusion in mice. Neurotox. Res. 2014, 26, 307–316. [Google Scholar] [CrossRef]
- Meyer, J.S.; Rauch, G.; Rauch, R.A.; Haque, A. Risk factors for cerebral hypoperfusion, mild cognitive impairment, and dementia. Neurobiol. Aging 2000, 21, 161–169. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Maestre, G.E.; Arizaga, R.; Friedland, R.P.; Galasko, D.; Hall, K.; Luchsinger, J.A.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef]
- Wimo, A.; Guerchet, M.; Ali, G.C.; Wu, Y.T.; Prina, A.M.; Winblad, B.; Jönsson, L.; Liu, Z.; Prince, M. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimer’s Dement. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Wang, M.; Zhang, W.; Bai, M.; Du, Y.; Zhang, Z.; Li, Z.; Miao, J. Neuronal damage, central cholinergic dysfunction and oxidative damage correlate with cognitive deficits in rats with chronic cerebral hypoperfusion. Neurobiol. Learn. Mem. 2014, 109, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Singh, P.; Sharma, B.M.; Sharma, B. Neuroprotective effects of agomelatine and vinpocetine against chronic cerebral hypoperfusion induced vascular dementia. Curr. Neurovascular Res. 2015, 12, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Bin-Jaliah, I.; Sakr, H.F. Melatonin ameliorates brain oxidative stress and upregulates senescence marker protein-30 and osteopontin in a rat model of vascular dementia. Physiol. Int. 2018, 105, 38–52. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Nagle, C.A.; Freire, F.; Rosner, J.M. Effects of melatonin on neurotransmitter uptake and release by synaptosome-rich homogenates of the rat hypothalamus. Neuroendocrinology 1975, 18, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Endo, H.; Chan, P.H. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 2009, 109 (Suppl. S1), 133–138. [Google Scholar] [CrossRef]
- Das, A.; McDowell, M.; Pava, M.J.; Smith, J.A.; Reiter, R.J.; Woodward, J.J.; Varma, A.K.; Ray, S.K.; Banik, N.L. The inhibition of apoptosis by melatonin in vsc4.1 motoneurons exposed to oxidative stress, glutamate excitotoxicity, or tnf-alpha toxicity involves membrane melatonin receptors. J. Pineal Res. 2010, 48, 157–169. [Google Scholar] [CrossRef]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates d-galactose-induced memory impairment, neuroinflammation and neurodegeneration via rage/nf-k b/jnk signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef]
- Mauriz, J.L.; Collado, P.S.; Veneroso, C.; Reiter, R.J.; González-Gallego, J. A review of the molecular aspects of melatonin’s anti-inflammatory actions: Recent insights and new perspectives. J. Pineal Res. 2013, 54, 1–14. [Google Scholar] [CrossRef]
- Ishigami, A.; Handa, S.; Maruyama, N.; Supakar, P.C. Nuclear localization of senescence marker protein-30, smp30, in cultured mouse hepatocytes and its similarity to rna polymerase. Biosci. Biotechnol. Biochem. 2003, 67, 158–160. [Google Scholar] [CrossRef][Green Version]
