Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers


Abstract
1. Introduction
2. Significance of Immunosuppressive Cells in Developing Skin Cancers
2.1. Significance of TAMs in Developing Skin Cancers
2.1.1. Chemokines from TAMs Determine Profiles of Tumor-Infiltrating Lymphocytes (TILs) in the Tumor Microenvironment
2.1.2. Angiogenic Factors from TAMs
2.2. Myeloid-Derived Suppressor Cells (MDSCs)
2.2.1. Significance of MDSCs in Developing Skin Cancers
2.2.2. MDSCs and ICIs
2.2.3. Cross-Talk between MDSCs and Other Immunosuppressive Cells
2.3. Regulatory T Cells: Tregs
2.3.1. Significance of Tregs in Developing Skin Cancers
2.3.2. Tregs and ICIs: Anti-PD1 Abs and Anti-CTLA4 Abs
2.4. TANs in Developing Skin Cancers
3. Immunosuppressive Myeloid Cells as a Target of Immunotherapy for Skin Cancer
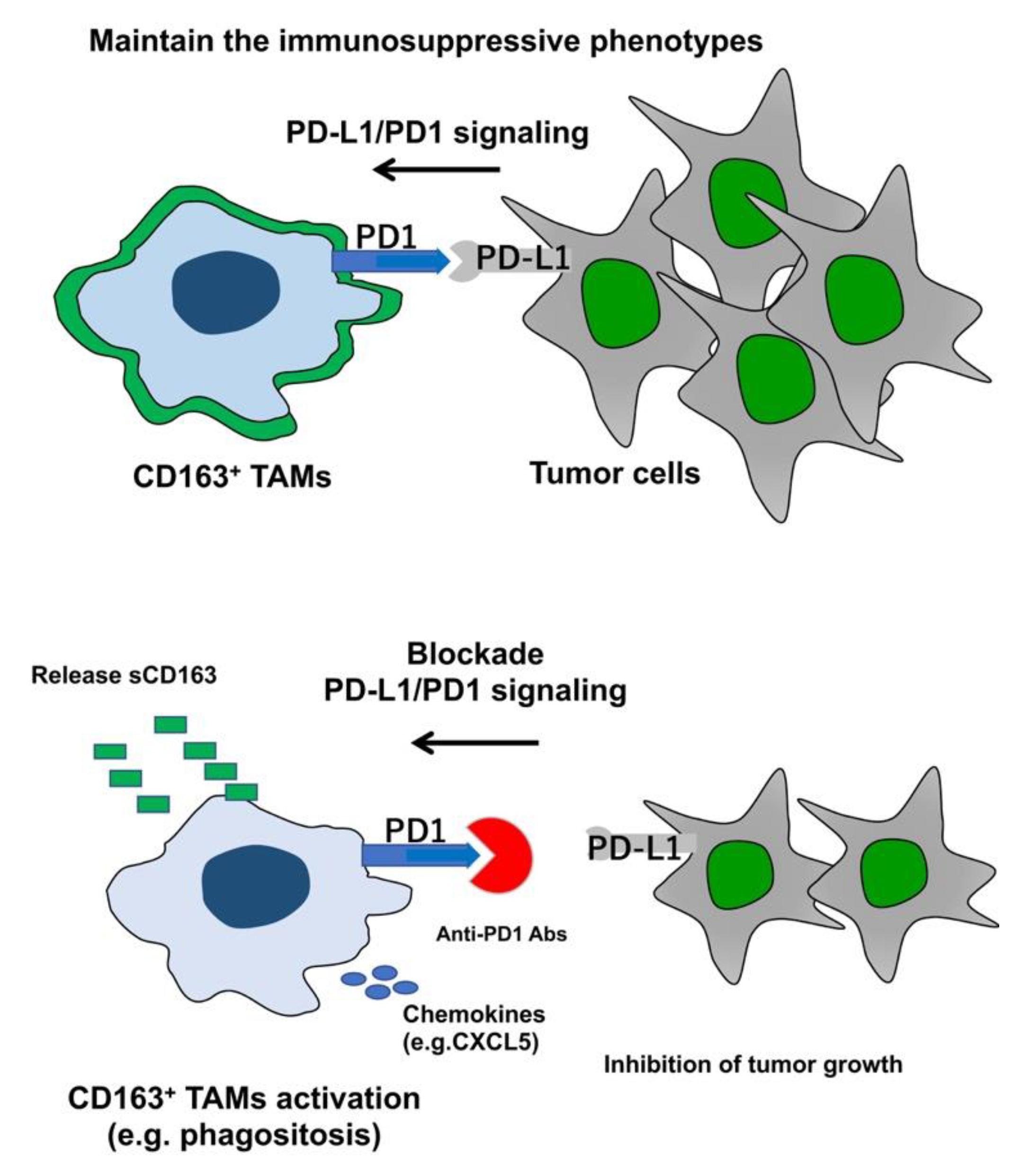
3.1. TAMs as a Target for Immunotherapy
3.2. MDSCs as a Target for Immunotherapy
4. TAM-Related Biomarkers for Predicting the Efficacy of ICIs
5. Roles of TAMs in Immune-Related Adverse Events irAEs
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Fujimura, T.; Kambayashi, Y.; Fujisawa, Y.; Hidaka, T.; Aiba, S. Tumor-associated macrophages: Therapeutic targets for skin cancer. Front. Oncol. 2018, 8, 3–9. [Google Scholar] [CrossRef]
- Baay, M.; Brouwer, A.; Pauwels, P.; Peeters, M.; Lardon, F. Tumor cells and tumor-associated macrophages: Secreted proteins as potential targets for therapy. Clin. Dev. Immunol. 2011, 2011, 565187–565199. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Ring, S.; Umansky, V.; Mahnke, K.; Enk, A.H. Regulatory T cells (Treg) stimulate B7-H1 expression in myeloid derived suppressor cells (MDSC) in ret melanomas. J. Investig. Dermatol. 2012, 132, 1239–1246. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-associated neutrophils in cancer: Going pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef]
- Thompson, A.K.; Kelley, B.F.; Prokop, L.J.; Murad, M.H.; Baum, C.L. Risk factors for cutaneous squamous cell carcinoma recurrence, metastasis, and disease-specific death: A systematic review and meta-analysis. JAMA Dermatol. 2016, 152, 419–428. [Google Scholar] [CrossRef]
- Hidaka, T.; Fujimura, T.; Aiba, S. Aryl hydrocarbon receptor modulates carcinogenesis and maintenance of skin cancers. Front. Med. 2019, 6, 194. [Google Scholar] [CrossRef]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. AhR links atopic dermatitis and air pollution 1 via Artemin induction. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef]
- Wang, F.; Li, B.; Wei, Y.; Zhao, Y.; Wang, L.; Zhang, P.; Yang, J.; He, W.; Chen, H.; Jiao, Z.; et al. Tumor-derived exosomes induce PD1+ macrophage population in human gastric cancer that promotes disease progression. Oncogenesis 2018, 7, 41–52. [Google Scholar] [CrossRef]
