POSCAbilities: The Application of the Prion Organotypic Slice Culture Assay to Neurodegenerative Disease Research



Abstract
1. Introduction
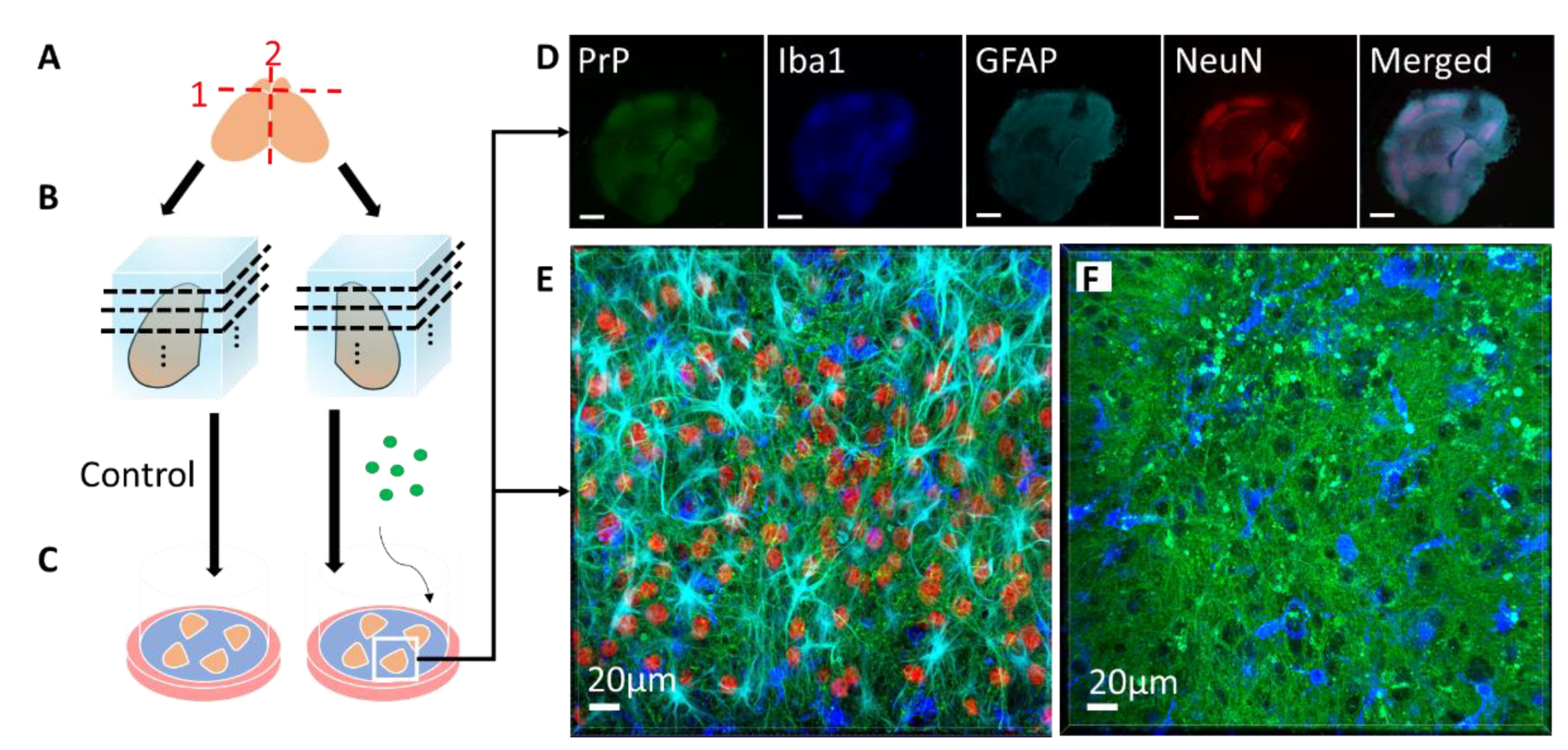
2. Prion Organotypic Slice Culture Assay (POSCA)
2.1. The Origin and Evolution of Prion Organotypic Slice Culture Assay
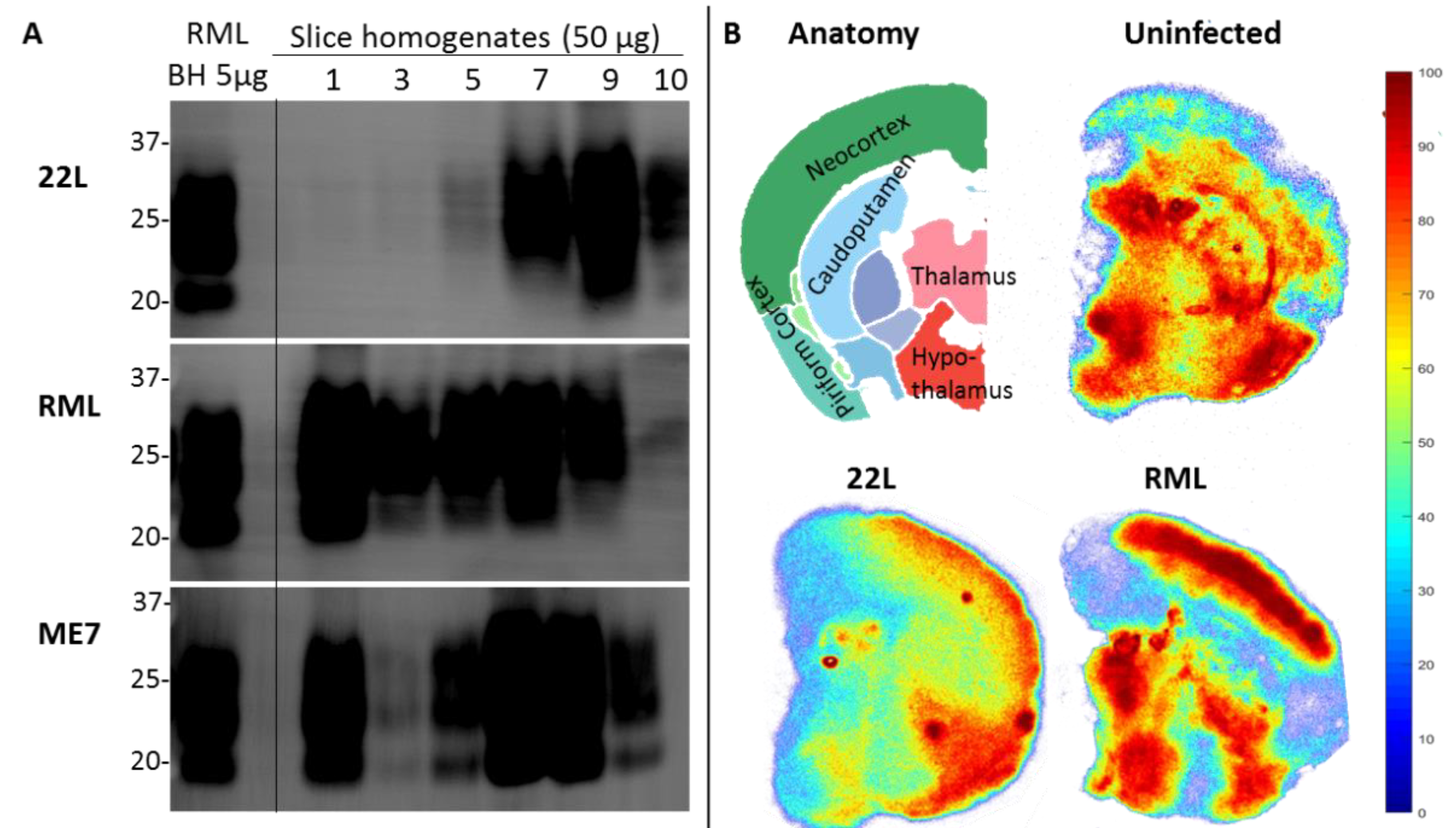
2.2. Pathology in Prion Organotypic Slice Culture Assay
2.3. Manipulation of Glia in Prion Organotypic Slice Culture Assay
2.4. Prion Organotypic Slice Culture Assay as a Tool for Drug Screening
3. Slice Culture to Study Prion-Like Mechanisms in Alzheimer’s Disease
3.1. Slice Culture from Alzheimer’s disease Transgenic Mouse Models
3.2. Application of Exogenous Amyloid Beta to Promote Aggregate Formation
3.3. Manipulation of Microglia
3.4. Drug Screening
3.5. Tau Aggregation in Slice Culture
3.6. Tau Spreading
4. Slice Culture to Study Prion-Like Mechanisms in Parkinson’s Disease
4.1. Induction of Rodent α-Syn Aggregation and Spreading
4.2. Induction of Human α-Syn Aggregation
4.3. Application of Exogenous α-Syn to Promote Aggregation
4.4. The Role of Cell Type in α-Syn Spreading
4.5.α- Syn Strains
5. Slice Culture to Study Prion-Like Mechanisms in Amyotrophic Lateral Sclerosis
5.1. Induction of TDP-43 Aggregation
5.2. Addition of Exogenous TDP-43
5.3. Application of Exogenous SOD1 to Promote Aggregation
5.4. Drug Screening
6. Slice Culture to Study Prion-Like Mechanisms in Huntington’s Disease
6.1. Induction of Human mHtt Aggregation
6.2. Prion-Like Spreading of mHtt
6.3. Manipulation of Microglia
6.4. Drug Screening
7. Advantages and Disadvantages of Organotypic Slice Culture for Neurodegenerative Disease Research
7.1. Economic and Ethical Advantages of Slice Culture
7.2. Seeded Aggregation
7.3. Cellular Manipulation
7.4. Genetic Manipulation
7.5. Prion-Like Spread
7.6. Drug Screening
7.7. Strain Features
8. Suggestions for Future Investigation
8.1. Strain Tropism
8.2. Slice Age
8.3. Transgenic vs. Wildtype Slices
8.4. Comorbidity of Protein Misfolding Diseases
9. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular biology and genetics of neurodegenerative diseases caused by prions. Adv. Virus Res. 1992, 41, 241–280. [Google Scholar] [CrossRef]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Diseas. 2020, 20, e2–e10. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef]
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Nat. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.Y. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Ayers, J.I.; Fromholt, S.E.; O’Neal, V.M.; Diamond, J.H.; Borchelt, D.R. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 2016, 131, 103–114. [Google Scholar] [CrossRef]
- Ast, A.; Buntru, A.; Schindler, F.; Hasenkopf, R.; Schulz, A.; Brusendorf, L.; Klockmeier, K.; Grelle, G.; McMahon, B.; Niederlechner, H.; et al. mHTT Seeding Activity: A Marker of Disease Progression and Neurotoxicity in Models of Huntington’s Disease. Mol. Cell 2018, 71, 675–688. [Google Scholar] [CrossRef]
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M.; et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016, 132, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Baekelandt, V.; Brundin, P. Prion-Like Propagation in Neurodegenerative Diseases. In The Molecular and Cellular Basis of Neurodegenerative Diseases: Underlying Mechanisms; Elsevier: Amsterdam, The Netherlands, 2018; pp. 189–242. ISBN 9780128113059. [Google Scholar]