- Yun, S.P.; Han, Y.S.; Lee, J.H.; Kim, S.M.; Lee, S.H. Melatonin rescues mesenchymal stem cells from senescence induced by the uremic toxin p-cresol via inhibiting mtor-dependent autophagy. Biomol. Ther. 2018, 26, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Meller, R.; Stevens, S.L.; Minami, M.; Cameron, J.A.; King, S.; Rosenzweig, H.; Doyle, K.; Lessov, N.S.; Simon, R.P.; Stenzel-Poore, M.P. Neuroprotection by osteopontin in stroke. J. Cereb. Blood Flow Metab. 2005, 25, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ayer, R.; Sugawara, T.; Chen, W.; Sozen, T.; Hasegawa, Y.; Kanamaru, K.; Zhang, J.H. Protective effects of recombinant osteopontin on early brain injury after subarachnoid hemorrhage in rats. Crit. Care Med. 2010, 38, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, Q.; Suzuki, H.; Hartman, R.; Tang, J.; Zhang, J.H. Osteopontin reduced hypoxia-ischemia neonatal brain injury by suppression of apoptosis in a rat pup model. Stroke 2011, 42, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, S.; Dong, Y.; Fan, C.; Zhao, L.; Yang, X.; Li, J.; Di, S.; Yue, L.; Liang, G.; et al. Melatonin prevents cell death and mitochondrial dysfunction via a sirt1-dependent mechanism during ischemic-stroke in mice. J. Pineal Res. 2015, 58, 61–70. [Google Scholar] [CrossRef]
- Feng, Z.; Cheng, Y.; Zhang, J.T. Long-term effects of melatonin or 17 beta-estradiol on improving spatial memory performance in cognitively impaired, ovariectomized adult rats. J. Pineal Res. 2004, 37, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Tian, X.; Sang, W.; Song, R. Effect of melatonin and resveratrol against memory impairment and hippocampal damage in a rat model of vascular dementia. Neuroimmunomodulation 2016, 23, 318–331. [Google Scholar] [CrossRef]
- Leal, G.; Comprido, D.; Duarte, C.B. Bdnf-induced local protein synthesis and synaptic plasticity. Neuropharmacology 2014, 76, 639–656. [Google Scholar] [CrossRef]
- Ninan, I. Synaptic regulation of affective behaviors; role of bdnf. Neuropharmacology 2014, 76, 684–695. [Google Scholar] [CrossRef]
- Imbesi, M.; Uz, T.; Dzitoyeva, S.; Manev, H. Stimulatory effects of a melatonin receptor agonist, ramelteon, on bdnf in mouse cerebellar granule cells. Neurosci. Lett. 2008, 439, 34–36. [Google Scholar] [CrossRef]
- Brusco, L.I.; Márquez, M.; Cardinali, D.P. Monozygotic twins with alzheimer’s disease treated with melatonin: Case report. J. Pineal Res. 1998, 25, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Khachiyants, N.; Trinkle, D.; Son, S.J.; Kim, K.Y. Sundown syndrome in persons with dementia: An update. Psychiatry Investig. 2011, 8, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Klaffke, S.; Staedt, J. Sundowning and circadian rhythm disorders in dementia. Acta Neurol. Belg. 2006, 106, 168–175. [Google Scholar] [PubMed]
- Lammers, M.; Ahmed, A.I. Melatonin for sundown syndrome and delirium in dementia: Is it effective? J. Am. Geriatrics Soc. 2013, 61, 1045–1046. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.N.; Jamieson, S.; Graham, A.J.; Shneerson, J.M. Rem sleep behaviour disorder treated with melatonin in a patient with alzheimer’s disease. Clin. Neurol. Neurosurg. 2008, 110, 492–495. [Google Scholar] [CrossRef]
- Jean-Louis, G.; Zizi, F.; von Gizycki, H.; Taub, H. Effects of melatonin in two individuals with alzheimer’s disease. Percept. Mot. Ski. 1998, 87, 331–339. [Google Scholar] [CrossRef]
- Wade, A.G.; Farmer, M.; Harari, G.; Fund, N.; Laudon, M.; Nir, T.; Frydman-Marom, A.; Zisapel, N. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate alzheimer’s disease: A 6-month, randomized, placebo-controlled, multicenter trial. Clin. Interv. Aging 2014, 9, 947–961. [Google Scholar]