- Li, B.; Song, T.N.; Wang, F.R.; Yin, C.; Li, Z.; Lin, J.P.; Meng, Y.Q.; Feng, H.M.; Jing, T. Tumor-derived exosomal HMGB1 promotes esophageal squamous cell carcinoma progression through inducing PD1+ TAM expansion. Oncogenesis 2019, 8, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.S.; Divino, C.M.; Chen, S. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Sato, Y.; Tanita, K.; Kambayashi, Y.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; Funakoshi, T.; Hata, H.; et al. Serum soluble CD163 and CXCL5 could be predictive markers for immune related adverse event in patients with advanced melanoma treated with nivolumab. Oncotarget 2018, 9, 15542–15551. [Google Scholar] [CrossRef] [PubMed]
- Furudate, S.; Fujimura, T.; Kakizaki, A.; Kambayashi, Y.; Asano, M.; Watabe, A.; Aiba, S. The possible interaction between periostin expressed by cancer stroma and tumor-associated macrophages in developing mycosis fungoides. Exp. Dermatol. 2016, 25, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Furudate, S.; Fujimura, T.; Kakizaki, A.; Hidaka, T.; Asano, M.; Aiba, S. Tumor-associated M2 macrophages in mycosis fungoides acquire immunomodulatory function by interferon alpha and interferon gamma. J. Dermatol. Sci. 2016, 83, 182–189. [Google Scholar] [CrossRef]
- Kakizaki, A.; Fujimura, T.; Furudate, S.; Kambayashi, Y.; Yamauchi, T.; Yagita, H.; Aiba, S. Immunomodulatory effect of peritumoral administration of interferon-beta on melanoma through tumor-associated macrophages. Oncoimmunology 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef]
- Fujimura, T.; Hidaka, T.; Kambayashi, Y.; Furudate, S.; Kakizaki, A.; Tono, H.; Tsukada, A.; Haga, T.; Hashimoto, A.; Morimoto, R.; et al. Phase I study of nivolumab combined with IFN-b for patients with advanced melanoma. Oncotarget 2017, 8, 71181–71187. [Google Scholar] [CrossRef][Green Version]
- Kambayashi, Y.; Fujimura, T.; Furudate, S.; Asano, M.; Kakizaki, A.; Aiba, S. The possible interaction between receptor activator of nuclear factor kappa-B ligand (RANKL) expressed by extramammary Paget cells and its ligand on dermal macrophages. J. Investig. Dermatol. 2015, 135, 2547–2550. [Google Scholar] [CrossRef]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.; Bhuvanendran, S.; Johnson-Huang, L.M.; et al. Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J. Investig. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Gaiser, M.R.; Weis, C.A.; Gaiser, T.; Jiang, H.; Buder-Bakhaya, K.; Herpel, E.; Warth, A.; Xiao, Y.; Miao, L.; Brownell, I. Merkel cell carcinoma expresses the immunoregulatory ligand CD200 and induces immunosuppressive macrophages and regulatory T cells. Oncoimmunology 2018, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujimura, T.; Tanita, K.; Chunbing, L.; Matsushita, S.; Fujisawa, Y.; Otsuka, A.; Yamamoto, Y.; Hidaka, T.; Aiba, S. Malassezia-derived aryl hydrocarbon receptor ligands enhance the CCL20/Th17/soluble CD163 pathogenic axis in extra-mammary Paget’s disease. Exp. Dermatol. 2019, 28, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Werchau, S.; Toberer, F.; Enk, A.; Dammann, R.; Helmbold, P. Merkel cell carcinoma induces lymphatic microvessel formation. J. Am. Acad. Dermatol. 2012, 67, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, Y.; Zhu, P.; Zhang, D.; Liu, S.; Tang, M.; Wang, Y.; Jin, Z.; Li, D.; Yan, D.; et al. TRIM59 loss in M2 macrophages promotes melanoma migration and invasion by upregulating MMP-9 and Madcam1. Aging 2019, 11, 8623–8641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Evans, K.; Xiao, C.; DeVito, N.; Theivanthiran, B.; Holtzhausen, A.; Siska, P.J.; Blobe, G.C.; Hanks, B.A. Stromal fibroblasts mediate anti-PD-1 resistance via MMP-9 and dictate TGFβ inhibitor sequencing in melanoma. Cancer Immunol. Res. 2018, 6, 1459–1471. [Google Scholar] [CrossRef]