- Bruce, M.E.; McConnell, I.; Fraser, H.; Dickinson, A.G. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: Implications for the nature of the agent and host control of pathogenesis. J. Gen. Virol. 1991, 72, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Solforosi, L.; Milani, M.; Mancini, N.; Clementi, M.; Burioni, R. A closer look at prion strains: Characterization and important implications. Prion 2013, 7, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc. Nat. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Nat. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Falsig, J.; Aguzzi, A. The prion organotypic slice culture assay-POSCA. Nat. Protoc. 2008, 3, 555–562. [Google Scholar] [CrossRef]
- Kanthasamy, A.; Kondru, N.; Manne, S.; Greenlee, J.; West Greenlee, H.; Anantharam, V.; Halbur, P.; Kanthasamy, A. Integrated Organotypic Slice Cultures and RT-QuIC (OSCAR) Assay: Implications for Translational Discovery in Protein Misfolding Diseases. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Halliez, S.; Jaumain, E.; Huor, A.; Douet, J.Y.; Lugan, S.; Cassard, H.; Lacroux, C.; Beŕingue, V.; Andreóletti, O.; Vilette, D. White blood cell-based detection of asymptomatic scrapie infection by Ex Vivo assays. PLoS ONE 2014, 9, e104287. [Google Scholar] [CrossRef] [PubMed]
- Kondru, N.; Manne, S.; Kokemuller, R.; Greenlee, J.; Greenlee, M.H.W.; Nichols, T.; Kong, Q.; Anantharam, V.; Kanthasamy, A.; Halbur, P.; et al. An Ex Vivo Brain Slice Culture Model of Chronic Wasting Disease: Implications for Disease Pathogenesis and Therapeutic Development. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Falsig, J.; Sonati, T.; Herrmann, U.S.; Saban, D.; Li, B.; Arroyo, K.; Ballmer, B.; Liberski, P.P.; Aguzzi, A. Prion Pathogenesis is Faithfully Reproduced in Cerebellar Organotypic Slice Cultures. PLoS Pathog. 2012, 8, e1002985. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; Hossinger, A.; Fehlinger, A.; Büttner, S.; Sim, V.; McKenzie, D.; Vorberg, I.M. Deposition pattern and subcellular distribution of disease-associated prion protein in cerebellar organotypic slice cultures infected with scrapie. Front. Neurosci. 2015, 9, 410. [Google Scholar] [CrossRef]
- Falsig, J.; Julius, C.; Margalith, I.; Schwarz, P.; Heppner, F.L.; Aguzzi, A. A versatile prion replication assay in organotypic brain slices. Nat. Neurosci. 2008, 11, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Campeau, J.L.; Wu, G.; Bell, J.R.; Rasmussen, J.; Sim, V.L. Early increase and late decrease of Purkinje cell dendritic spine density in prion-infected organotypic mouse cerebellar cultures. PLoS ONE 2013, 8, e81776. [Google Scholar] [CrossRef]
- Harischandra, D.S.; Kondru, N.; Martin, D.P.; Kanthasamy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.G. Role of proteolytic activation of protein kinase Cδ in the pathogenesis of prion disease. Prion 2014, 8, 143–153. [Google Scholar] [CrossRef]
- Herrmann, U.S.; Sonati, T.; Falsig, J.; Reimann, R.R.; Dametto, P.; O’Connor, T.; Li, B.; Lau, A.; Hornemann, S.; Sorce, S.; et al. Prion Infections and Anti-PrP Antibodies Trigger Converging Neurotoxic Pathways. PLoS Pathog. 2015, 11, 1–22. [Google Scholar] [CrossRef]
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia in prion diseases. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef]
- Hofmann, J.P.; Denner, P.; Nussbaum-Krammer, C.; Kuhn, P.H.; Suhre, M.H.; Scheibel, T.; Lichtenthaler, S.F.; Schätzl, H.M.; Bano, D.; Vorberg, I.M. Cell-to-cell propagation of infectious cytosolic protein aggregates. Proc. Nat. Acad. Sci. USA 2013, 110, 5951–5956. [Google Scholar] [CrossRef]
- Cortez, L.M.; Campeau, J.; Norman, G.; Kalayil, M.; van der Merwe, J.; McKenzie, D.; Sim, V.L. Bile Acids Reduce Prion Conversion, Reduce Neuronal Loss, and Prolong Male Survival in Models of Prion Disease. J. Virol. 2015, 89, 7660–7672. [Google Scholar] [CrossRef] [PubMed]