- Alves, G.S.; Carvalho, A.F.; de Amorim de Carvalho, L.; Sudo, F.K.; Siqueira-Neto, J.I.; Oertel-Knochel, V.; Jurcoane, A.; Knochel, C.; Boecker, H.; Laks, J.; et al. Neuroimaging findings related to behavioral disturbances in alzheimer’s disease: A systematic review. Curr. Alzheimer Res. 2017, 14, 61–75. [Google Scholar] [CrossRef]
- Serfaty, M.; Kennell-Webb, S.; Warner, J.; Blizard, R.; Raven, P. Double blind randomised placebo controlled trial of low dose melatonin for sleep disorders in dementia. Int. J. Geriatr. Psychiatry 2002, 17, 1120–1127. [Google Scholar] [CrossRef]
- Singer, C.; Tractenberg, R.E.; Kaye, J.; Schafer, K.; Gamst, A.; Grundman, M.; Thomas, R.; Thal, L.J. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in alzheimer’s disease. Sleep 2003, 26, 893–901. [Google Scholar] [CrossRef]
- Asayama, K.; Yamadera, H.; Ito, T.; Suzuki, H.; Kudo, Y.; Endo, S. Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in alzheimer type dementia. J. Nippon Med. Sch. 2003, 70, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Fainstein, I.; Bonetto, A.J.; Brusco, L.I.; Cardinali, D.P. Effects of melatonin in elderly patients with sleep disturbance: A pilot study. Curr. Ther. Res. 1997, 58, 990–1000. [Google Scholar] [CrossRef]
- Cohen-Mansfield, J.; Garfinkel, D.; Lipson, S. Melatonin for treatment of sundowning in elderly persons with dementia—A preliminary study. Arch. Gerontol Geriatr. 2000, 31, 65–76. [Google Scholar] [CrossRef]
- Mishima, K.; Okawa, M.; Hozumi, S.; Hishikawa, Y. Supplementary administration of artificial bright light and melatonin as potent treatment for disorganized circadian rest-activity and dysfunctional autonomic and neuroendocrine systems in institutionalized demented elderly persons. Chronobiol. Int. 2000, 17, 419–432. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Brusco, L.I.; Liberczuk, C.; Furio, A.M. The use of melatonin in alzheimer’s disease. Neuro Endocrinol. Lett. 2002, 23 (Suppl. S1), 20–23. [Google Scholar]
- Mahlberg, R.; Walther, S. Actigraphy in agitated patients with dementia. Monitoring treatment outcomes. Z. Gerontol. Geriatr. 2007, 40, 178–184. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Furio, A.M.; Brusco, L.I. Clinical aspects of melatonin intervention in alzheimer’s disease progression. Curr. Neuropharmacol. 2010, 8, 218–227. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Vigo, D.E.; Olivar, N.; Vidal, M.F.; Brusco, L.I. Melatonin therapy in patients with alzheimer’s disease. Antioxidants 2014, 3, 245–277. [Google Scholar] [CrossRef]
- de Jonghe, A.; Korevaar, J.C.; van Munster, B.C.; de Rooij, S.E. Effectiveness of melatonin treatment on circadian rhythm disturbances in dementia. Are there implications for delirium? A systematic review. Int. J. Geriatr. Psychiatry 2010, 25, 1201–1208. [Google Scholar] [CrossRef]
- Sanchez-Barcelo, E.J.; Rueda, N.; Mediavilla, M.D.; Martinez-Cue, C.; Reiter, R.J. Clinical uses of melatonin in neurological diseases and mental and behavioural disorders. Curr. Med. Chem. 2017, 24, 3851–3878. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; BaHammam, A.S.; Brown, G.M.; Spence, D.W.; Bharti, V.K.; Kaur, C.; Hardeland, R.; Cardinali, D.P. Melatonin antioxidative defense: Therapeutical implications for aging and neurodegenerative processes. Neurotox. Res. 2013, 23, 267–300. [Google Scholar] [CrossRef] [PubMed]
