Against Repurposing Methadone for Glioblastoma Therapy

 and
and 
Abstract
1. Introduction
2. Methadone Pharmacokinetics
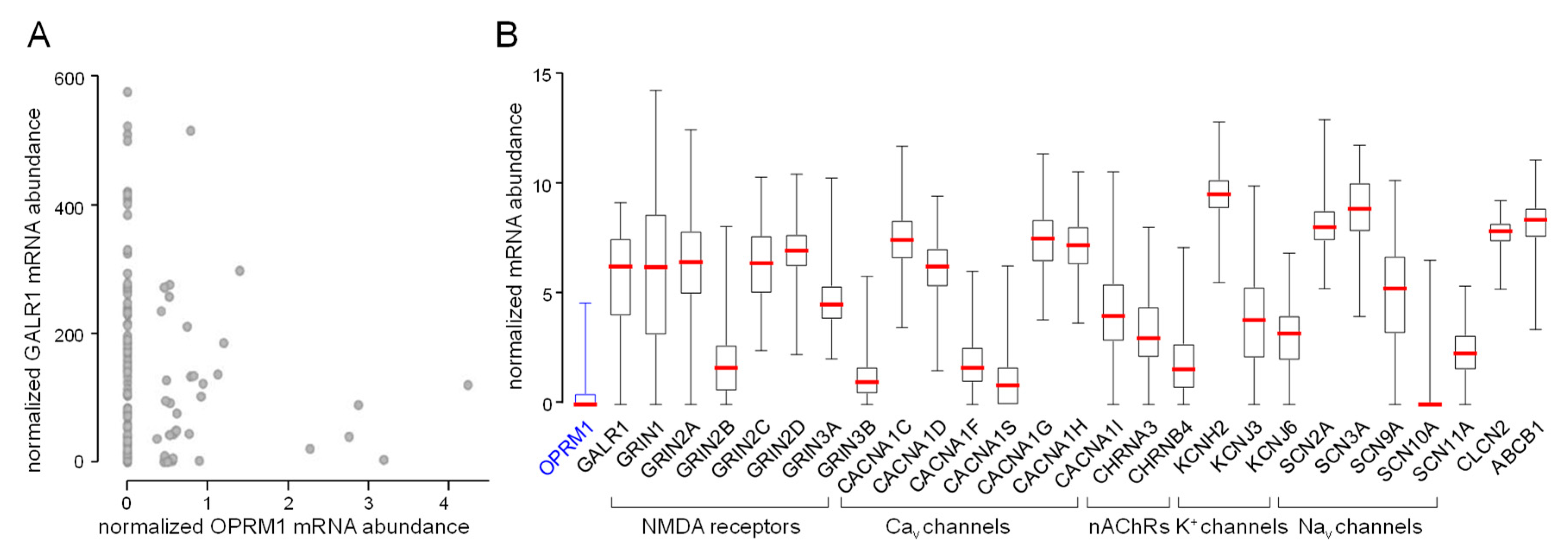
3. Methadone Target Molecules
4. Evidence for a Tumoricidal Activity of Methadone
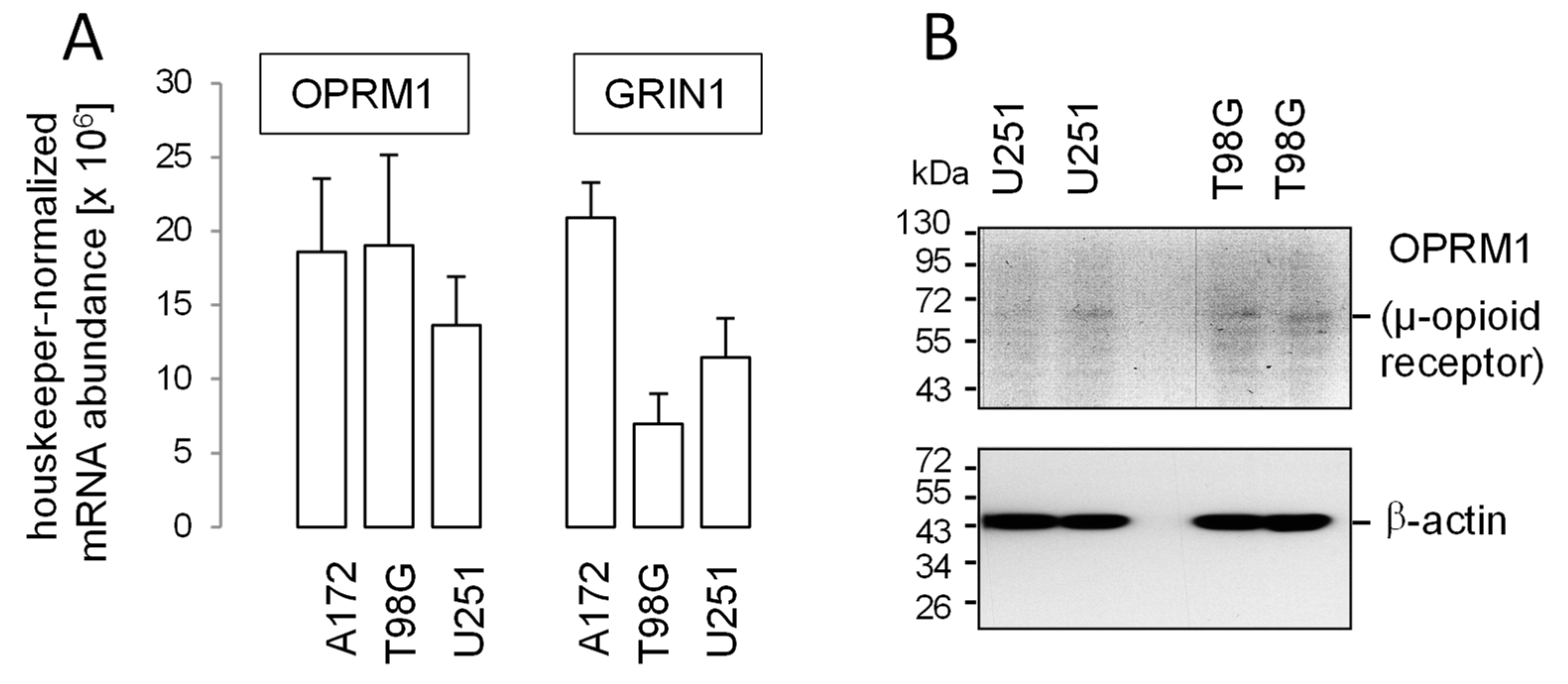
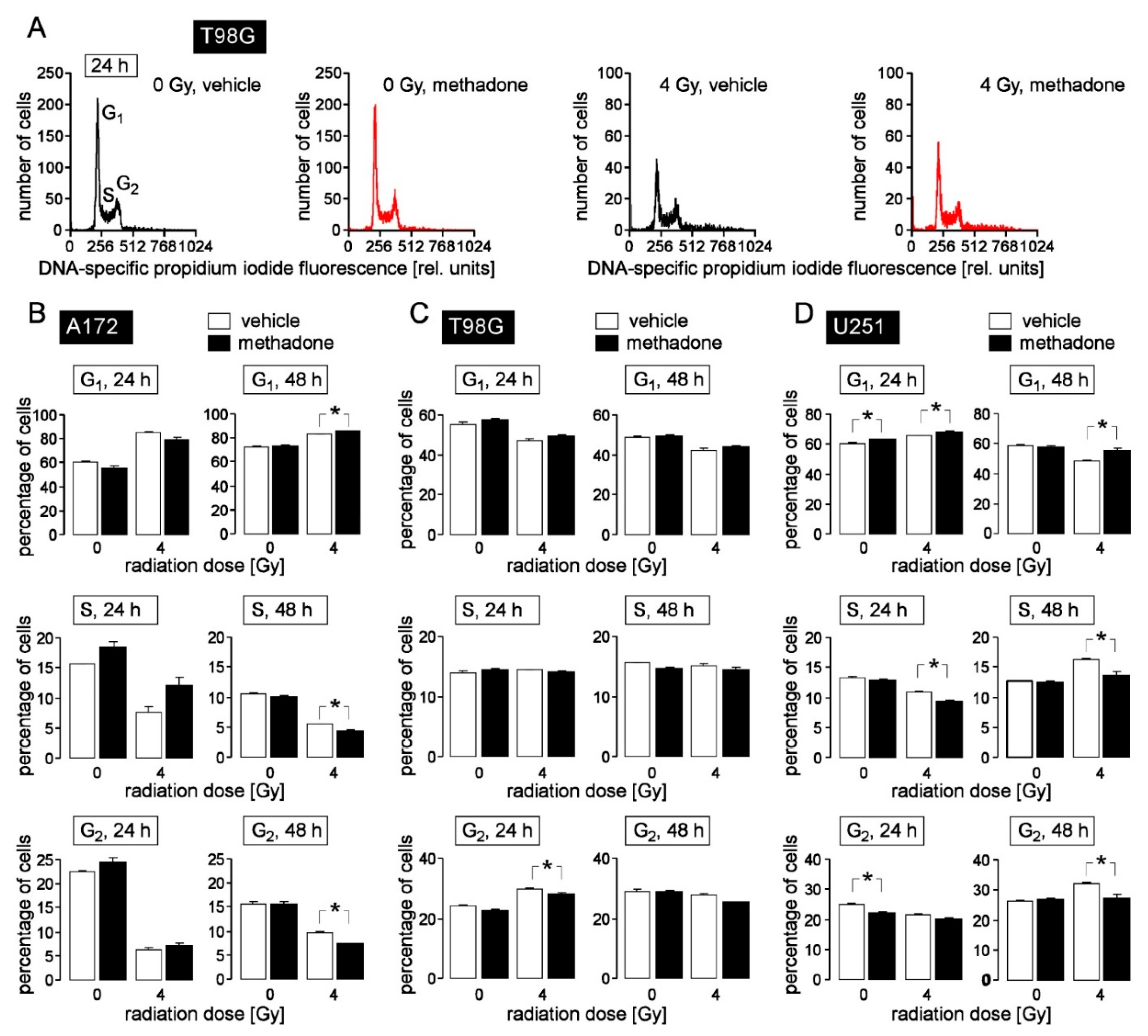
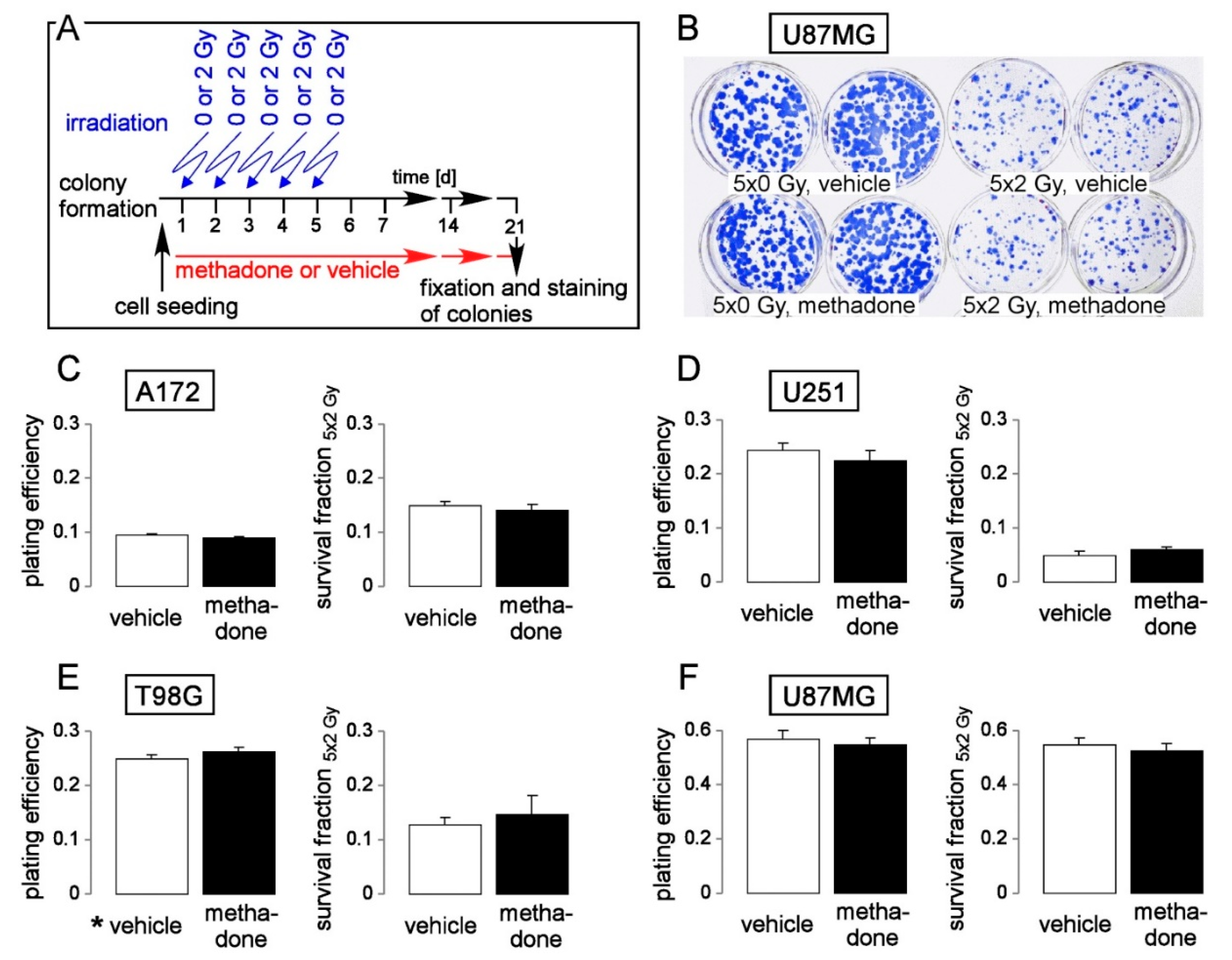
5. Methadone in “Supratherapeutic” Concentrations May Modify Cell Cycle but Fails to Impair Clonogenic Survival or Radioresistance of Human Glioblastoma Cells in Clinically Relevant Concentrations In Vitro
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Wolff, P. On pethidine and methadone derivatives. Bull. World Health Organ. 1949, 2, 193. [Google Scholar]