- Margalith, I.; Suter, C.; Ballmer, B.; Schwarz, P.; Tiberi, C.; Sonati, T.; Falsig, J.; Nyström, S.; Hammarström, P.; Åslund, A.; et al. Polythiophenes inhibit prion propagation by stabilizing prion protein (PrP) aggregates. J. Biolog. Chem. 2012, 287, 18872–18887. [Google Scholar] [CrossRef] [PubMed]
- Goniotaki, D.; Lakkaraju, A.K.K.; Shrivastava, A.N.; Bakirci, P.; Sorce, S.; Senatore, A.; Marpakwar, R.; Hornemann, S.; Gasparini, F.; Triller, A.; et al. Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog. 2017, 13, e1006733. [Google Scholar] [CrossRef] [PubMed]
- Harwell, C.S.; Coleman, M.P. Synaptophysin depletion and intraneuronal Aβ in organotypic hippocampal slice cultures from huAPP transgenic mice. Mol. Neurodegener. 2016, 11, 44. [Google Scholar] [CrossRef]
- Novotny, R.; Langer, F.; Mahler, J.; Skodras, A.; Vlachos, A.; Wegenast-Braun, B.M.; Kaeser, S.A.; Neher, J.J.; Eisele, Y.S.; Pietrowski, M.J.; et al. Conversion of synthetic Aβ to In Vivo active seeds and amyloid plaque formation in a hippocampal slice culture model. J. Neurosci. 2016, 36, 5084–5093. [Google Scholar] [CrossRef]
- Croft, C.L.; Noble, W. Preparation of organotypic brain slice cultures for the study of Alzheimer’s disease. F1000Research 2018, 7, 592. [Google Scholar] [CrossRef]
- Croft, C.L.; Wade, M.A.; Kurbatskaya, K.; Mastrandreas, P.; Hughes, M.M.; Phillips, E.C.; Pooler, A.M.; Perkinton, M.S.; Hanger, D.P.; Noble, W. Membrane association and release of wild-type and pathological tau from organotypic brain slice cultures. Cell Death Dis. 2017, 8, e2671. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Beneventano, M.; Sortino, M.A. The contribution of microglia to early synaptic compensatory responses that precede β-amyloid-induced neuronal death. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Hellwig, S.; Masuch, A.; Nestel, S.; Katzmarski, N.; Meyer-Luehmann, M.; Biber, K. Forebrain microglia from wild-type but not adult 5xFAD mice prevent amyloid-β plaque formation in organotypic hippocampal slice cultures. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef]
- Richter, M.; Vidovic, N.; Biber, K.; Dolga, A.; Culmsee, C.; Dodel, R. The neuroprotective role of microglial cells against amyloid beta-mediated toxicity in organotypic hippocampal slice cultures. Brain Pathol. 2019. [Google Scholar] [CrossRef]
- Alberdi, E.; Sánchez-Gómez, M.V.; Ruiz, A.; Cavaliere, F.; Ortiz-Sanz, C.; Quintela-López, T.; Capetillo-Zarate, E.; Solé-Domènech, S.; Matute, C. Mangiferin and Morin Attenuate Oxidative Stress, Mitochondrial Dysfunction, and Neurocytotoxicity, Induced by Amyloid Beta Oligomers. Oxid. Med. Cell. Longev. 2018, 2018, 2856063. [Google Scholar] [CrossRef] [PubMed]
- Arbo, B.D.; Hoppe, J.B.; Rodrigues, K.; Garcia-Segura, L.M.; Salbego, C.G.; Ribeiro, M.F. 4′-Chlorodiazepam is neuroprotective against amyloid-beta in organotypic hippocampal cultures. J. Steroid Biochem. Mol. Biol. 2017, 171, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [CrossRef]
- Hoppe, J.B.; Haag, M.; Whalley, B.J.; Salbego, C.G.; Cimarosti, H. Curcumin protects organotypic hippocampal slice cultures from Aβ1-42-induced synaptic toxicity. Toxicol. In Vitro 2013, 27, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Barucker, C.; Bittner, H.J.; Chang, P.K.Y.; Cameron, S.; Hancock, M.A.; Liebsch, F.; Hossain, S.; Harmeier, A.; Shaw, H.; Charron, F.M.; et al. Aβ42-oligomer Interacting Peptide (AIP) neutralizes toxic amyloid-β42 species and protects synaptic structure and function. Sci. Rep. 2015, 5, 1–15. [Google Scholar] [CrossRef]
- Mendes, N.D.; Fernandes, A.; Almeida, G.M.; Santos, L.E.; Selles, M.C.; Lyra e Silva, N.M.; Machado, C.M.; Horta-Júnior, J.A.C.; Louzada, P.R.; de Felice, F.G.; et al. Free-floating adult human brain-derived slice cultures as a model to study the neuronal impact of Alzheimer’s disease-associated Aβ oligomers. J. Neurosci. Methods 2018, 307, 203–209. [Google Scholar] [CrossRef]