- Dowling, G.A.; Burr, R.L.; Van Someren, E.J.; Hubbard, E.M.; Luxenberg, J.S.; Mastick, J.; Cooper, B.A. Melatonin and bright-light treatment for rest-activity disruption in institutionalized patients with alzheimer’s disease. J. Am. Geriatr. Soc. 2008, 56, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Riemersma-van der Lek, R.F.; Swaab, D.F.; Twisk, J.; Hol, E.M.; Hoogendijk, W.J.; Van Someren, E.J. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: A randomized controlled trial. JAMA 2008, 299, 2642–2655. [Google Scholar] [CrossRef] [PubMed]
- Bordet, R.; Devos, D.; Brique, S.; Touitou, Y.; Guieu, J.D.; Libersa, C.; Destée, A. Study of circadian melatonin secretion pattern at different stages of parkinson’s disease. Clin. Neuropharmacol. 2003, 26, 65–72. [Google Scholar] [CrossRef]
- Dowling, G.A.; Mastick, J.; Colling, E.; Carter, J.H.; Singer, C.M.; Aminoff, M.J. Melatonin for sleep disturbances in parkinson’s disease. Sleep Med. 2005, 6, 459–466. [Google Scholar] [CrossRef]
- Medeiros, C.A.; Carvalhedo de Bruin, P.F.; Lopes, L.A.; Magalhães, M.C.; de Lourdes Seabra, M.; de Bruin, V.M. Effect of exogenous melatonin on sleep and motor dysfunction in parkinson’s disease. A randomized, double blind, placebo-controlled study. J. Neurol. 2007, 254, 459–464. [Google Scholar] [CrossRef]
- Dowling, G.; Mastick, J.; Aminoff, M. Melatonin for sleep disturbances in parkinson’s disease: A pilot study. Sleep Res. Online 2003, 5, 99–103. [Google Scholar]
- Litvinenko, I.V.; Krasakov, I.V.; Tikhomirova, O.V. [sleep disorders in parkinson’s disease without dementia: A comparative randomized controlled study of melatonin and clonazepam]. Zhurnal Nevrologii Psikhiatrii Imeni S.S. Korsakova 2012, 112, 26–30. [Google Scholar]
- Datieva, V.K.; Rosinskaia, A.V.; Levin, O.S. [the use of melatonin in the treatment of chronic fatigue syndrome and circadian rhythm disorders in parkinson’s disease]. Zhurnal Nevrologii Psikhiatrii Imeni S.S. Korsakova 2013, 113, 77–81. [Google Scholar]
- Jacob, S.; Poeggeler, B.; Weishaupt, J.H.; Sirén, A.L.; Hardeland, R.; Bähr, M.; Ehrenreich, H. Melatonin as a candidate compound for neuroprotection in amyotrophic lateral sclerosis (als): High tolerability of daily oral melatonin administration in als patients. J. Pineal Res. 2002, 33, 186–187. [Google Scholar] [CrossRef]
- Adamczyk-Sowa, M.; Pierzchala, K.; Sowa, P.; Polaniak, R.; Kukla, M.; Hartel, M. Influence of melatonin supplementation on serum antioxidative properties and impact of the quality of life in multiple sclerosis patients. J. Physiol. Pharmacol. 2014, 65, 543–550. [Google Scholar] [PubMed]
- Skarlis, C.; Anagnostouli, M. The role of melatonin in multiple sclerosis. Neurol. Sci. 2020, 41, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin in aging and disease -multiple consequences of reduced secretion, options and limits of treatment. Aging Dis. 2012, 3, 194–225. [Google Scholar] [PubMed]
- Turek, F.W.; Gillette, M.U. Melatonin, sleep, and circadian rhythms: Rationale for development of specific melatonin agonists. Sleep Med. 2004, 5, 523–532. [Google Scholar] [CrossRef]
- Hardeland, R. Investigational melatonin receptor agonists. Expert Opin. Investig. Drugs 2010, 19, 747–764. [Google Scholar] [CrossRef]