- Wang, T.; Ge, Y.; Xiao, M.; Lopez-Coral, A.; Azuma, R.; Somasundaram, R.; Zhang, G.; Wei, Z.; Xu, X.; Rauscher, F.J., 3rd; et al. Melanoma-derived conditioned media efficiently induce the differentiation of monocytes to macrophages that display a highly invasive gene signature. Pigment Cell Melanoma Res. 2012, 25, 493–505. [Google Scholar] [CrossRef]
- Bergenfelz, C.; Leandersson, K. The generation and identity of human myeloid-derived suppressor cells. Front. Oncol. 2020, 10, 109–121. [Google Scholar] [CrossRef]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef]
- Mengos, A.E.; Gastineau, D.A.; Gustafson, M.P. The CD14+HLA-DRlo/neg monocyte: An immunosuppressive phenotype that restrains responses to cancer immunotherapy. Front. Immunol. 2019, 10, 1147–1161. [Google Scholar] [CrossRef]
- Jahchan, N.S.; Mujal, A.M.; Pollack, J.L.; Binnewies, M.; Sriram, V.; Reyno, L.; Krummel, M.F. Tuning the tumor myeloid microenvironment to fight cancer. Front. Immunol. 2019, 10, 1611–1631. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulos, K.V.; Pulitzer, M.; Perez, S.; Korkolopoulou, P.; Angelopoulou, M.; Baxevanis, C.; Palomba, M.L.; Siakantaris, M. Tumor-infiltrating and circulating granulocytic myeloid-derived suppressor cells correlate with disease activity and adverse clinical outcomes in mycosis fungoides. Clin. Transl. Oncol. 2019. In Press. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Hu, S.; Wu, J.C.; Martelli, F.; Bronte, V.; Chouaib, S. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res. 2015, 75, 3771–3787. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 2018, 9, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The role of CXC chemokine receptors 1-4 on immune cells in the tumor microenvironment. Front. Immunol. 2018, 9, 2159–2168. [Google Scholar] [CrossRef]
- Han, X.; Shi, H.; Sun, Y.; Shang, C.; Luan, T.; Wang, D.; Ba, X.; Zeng, X. CXCR2 expression on granulocyte and macrophage progenitors under tumor conditions contributes to mo-MDSC generation via SAP18/ERK/STAT3. Cell Death Dis. 2019, 10, 598–613. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ma, X.L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Shi, L.; Venkataramani, M.; Abdelaal, A.M.; Culpepper, C.; Kidder, K.; Liang, H.; Zen, K.; Liu, Y. Tumor conditions induce bone marrow expansion of granulocytic, but not monocytic, immunosuppressive leukocytes with increased CXCR2 expression in mice. Eur. J. Immunol. 2018, 48, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Samaniego, R.; Gutiérrez-González, A.; Gutiérrez-Seijo, A.; Sánchez-Gregorio, S.; García-Giménez, J.; Mercader, E.; Márquez-Rodas, I.; Avilés, J.A.; Relloso, M.; Sánchez-Mateos, P. CCL20 expression by tumor-associated macrophages predicts progression of human primary cutaneous melanoma. Cancer Immunol. Res. 2018, 6, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef]