- Messing, L.; Decker, J.M.; Joseph, M.; Mandelkow, E.; Mandelkow, E.M. Cascade of tau toxicity in inducible hippocampal brain slices and prevention by aggregation inhibitors. Neurobiol. Aging 2013, 34, 1343–1354. [Google Scholar] [CrossRef]
- Stancu, I.C.; Vasconcelos, B.; Ris, L.; Wang, P.; Villers, A.; Peeraer, E.; Buist, A.; Terwel, D.; Baatsen, P.; Oyelami, T.; et al. Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. 2015, 129, 875–894. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 1–25. [Google Scholar] [CrossRef]
- Suttkus, A.; Holzer, M.; Morawski, M.; Arendt, T. The neuronal extracellular matrix restricts distribution and internalization of aggregated Tau-protein. Neuroscience 2016, 313, 225–235. [Google Scholar] [CrossRef]
- Halliday, G.M.; McCann, H. Human-based studies on α-synuclein deposition and relationship to Parkinson’s disease symptoms. Exp. Neurol. 2008, 209, 12–21. [Google Scholar] [CrossRef]
- Cavaliere, F.; Vicente, E.S.; Matute, C. An organotypic culture model to study nigro-striatal degeneration. J. Neurosci. Methods 2010, 188, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Tuschl, K.; Mills, P.B.; Clayton, P.T. Manganese and the Brain. Int. Rev. Neurobiol. 2013, 110, 277–312. [Google Scholar] [CrossRef]
- Xu, B.; Wu, S.W.; Lu, C.W.; Deng, Y.; Liu, W.; Wei, Y.G.; Yang, T.Y.; Xu, Z.F. Oxidative stress involvement in manganese-induced alpha-synuclein oligomerization in organotypic brain slice cultures. Toxicology 2013, 305, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Jin, C.H.; Deng, Y.; Liu, W.; Yang, T.Y.; Feng, S.; Xu, Z.F. Alpha-Synuclein Oligomerization in Manganese-Induced Nerve Cell Injury in Brain Slices: A Role of NO-Mediated S-Nitrosylation of Protein Disulfide Isomerase. Mol. Neurobiol. 2014, 50, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Liu, W.; Deng, Y.; Yang, T.Y.; Feng, S.; Xu, Z.F. Inhibition of Calpain Prevents Manganese-Induced Cell Injury and Alpha-Synuclein Oligomerization in Organotypic Brain Slice Cultures. PLoS ONE 2015, 10, e0119205. [Google Scholar] [CrossRef] [PubMed]
- Ponsen, M.M.; Stoffers, D.; Twisk, J.W.R.; Wolters, E.C.; Berendse, H.W. Hyposmia and executive dysfunction as predictors of future Parkinson’s disease: A prospective study. Mov. Disord. 2009, 24, 1060–1065. [Google Scholar] [CrossRef]
- Park, H.; Lee, J.Y.; Shin, C.M.; Kim, J.M.; Kim, T.J.; Kim, J.W. Characterization of gastrointestinal disorders in patients with parkinsonian syndromes. Parkinsonism Relat. Disord. 2015, 21, 455–460. [Google Scholar] [CrossRef]
- Passali, G.C.; Bove, F.; Vargiu, L.; Bentivoglio, A.R.; Anzivino, R.; de Corso, E.; Galli, J.; Rigante, M.; Pandolfini, M.; Sergi, B.; et al. New olfactometric findings in Parkinson’s disease. Clin. Otolaryng. 2017, 42, 837–843. [Google Scholar] [CrossRef]
- Sharrad, D.F.; Chen, B.N.; Gai, W.P.; Vaikath, N.; El-Agnaf, O.M.; Brookes, S.J.H. Rotenone and elevated extracellular potassium concentration induce cell-specific fibrillation of α-synuclein in axons of cholinergic enteric neurons in the guinea-pig ileum. Neurogastroenter. Motil. 2017, 29, e12985. [Google Scholar] [CrossRef]
- Croft, C.L.; Cruz, P.E.; Ryu, D.H.; Ceballos-Diaz, C.; Strang, K.H.; Woody, B.M.; Lin, W.L.; Deture, M.; Rodríguez-Lebrón, E.; Dickson, D.W.; et al. rAAV-based brain slice culture models of Alzheimer’s and Parkinson’s disease inclusion pathologies. J. Exp. Med. 2019, 216, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.L.; Vercruysse, F.; MacO, B.; Ait-Bouziad, N.; de Roo, M.; Muller, D.; Lashuel, H.A. Fibril growth and seeding capacity play key roles in α-synuclein-mediated apoptotic cell death. Cell Death Differ. 2015, 22, 2107–2122. [Google Scholar] [CrossRef]