- Spadoni, G.; Bedini, A.; Rivara, S.; Mor, M. Melatonin receptor agonists: New options for insomnia and depression treatment. CNS Neurosci. Ther. 2011, 17, 733–741. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Effects | Model | Signaling Pathway | Concentrations | Reference |
|---|---|---|---|---|
| Inhibiting apoptosis | Cell | Bax/bcl-2/caspase-3 | Pharma | [21] |
| Inhibiting Aβ neurotoxicity | Cell | Pin1/GSK3β/NF-κB | Physio | [57] |
| Inhibiting amyloid fibrils | Cell | Apoe4 | Pharma | [59] |
| Inhibiting apoptosis | Animal | Bax/caspase-3/Par-4 | Pharma | [60] |
| Inhibiting tau hyperphosphorylation | Cell | GSK-3β | Pharma | [61] |
| Inhibiting tau hyperphosphorylation | Animal | PI3K/Akt/GSK3β | Pharma | [62] |
| Inhibiting phosphorylation and accumulation of neurofilaments | Cell | PP-2A/PP-1 | Pharma | [67] |
| Inhibiting tau hyperphosphorylation | Animal | PP-2A/PP-1 | Pharma | [68] |
| Inhibiting tau hyperphosphorylation | Animal | GSK3β/PKA/ PP-2A/PP-1 and ER Stress | Pharma | [69] |
| Inhibiting tau hyperphosphorylation | Cell and Animal | ER Stress/GSK-3β/CDK5 | Pharma | [70] |
| Inhibiting tau hyperphosphorylation | Animal | PKA | Pharma | [71] |
| Inhibiting tau hyperphosphorylation and oxidative stress | Cell | GSK-3β | Pharma | [72] |
| Inhibiting tau hyperphosphorylation and Aβ neurotoxicity | Animal | GSK-3β | Pharma | [74] |
| Inhibiting phosphorylation of neurofilaments | Animal | CDK5 | Pharma | [75] |
| Regulating circadian rhythms | Cell | PKC | Physio | [76] |
| Inhibiting tau hyperphosphorylation and oxidative stress | Cell | PP-2A/GSK-3β | Pharma | [77] |
| Inhibiting tau hyperphosphorylation | Animal | PP-2A | Pharma | [78] |
| Inhibiting apoptosis | Cell | Calpain/CDK5 | Pharma | [79] |
| Inhibiting tau hyperphosphorylation | Cell | DAPK1/Pin1 | Physio | [80] |
| Effects | Model | Signaling Pathway | Concentrations | Reference |
|---|---|---|---|---|
| Inhibiting apoptosis and oxidative stress | Animal | CYP2E1/GST/p53/Bax/caspase-9 | Pharma | [91] |
| Inhibiting autophagy and α-synuclein aggregation | Animal | Caspase-3/12 and LC3-II/LAMP-2/cathepsin B | Pharma | [95] |
| Inhibiting apoptosis | Animal | ER stress/Bcl2/caspase-3 | Pharma | [96] |
| Inhibiting autophagy and α-synuclein | Cell | CDK5 | Pharma | [97] |
| Effects | Model | Signaling Pathway | Concentrations | Reference |
|---|---|---|---|---|
| Inhibiting apoptosis | Animal | ER Stress | Pharma | [109] |
| Inhibiting cell death | Cell and Animal | MT1 receptor | Pharma | [116] |
| Improving mitochondrial dysfunction | Animal | Apelin 13 | Pharma | [117] |
| Inhibiting apoptosis | Animal | Caspase-3 | Pharma | [121] |
| Inhibiting apoptosis | Animal | Bax/bcl-2 | Pharma | [123] |
| Inhibiting apoptosis | Cell | Bax/bcl-2 | Pharma | [124] |
| Inhibiting apoptosis | Cell | Bax/bcl-2 | Physio and Pharma | [125] |
| Effects | Model | Signaling Pathway | Concentrations | Reference |
|---|---|---|---|---|
| Inhibiting apoptosis | Animal | NF-κB/bax/bcl-2 | Pharma | [135] |
| Anti-inflammatory | Animal | MT1/Erk1/2 | Pharma | [139] |
| Effects | Model | Signaling Pathway | Concentrations | Reference |
|---|---|---|---|---|
| Inhibiting apoptosis | Animal | Caspase-1/cytochrome c/caspase-3 | Pharma | [141] |
| Inhibiting oxidative stress | Animal | SOD1/SOD2/nNOS | Pharma | [142] |