- Nagase, H.; Takeoka, T.; Urakawa, S.; Morimoto-Okazawa, A.; Kawashima, A.; Iwahori, K.; Takiguchi, S.; Nishikawa, H.; Sato, E.; Sakaguchi, S.; et al. ICOS+ Foxp3+ TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int. J. Cancer 2017, 140, 686–695. [Google Scholar] [CrossRef]
- Walker, L.S.; Sansom, D.M. Confusing signals: Recent progress in CTLA-4 biology. Trends Immunol. 2015, 36, 63–70. [Google Scholar] [CrossRef]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Gambichler, T.; Schröter, U.; Höxtermann, S.; Susok, L.; Stockfleth, E.; Becker, J.C. Decline of programmed death-1-positive circulating T regulatory cells predicts more favourable clinical outcome of patients with melanoma under immune checkpoint blockade. Br. J. Dermatol. 2020, 182, 1214–1220. [Google Scholar] [CrossRef]
- Gambichler, T.; Schröter, U.; Höxtermann, S.; Susok, L.; Stockfleth, E.; Becker, J.C. A brief communication on circulating PD-1-positive T-regulatory lymphocytes in melanoma patients undergoing adjuvant immunotherapy with nivolumab. J. Immunother. 2019, 42, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Schoonderwoerd, M.J.; Koops, M.F.; Angela, R.A.; Koolmoes, B.; Toitou, M.; Paauwe, M.; Barnhoorn, M.C.; Liu, Y.; Sier, C.F.; Hardwick, J.C.; et al. Targeting endoglin expressing regulatory T cells in the tumor microenvironment enhances the effect of PD1 checkpoint inhibitor immunotherapy. Clin. Cancer Res. 2020. In Press. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef] [PubMed]
- Forsthuber, A.; Lipp, K.; Andersen, L.; Ebersberger, S.; Graña-Castro, O.; Ellmeier, W.; Petzelbauer, P.; Lichtenberger, B.M.; Loewe, R. CXCL5 as regulator of neutrophil function in cutaneous melanoma. J. Investig. Dermatol. 2019, 139, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, J.; Wu, C.F.; Andzinski, L.; Leschner, S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef]
- Simon, N.; Antignani, A.; Hewitt, S.M.; Gadina, M.; Alewine, C.; FitzGerald, D. Tofacitinib enhances delivery of antibody-based therapeutics to tumor cells through modulation of inflammatory cells. JCI Insight 2019, 4, 1–18. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Neutrophils in cancer: Two sides of the same coin. J. Immunol. Res. 2015, 2015, 983698–983719. [Google Scholar] [CrossRef]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Wu, L.; Chen, X.; Zhao, J.; Martin, B.; Zepp, J.A.; Ko, J.S.; Gu, C.; Cai, G.; Ouyang, W.; Sen, G.; et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J. Exp. Med. 2015, 212, 1571–1587. [Google Scholar] [CrossRef] [PubMed]