- Elfarrash, S.; Jensen, N.M.; Ferreira, N.; Betzer, C.; Thevathasan, J.V.; Diekmann, R.; Adel, M.; Omar, N.M.; Boraie, M.Z.; Gad, S.; et al. Organotypic slice culture model demonstrates inter-neuronal spreading of alpha-synuclein aggregates. Acta Neuropathol. Commun. 2019, 7, 213. [Google Scholar] [CrossRef]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.N.; Bousset, L.; Renner, M.; Redeker, V.; Savistchenko, J.; Triller, A.; Melki, R. Differential Membrane Binding and Seeding of Distinct α-Synuclein Fibrillar Polymorphs. Biophys. J. 2020, 118, 1301–1320. [Google Scholar] [CrossRef]
- Leggett, C.; McGehee, D.S.; Mastrianni, J.; Yang, W.; Bai, T.; Brorson, J.R. Tunicamycin produces TDP-43 cytoplasmic inclusions in cultured brain organotypic slices. J. Neurol. Sci. 2012, 317, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ayala, V.; Granado-Serrano, A.B.; Cacabelos, D.; Naudí, A.; Ilieva, E.v.; Boada, J.; Caraballo-Miralles, V.; Lladó, J.; Ferrer, I.; Pamplona, R.; et al. Cell stress induces TDP-43 pathological changes associated with ERK1/2 dysfunction: Implications in ALS. Acta Neuropathol. 2011, 122, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Leal-Lasarte, M.M.; Franco, J.M.; Labrador-Garrido, A.; Pozo, D.; Roodveldt, C. Extracellular TDP-43 aggregates target MAPK/MAK/MRK overlapping kinase (MOK) and trigger caspase-3/IL-18 signaling in microglia. FASEB J. 2017, 31, 2797–2816. [Google Scholar] [CrossRef]
- Ayers, J.I.; Diamond, J.; Sari, A.; Fromholt, S.; Galaleldeen, A.; Ostrow, L.W.; Glass, J.D.; Hart, P.J.; Borchelt, D.R. Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol. 2016, 132, 827–840. [Google Scholar] [CrossRef]
- Rasouli, S.; Abdolvahabi, A.; Croom, C.M.; Plewman, D.L.; Shi, Y.; Ayers, J.I.; Shaw, B.F. Lysine acylation in superoxide dismutase-1 electrostatically inhibits formation of fibrils with prion-like seeding. J. Biolog. Chem. 2017, 292, 19366–19380. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Portier, R.; Woodman, B.; Hockly, E.; Mahal, A.; Klunk, W.E.; Li, X.J.; Wanker, E.; Murray, K.D.; Bates, G.P. Inhibition of polyglutamine aggregation in R6/2 HD brain slices-Complex dose-response profiles. Neurobiol. Dis. 2001, 8, 1017–1026. [Google Scholar] [CrossRef][Green Version]
- Reinhart, P.H.; Kaltenbach, L.S.; Essrich, C.; Dunn, D.E.; Eudailey, J.A.; DeMarco, C.T.; Turmel, G.J.; Whaley, J.C.; Wood, A.; Cho, S.; et al. Identification of anti-inflammatory targets for Huntington’s disease using a brain slice-based screening assay. Neurobiol. Dis. 2011, 43, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Pecho-Vrieseling, E.; Rieker, C.; Fuchs, S.; Bleckmann, D.; Esposito, M.S.; Botta, P.; Goldstein, C.; Bernhard, M.; Galimberti, I.; Müller, M.; et al. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 2014, 17, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Khoshnan, A.; Sabbaugh, A.; Calamini, B.; Marinero, S.A.; Dunn, D.E.; Yoo, J.H.; Ko, J.; Lo, D.C.; Patterson, P.H. IKK β and mutant huntingtin interactions regulate the expression of IL-34: Implications for microglialmediated neurodegeneration in HD. Human Mol. Genet. 2017, 26, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.C.; Messer, A. A single-chain Fv intrabody provides functional protection against the effects of mutant protein in an organotypic slice culture model of Huntington’s disease. Mol. Brain Res. 2004, 121, 141–145. [Google Scholar] [CrossRef]
- Lecerf, J.M.; Shirley, T.L.; Zhu, Q.; Kazantsev, A.; Amersdorfer, P.; Housman, D.E.; Messer, A.; Huston, J.S. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Nat. Acad. Sci. USA 2001, 98, 4764–4769. [Google Scholar] [CrossRef]