| Effects | Model | Signaling Pathway | Concentrations | Reference |
|---|---|---|---|---|
| Inhibiting oxidative stress | Animal | SMP30/OPN | Pharma | [154] |
| Inhibiting oxidative stress | Cell | MT1/MT2 | Physio | [157] |
| Inhibiting oxidative stress | Animal | RAGE/NF-κB/JNK | Pharma | [158] |
| Inhibiting autophagy | Cell | MTOR | Pharma | [161] |
| Inhibiting apoptosis | Animal | SIRT1/bax/bcl-2 | Pharma | [165] |
| Design | Subjects | Treatment | Assessment | Results | Reference |
|---|---|---|---|---|---|
| CR | 2 AD patients (age: 79 years) | 6 mg at bedtime for 36 months | Cognitive evaluation by FAST; neuroimaging evaluation by NMR | Significant improvement of sleep quality, reduction of sundowning, and lack of progression of cognitive and behavioral disorders | [171] |
| CR | 1 AD patient (age: 81 years) | 2 mg at 8 p.m. for 1 week, 2 mg at 3 p.m. and 8 p.m. for 2 weeks | Cognitive evaluation by MMSE; neuropsychiatric evaluation by NPI | Significant improvement of sleep quality and behavioral symptoms after the first week, and gradual improvement over the subsequent two weeks | [174] |
| CR | 1 AD patient (age: 68 years) | 5–10 mg at bedtime for 20 months | Sleep evaluation by PSG | Significant effects on suppression of REM sleep behavior disorder | [175] |
| CR | 2 AD patients (age: 72 and 75 years) | 6 mg (2 h before bedtime) for 35 days | Sleep evaluation by actigraphy; cognitive evaluation by ADAS and MMSE | Significant improvement of the circadian rest–activity rhythm and mood and reduction of daytime sleepiness in one of them | [176] |
| R, DB, PC | 73 AD patients (mean age: 75.3 years) | 2 mg (slow-release, 1–2 h before bedtime) for 24 weeks | Sleep evaluation by PSQI; cognitive evaluation by ADAS, MMSE and IADL | Significant improvement of sleep efficiency and cognitive performance | [177] |
| R, DB, PC | 41 AD patients (age: 61–95 years) | 1.5 mg (slow-release) and 8.5 mg (fast-release) at 10 p.m. for 10 days | Sleep evaluation by actigraphy | No significant effects on sleep, circadian rhythms or agitated behaviors | [178] |
| R, DB, PC | 25 patients with dementia (21 AD patients, age: over 65 years) | 6 mg (slow-release) at bedtime for 2 weeks | Sleep evaluation by actigraphy; cognitive evaluation by MMSE | No significant effects on sleep or cognitive function | [179] |
| R, DB, PC | 20 AD patients (mean age: 79.2 years) | 3 mg at 8.5 p.m. for 4 weeks | Sleep evaluation by actigraphy; cognitive evaluation by ADAS, MMSE and CDRS | Significant improvement of the sleep–wake rhythm, cognitive dysfunction and behavioral problems | [181] |
| R, PC | 157 AD patients (mean age: 77.4 years) | 2.5 mg (slow-release) or 10 mg (fast-release),1 h before bedtime for 2 months | Sleep evaluation by actigraphy and diary; cognitive evaluation by ADAS, MMSE and IADL; neuropsychiatric evaluation by NPI and SDI | No significant effects on sleep disturbances by actigraphy; slightly improvement of sleep quality by diary; no effects on cognitive function | [180] |
| R, PC | 24 AD patients (mean age: 78.6 years) | 3 mg at bedtime for 2 weeks | Sleep evaluation by actigraphy; neuropsychiatric evaluation by NPI | Significant improvement of circadian rhythm disturbances, agitation and behavioral symptoms | [186] |