- Gasparoto, T.H.; de Oliveira, C.E.; de Freitas, L.T.; Pinheiro, C.R.; Ramos, R.N.; da Silva, A.L.; Cavassani, K.A.; Campanelli, A.P. Inflammatory events during murine squamous cell carcinoma development. J. Inflamm. 2012, 9, 46–57. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Pascual-García, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martínez-Ricarte, F.; et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8+ T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 2019, 10, 2416–2427. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Okuyama, R.; Ohtani, T.; Ito, Y.; Haga, T.; Hashimoto, A.; Aiba, S. Perilesional treatment of metastatic melanoma with interferon-beta. Clin. Exp. Dermatol. 2009, 34, 793–799. [Google Scholar] [CrossRef]
- Antsiferova, M.; Piwko-Czuchra, A.; Cangkrama, M.; Wietecha, M.; Sahin, D.; Birkner, K.; Amann, V.C.; Levesque, M.; Hohl, D.; Dummer, R.; et al. Activin promotes skin carcinogenesis by attraction and reprogramming of macrophages. EMBO Mol. Med. 2017, 9, 27–45. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974–3990. [Google Scholar] [CrossRef]
- Nam, S.; Kang, K.; Cha, J.S.; Kim, J.W.; Lee, H.G.; Kim, Y.; Yang, Y.; Lee, M.S.; Lim, J.S. Interferon regulatory factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J. Leukoc. Biol. 2016, 100, 1273–1284. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity 2017, 47, 284–297. [Google Scholar] [CrossRef]
- Sierra, R.A.; Trillo-Tinoco, J.; Mohamed, E.; Yu, L.; Achyut, B.R.; Arbab, A.; Bradford, J.W.; Osborne, B.A.; Miele, L.; Rodriguez, P.C. Anti-Jagged immunotherapy inhibits MDSCs and overcomes tumor-induced tolerance. Cancer Res. 2017, 77, 5628–5638. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. CCR5 in recruitment and activation of myeloid-derived suppressor cells in melanoma. Cancer Immunol. Immunother. 2017, 66, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Han, X.; Sun, Y.; Shang, C.; Wei, M.; Ba, X.; Zeng, X. Chemokine (C-X-C motif) ligand 1 and CXCL2 produced by tumor promote the generation of monocytic myeloid-derived suppressor cells. Cancer Sci. 2018, 109, 3826–3839. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, Y.; Fujimura, T.; Hidaka, T.; Aiba, S. Biomarkers for the prediction of efficacies of anti-PD1 antibodies: Mini reviews. Front. Med. 2019, 6, 174. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Ho, W.J.; Yarchoan, M.; Hopkins, A.; Mehra, R.; Grossman, S.; Kang, H. Association between pretreatment lymphocyte count and response to PD1 inhibitors in head and neck squamous cell carcinomas. J. Immunother. Cancer 2018, 6, 84–92. [Google Scholar] [CrossRef]
- Fujisawa, Y.; Yoshino, K.; Otsuka, A.; Funakoshi, T.; Fujimura, T.; Yamamoto, Y.; Hata, H.; Tanaka, R.; Yamaguchi, K.; Nonomura, Y.; et al. Baseline neutrophil to lymphocyte ratio and serum LDH level associated with outcome of nivolumab immunotherapy in Japanese advanced melanoma population. Br. J. Dermatol. 2018, 179, 213–215. [Google Scholar] [CrossRef]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Yagi, T.; Baba, Y.; Okadome, K.; Kiyozumi, Y.; Hiyoshi, Y.; Ishimoto, T.; Iwatsuki, M.; Miyamoto, Y.; Yoshida, N.; Watanabe, M.; et al. Tumour-associated macrophages are associated with poor prognosis and programmed death ligand 1 expression in oesophageal cancer. Eur. J. Cancer 2019, 111, 38–49. [Google Scholar] [CrossRef]