- Croft, C.L.; Kurbatskaya, K.; Hanger, D.P.; Noble, W. Inhibition of glycogen synthase kinase-3 by BTA-EG 4 reduces tau abnormalities in an organotypic brain slice culture model of Alzheimer’s disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Hung, C.W.; Chen, Y.C.; Hsieh, W.L.; Chiou, S.H.; Kao, C.L. Ageing and neurodegenerative diseases. Ageing Res. Rev. 2010, 9, S36–S46. [Google Scholar] [CrossRef]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017, 36, 583–603. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic vibrosections from whole brain adult Alzheimer mice (overexpressing amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as a model to study clearance of beta-amyloid plaques. Front. Aging Neurosci. 2015, 7, 47. [Google Scholar] [CrossRef]
- Kim, H.; Kim, E.; Park, M.; Lee, E.; Namkoong, K. Organotypic hippocampal slice culture from the adult mouse brain: A versatile tool for translational neuropsychopharmacology. Prog. Neuro Psychopharmac. Biol. Psychiat. 2013, 41, 36–43. [Google Scholar] [CrossRef]
- Jang, S.; Kim, H.; Kim, H.J.; Lee, S.K.; Kim, E.W.; Namkoong, K.; Kim, E. Long-term culture of organotypic hippocampal slice from old 3xTg-AD mouse: An ex vivo model of alzheimer’s disease. Psychiat. Investig. 2018, 15, 205–213. [Google Scholar] [CrossRef]
- Su, T.; Paradiso, B.; Long, Y.S.; Liao, W.P.; Simonato, M. Evaluation of cell damage in organotypic hippocampal slice culture from adult mouse: A potential model system to study neuroprotection. Brain Res. 2011, 1385, 68–76. [Google Scholar] [CrossRef]
- Guo, W.; Fumagalli, L.; Prior, R.; van den Bosch, L. Current advances and limitations in modeling ALS/FTD in a dish using induced pluripotent stem cells. Front. Neurosci. 2017, 11, 1–20. [Google Scholar] [CrossRef]
- Marsh, S.E.; Blurton-Jones, M. Examining the mechanisms that link β-amyloid and α-synuclein pathologies. Alzheimer’s Res.Ther. 2012, 4, 11. [Google Scholar] [CrossRef]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset Amyloid Deposition and Cognitive Deficits in Transgenic Mice Expressing a Double Mutant Form of Amyloid Precursor Protein 695. J. Biolog. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Nat. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Sydow, A.; van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E.; et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic tau mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Wang, J.; Farr, G.W.; Zeiss, C.J.; Rodriguez-Gil, D.J.; Wilson, J.H.; Furtak, K.; Rutkowski, D.T.; Kaufman, R.J.; Ruse, C.I.; Yates, J.R.; et al. Progressive aggregation despite chaperone associations of a mutant SOD1-YFP in transgenic mice that develop ALS. Proc. Nat. Acad. Sci. USA 2009, 106, 1392–1397. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon I of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| In Vivo | Slice Culture | Primary Culture | Simple Cell Culture | |
|---|---|---|---|---|
| Cost | + Housing for duration of experiment, high animal numbers | ++ Breeding costs, fewer animals | ++ Breeding or animal purchase costs, fewer animals | +++ |
| Time | + Months | ++ Weeks | ++ Weeks | +++ Days |
| Ethics of animal use | + High animal numbers, induction of symptomatic disease | ++ Fewer animals, no need to induce disease | ++ Fewer animals, no need to induce disease | +++ No animals required |
| Technical difficulty | + Inoculation, clinical assessment | ++ Fast, accurate dissection, orientation for slicing | ++ Fast, accurate dissection | +++ Sterile technique |
| Cell types, cyto-architecture | +++ All cell types, connections intact | ++ All cell types, some connectivity disrupted | - Often one cell type No cytoarchitecture | - Often one cell type No cytoarchitecture |