| R, PC | 50 AD patients (mean age: 86 years) | 5 mg melatonin and 1 h morning light (≥2500 lux) for 10 weeks | Sleep evaluation by actigraphy | Significant improvement of the rest–activity rhythm | [192] |
| OL | 14 AD patients (mean age: 72 years) | 9 mg at bedtime for 22–35 months | Sleep evaluation by diary; cognitive evaluation by FAST, ADAS, MMSE and Mattis’ and Blessed’s scales | Significant improvement of sleep quality; no cognitive or behavioral deterioration and loss of sundown syndrome | [171] |
| OL | 10 AD patients (mean age: 74 years) | 3 mg at bedtime for 3 weeks | Sleep evaluation by diary | Significant improvement of sleep disturbances and sundowning | [182] |
| OL | 11 AD patients (mean age: 85 years) | 3 mg at bedtime for 3 weeks | Sleep evaluation by diary | Significant attenuation of daytime sleepiness and agitation | [183] |
| OL, PC | 14 AD patients | 6 mg at 9 p.m. for 4 weeks | Sleep evaluation by actigraphy and diary | Significant improvement of insomnia | [184] |
| OL | 45 AD patients (mean age: 73 years) | 6–9 mg at bedtime for 4 months | Sleep evaluation by diary; cognitive evaluation by FAST | Significant improvement of sleep quality, sundowning, and cognitive and behavioral impairment | [185] |
| OL | 7 AD patients (mean age: 75.6 years) | 3 mg at around 9 p.m. for 3 weeks | Sleep evaluation by actigraphy; cognitive evaluation by MMSE and GDS | Significant improvement of circadian rhythm dysfunction and sundown syndrome | [51] |
| R, DB, PC | 40 PD patients (age: 40–80 years) | 5–50 mg at bedtime for 2 weeks | Sleep evaluation by actigraphy and diary, ESS, SSS and GSDS | Significant increased nighttime sleep with 50 mg by objective; significant improvement of sleep quality with 5 mg only by subjective but not objective | [195] |
| R, DB, PC | 18 PD patients (mean age: 61.8 years) | 3 mg at bedtime for 4 weeks | Sleep evaluation by PSG, PSQI and ESS; motor evaluation by UPDRS | Significant improvement of sleep quality; no improvement of motor dysfunction | [196] |
| R | 38 PD patients (mean age: 67.3 years) | 3 mg (30 min before bedtime) for 6 weeks | Sleep evaluation by PSG, PDSS and ESS; cognitive evaluation by MMSE, five-word test, digit span and the Hamilton scale | Significant improvement of sleep quality, daytime sleepiness and cognitive dysfunction | [198] |
| R | 30 PD patients (mean age: 64.1 years) | 3 mg at bedtime for 2 months | Sleep evaluation by PDSS and ESS; neuropsychiatric evaluation by Beck’s scale and Spielberger’s scale | Significant improvement of sleep quality and anxiety status; no significant changes in motor, cognitive or autonomic dysfunction or depression status | [199] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Zhang, T.; Lee, T.H. Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases. Biomolecules 2020, 10, 1158. https://doi.org/10.3390/biom10081158
Chen D, Zhang T, Lee TH. Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases. Biomolecules. 2020; 10(8):1158. https://doi.org/10.3390/biom10081158
Chicago/Turabian StyleChen, Dongmei, Tao Zhang, and Tae Ho Lee. 2020. "Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases" Biomolecules 10, no. 8: 1158. https://doi.org/10.3390/biom10081158
APA StyleChen, D., Zhang, T., & Lee, T. H. (2020). Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases. Biomolecules, 10(8), 1158. https://doi.org/10.3390/biom10081158