- Fujimura, T.; Sato, Y.; Tanita, K.; Kambayash, Y.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; Funakoshi, T.; Hata, H.; et al. Serum level of soluble CD163 may be a predictive marker of the effectiveness of nivolumab in patients with advanced cutaneous melanoma. Front. Oncol. 2018, 8, 530–538. [Google Scholar] [CrossRef]
- Fujimura, T.; Sato, Y.; Tanita, K.; Lyu, C.; Kambayash, Y.; Amagai, R.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; et al. Association of baseline serum levels of CXCL5 with the efficacy of nivolumab in advanced melanoma. Front. Med. 2019, 6, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Tanita, K.; Sato, Y.; Lyu, C.; Kambayashi, Y.; Fujisawa, Y.; Uchi, H.; Yamamoto, Y.; Otsuka, A.; Yoshino, K.; et al. Immune checkpoint inhibitor-induced vitiligo in advanced melanoma could be related to increased levels of CCL19. Br. J. Dermatol. 2020, 182, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Darnell, E.P.; Mooradian, M.J.; Baruch, E.N.; Yilmaz, M.; Reynolds, K.L. Immune-related adverse events (irAEs): Diagnosis, management, and clinical pearls. Curr. Oncol. Rep. 2020, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Furudate, S.; Kakizaki, A.; Hidaka, T.; Haga, T.; Hashimoto, A.; Morimoto, R.; Aiba, S. Isolated ACTH deficiency possibly caused by nivolumab in a metastatic melanoma patient. J. Dermatol. 2017, 44, 13–14. [Google Scholar] [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Tanita, K.; Sato, Y.; Hidaka, T.; Otsuka, A.; Tanaka, H.; Furudate, S.; Hashimoto, A.; Aiba, S. Two cases of Vogt-Koyanagi Harada disease-like uveitis developing from an advanced melanoma patient treated by sequential administration of nivolumab and dabrafenib/trametinib therapy. J. Dermatol. 2018, 45, 735–737. [Google Scholar] [CrossRef]
- Nelson, C.A.; Singer, S.; Chen, T.; Puleo, A.E.; Lian, C.G.; Wei, E.X.; Giobbie-Hurder, A.; Mostaghimi, A.; LeBoeuf, N.R. Bullous pemphigoid after anti-PD-1 therapy: A retrospective case-control study evaluating impact on tumor response and survival outcomes. J. Am. Acad. Dermatol. 2020. In Press. [Google Scholar] [CrossRef]
- Voudouri, D.; Nikolaou, V.; Laschos, K.; Charpidou, A.; Soupos, N.; Triantafyllopoulou, I.; Panoutsopoulou, I.; Aravantinos, G.; Syrigos, K.; Stratigos, A. Anti-PD1/PDL1 induced psoriasis. Curr. Probl. Cancer 2017, 41, 407–412. [Google Scholar] [CrossRef]
- Yilmaz, M.; Mese, S.G.; Celik, U. Nivolumab-induced lichen planus. J. Oncol. Pharm. Pract. 2019, 23, 758–760. [Google Scholar] [CrossRef]
- Tojo, G.; Fujimura, T.; Kawano, M.; Ogasawara, K.; Kambayashi, Y.; Furudate, S.; Mizuashi, M.; Aiba, S. Comparison of IL-17 producing cells in different clinical types of alopecia areata. Dermatology 2013, 227, 77–82. [Google Scholar] [CrossRef]
- Furudate, S.; Fujimura, T.; Kambayashi, Y.; Kakizaki, A.; Aiba, S. Comparison of CD163+ CD206+ M2 macrophages in the lesional skin of bullous pemphigoid and pemphigus vulgaris: The possible pathogenesis of bullous pemphigoid. Dermatology 2014, 229, 369–378. [Google Scholar] [CrossRef]