| Genetics | ++ Many models | +++ Can use any mouse models PLUS transgenic models that are lethal beyond the perinatal period, can transfect with recombinant adeno-associated viruses, can have many genetically identical slices from same mouse | + Many genetically identical cell populations can be obtained from a single animal | - Immortalized cell lines are often cancer-based and can be unstable genetically |
| Real time monitoring | ++ Closed system, monitor behavioural/clinical phenotype | +++ Open system, more amenable to live-cell imaging, no phenotype | +++ Open system amenable to live-cell imaging, no phenotype | +++ Open system amenable to live-cell imaging, no phenotype |
| Animal age | +++ Any age | ++ Most viable cultures are from neonatal animals | + Tissue must be taken from prenatal or neonatal animals | n/a |
| Vasculature | +++ Intact, blood–brain barrier present | ++ No blood–brain barrier—better for drug testing | - None | n/a |
| Prion Feature | Amyloid Beta | Tau | α-Synuclein | TDP-43 | SOD1 | Huntingtin |
|---|---|---|---|---|---|---|
| Seeded aggregation | [36,40,41] | [49] | [63,64] | [71] | ||
| Prion-like spreading | [50,51] | [64,65] | [75] | |||
| Pathology induced by seeding | [36,47] | [63] | [70] | [75] | ||
| Strains | [36] | [66] | [71] | |||
| Drug screening (to prevent aggregation/spread) | [45,46] | [48] | [63] | [72] | [73,76,77] |
| Ref. | Animal Model | In Vivo Pathology | Unseeded Slice Pathology | Prion-Like Seeding Agent | Seeded Slice Pathology |
|---|---|---|---|---|---|
| Amyloid beta | |||||
| [35] | CRND8 mice: 5x expression of human APP with Swedish and Indiana mutations [89] | ThS-positive plaques by 3 months Dense-cored plaques by 5 months [89] | No ThS-positive Aβ aggregates by 9 weeks of culture Accumulation of intracellular Aβ in axonal swellings | Synthetic Aβ | No ThS-positive Aβ aggregates by 9 weeks of culture |
| [36] | APPPS1 mice: 3x expression of human APP with Swedish mutation human PS1 with L166P mutation [90] | Aβ plaques by at 6 weeks [90] | No evidence of Aβ aggregates by 10 weeks | Single treatment with aged APPPS1 or APP23 brain homogenate and continuous supplementation with synthetic Aβ | Extensive Aβ aggregates after 1 week Aggregate morphology dependent on source of brain homogenate (APPPS1 or APP23) |
| APP23 mice: 7x expression of human APP with Swedish mutation [91] | Aβ plaques by 6 months [91] | ||||
| WT mice | N/A | ||||
| APP-null mice | N/A | ||||
| [40] | WT mice | N/A | No evidence of Aβ aggregates | Treatment with Clodronate to remove microglia and 4 treatments with synthetic Aβ42 | ThS-positive Aβ aggregates after two weeks |
| [41] | WT mice | N/A | No evidence of Aβ aggregates | Treatment with clodronate to remove microglia and addition of Aβ42 oligomer solution | Increased ThT fluorescence after 1 week (suggests Aβ aggregate formation) |
| Amyloid beta and tau | |||||
| [37] | 3xTg-AD mice: Express human APP with Swedish mutation, human mutant P301L tau, and human PSEN1 with M146V mutation [92] | Extracellular Aβ deposits by 6 months Aggregates of hyperphosphor-ylated tau by 12–15 months [92] | No evidence of Aβ or tau aggregates after 28 days of culture Accelerated accumulation of Aβ42 and phosphorylated tau compared to in vivo | N/A | N/A |
| Tau | |||||
| [48] | Tg Mice expressing human 4R tau with the ΔK280 FTD mutation [93] | Hyperphosphor-ylation and aggregation of tau by 5–10 months [93] | ThS-positive cell bodies by 20 days | N/A | N/A |
| [49] | PS19 tau mice: Express human 1N4R Tau with P301S mutation [94] | PHF1-positive neuronal staining by 6 months in hippocampus, amygdala and spinal cord [94] | No evidence of tau aggregation after 13 days of culturing | Recombinant tau fibrils added on days 3 and 6 of culturing | Tau aggregation in hippocampal CA1 neurons 10 days after first seeding |