- Fujimura, T.; Kakizaki, A.; Furudate, S.; Aiba, S. A possible interaction between periostin and CD163+ skin-resident macrophages in pemphigus vulgaris and bullous pemphigoid. Exp. Dermatol. 2017, 26, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Riani, M.; Muller, C.; Bour, C.; Bernard, P.; Antonicelli, F.; Le Jan, S. Blister fluid induces MMP-9-associated M2-type macrophages in bullous pemphigoid. Front. Immunol. 2019, 10, 1858–1869. [Google Scholar] [CrossRef]
- Fuentes-Duculan, J.; Suárez-Fariñas, M.; Zaba, L.C.; Nograles, K.E.; Pierson, K.C.; Mitsui, H.; Pensabene, C.A.; Kzhyshkowska, J.; Krueger, J.G.; Lowes, M.A. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J. Investig. Dermatol. 2010, 130, 2412–2422. [Google Scholar] [CrossRef]
- Van Gorp, H.; Delputte, P.L.; Nauwynck, H.J. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol. Immunol. 2010, 47, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Keller, M.; Kim, Y.; Cronin, H.; Richards, A.; Gibney, G.; Weber, J.S. Nivolumab in resected and unresectable metastatic melanoma: Characteristics of immune-related adverse events and association with outcomes. Clin. Cancer Res. 2016, 22, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Sato, T.; Tanita, K.; Amagai, R.; Hashimoto, A.; Hidaka, T.; Aiba, S. Successful treatment of unresectable advanced melanoma with pre-surgical administration of nivolumab with ipilimumab. Front. Med. 2019, 6, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Sugihara, S.; Mori, S.; Hamada, K.; Sasaki, Y.; Yoshikawa, S.; Kiyohara, Y. Immune-related adverse events correlate with improved survival in patients with advanced mucosal melanoma treated with nivolumab: A single-center retrospective study in Japan. J. Dermatol. 2020. In Press. [Google Scholar] [CrossRef]
- Okada, N.; Kawazoe, H.; Takechi, K.; Matsudate, Y.; Utsunomiya, R.; Zamami, Y.; Goda, M.; Imanishi, M.; Chuma, M.; Hidaka, N.; et al. Association between immune-related adverse events and clinical efficacy in patients with melanoma treated with nivolumab: A multicenter retrospective study. Clin. Ther. 2019, 41, 59–67. [Google Scholar] [CrossRef]


{kind=link}
| Subtypes | Positive | Negative | |
|---|---|---|---|
| TAMs | M1 | CD68, CD86, CD169, HLA-DR, CCR7 | |
| M2 | CD163, CD204, CD206, PD-L1, ARG1 | ||
| MDSCs | MoMDSC | CD11b, PGE2, IL-10, TGFb, iNOS, ARG1 | HLA-DR, CD14 |
| G-MDSC | CD15, CD33, CD66b, ROS, G-CSF, ARG1 | HLA-DR, CD14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimura, T.; Aiba, S. Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules 2020, 10, 1087. https://doi.org/10.3390/biom10081087
Fujimura T, Aiba S. Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules. 2020; 10(8):1087. https://doi.org/10.3390/biom10081087
Chicago/Turabian StyleFujimura, Taku, and Setsuya Aiba. 2020. "Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers" Biomolecules 10, no. 8: 1087. https://doi.org/10.3390/biom10081087
APA StyleFujimura, T., & Aiba, S. (2020). Significance of Immunosuppressive Cells as a Target for Immunotherapies in Melanoma and Non-Melanoma Skin Cancers. Biomolecules, 10(8), 1087. https://doi.org/10.3390/biom10081087