| WT mice | N/A | No evidence of tau aggregation 10 days after first seeding | |||
| α-synuclein | |||||
| [63] | WT rats | N/A | N/A | Treated on day 13/14 with monomeric α-syn, fibrillar α-syn, or a mixture of both exogenous forms | α-syn monomers did not cause cell death Mixture of α-syn monomers and fibrils significantly more toxic than α-syn fibrils alone |
| [64] | WT mice | N/A | N/A | α-syn fibrils microinjected into the dentate gyrus | α-syn aggregates appeared in the dentate gyrus after 3 days and in CA1 and CA3 by 3–5 days (no evidence of aggregates when monomeric α-syn injected) |
| SNCA knockout mice | No evidence of α-syn aggregates | ||||
| [65] | WT mice | N/A | N/A | Primary ROSAmT/mG astrocytes were incubated with Alexa-488-labeled α-syn fibrils for 16 h. The astrocytes were then added on top of slice culture | α-syn inclusions observed in slice culture astrocytes but not neurons after 3–6 days |
| [66] | WT mice | N/A | N/A | α-syn fibrils of 5 different polymorphs were added to slice cultures | α-syn aggregates were observed after 4–7 days. Extent and rate of aggregation depended on fibril polymorph |
| SOD1 | |||||
| [9] | Tg mice expressing human G85R mutant SOD1-YFP fusion protein [95] | Develop fluorescent SOD1 puncta in anterior horn of spinal cord by 9 months in cell bodies and neuropil [95] | (spinal cord culture) N/A | Treatment with spinal cord homogenates from paralyzed G85R SOD1:YFP mice that had been occulated with different “mouse-adapted” ALS strains1 | SOD1 aggregates induced in spinal cord slices Morphology of aggregates were dependent on strain |
| Treatment with spinal cord homogenates from sporadic or familial ALS patients | SOD1-YFP punctate inclusions by 7 days only when treated with A4V SOD1 mutation | ||||
| [72] | Tg mice expressing human G85R mutant SOD1-YFP fusion protein [95] | Develop fluorescent SOD1 puncta in anterior horn of spinal cord by 9 months in both cell bodies and neuropil [95] | (spinal cord culture) N/A | WT SOD1 was modified with various acyl groups and aggregated in vitro. The resulting ThT-positive or negative fibrils were then added to slice culture. | After 1 month of culture, only treatment with ThT-positive fibrils led to the formation of SOD1-YFP inclusions |
| Huntingtin | |||||
| [73] | R6/2 mice: Express mHtt with ~115–150 CAG repeats [96] | Develop mHtt aggregates in CA1 of hippocampus by 3 weeks and in CA3 by 5 weeks [73] | mHtt aggregates are observed in CA1 of the hippocampus by 2 weeks and in CA3 and dentate gyrus by 3 weeks [73] | N/A | N/A |
| [75] | WT mice | N/A | Cortex or Striatum culture N/A | WT-R6/2 Cocultures: WT striatum with R6/2 cortex WT cortex with R6/2 striatum | mHtt aggregates spread from R6/2 cortex to MSNs in WT striatum by 4 weeks, but not from R6/2 striatum to WT cortex |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pineau, H.; Sim, V. POSCAbilities: The Application of the Prion Organotypic Slice Culture Assay to Neurodegenerative Disease Research. Biomolecules 2020, 10, 1079. https://doi.org/10.3390/biom10071079
Pineau H, Sim V. POSCAbilities: The Application of the Prion Organotypic Slice Culture Assay to Neurodegenerative Disease Research. Biomolecules. 2020; 10(7):1079. https://doi.org/10.3390/biom10071079
Chicago/Turabian StylePineau, Hailey, and Valerie Sim. 2020. "POSCAbilities: The Application of the Prion Organotypic Slice Culture Assay to Neurodegenerative Disease Research" Biomolecules 10, no. 7: 1079. https://doi.org/10.3390/biom10071079
APA StylePineau, H., & Sim, V. (2020). POSCAbilities: The Application of the Prion Organotypic Slice Culture Assay to Neurodegenerative Disease Research. Biomolecules, 10(7), 1079. https://doi.org/10.3390/biom10071079