- Bockmühl, M.; Ehrhart, G. Über eine neue Klasse von spasmolytisch und analgetisch wirkenden Verbindungen, I. Justus Liebigs Ann. Chem. 1949, 561, 52–85. [Google Scholar] [CrossRef]
- Scott, C.C.; Chen, K. The action of 1, 1-diphenyl-1-(dimethylaminoisopropyl)-butanone-2, a potent analgesic agent. J. Pharmacol. Exp. Ther. 1946, 87, 63–71. [Google Scholar] [CrossRef]
- Kreek, M.J.; Vocci, F.J. History and current status of opioid maintenance treatments: Blending conference session. J. Subst. Abuse Treat. 2002, 23, 93–105. [Google Scholar] [CrossRef]
- Dole, V.P.; Nyswander, M. A medical treatment for diacetylmorphine (heroin) addiction. A clinical trial with methadone hydrochloride. JAMA 1965, 193, 646–650. [Google Scholar] [CrossRef]
- Davis, A.M.; Inturrisi, C.E. d-Methadone blocks morphine tolerance and N-methyl-d-aspartate-induced hyperalgesia. J. Pharmacol. Exp. Ther. 1999, 289, 1048–1053. [Google Scholar]
- Inturrisi, C.E. Pharmacology of methadone and its isomers. Minerva Anestesiol. 2005, 71, 435–437. [Google Scholar]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- American-Cancer-Society. Survival Rates for Selected Adult Brain and Spinal Cord Tumors. Available online: https://www.cancer.org/cancer/brain-spinal-cord-tumors-adults/detection-diagnosis-staging/survival-rates.html (accessed on 5 May 2020).
- Eckert, M.; Klumpp, L.; Huber, S.M. Cellular Effects of the Antiepileptic Drug Valproic Acid in Glioblastoma. Cell Physiol. Biochem. 2017, 44, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Maneckjee, R.; Minna, J.D. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc. Natl. Acad. Sci. USA 1990, 87, 3294–3298. [Google Scholar] [CrossRef]
- Maneckjee, R.; Minna, J.D. Nonconventional opioid binding sites mediate growth inhibitory effects of methadone on human lung cancer cells. Proc. Natl. Acad. Sci. USA 1992, 89, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Maneckjee, R.; Minna, J.D. Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ. 1994, 5, 1033–1040. [Google Scholar] [PubMed]
- Friesen, C.; Hormann, I.; Roscher, M.; Fichtner, I.; Alt, A.; Hilger, R.; Debatin, K.M.; Miltner, E. Opioid receptor activation triggering downregulation of cAMP improves effectiveness of anti-cancer drugs in treatment of glioblastoma. Cell Cycle 2014, 13, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Friesen, C.; Roscher, M.; Alt, A.; Miltner, E. Methadone, commonly used as maintenance medication for outpatient treatment of opioid dependence, kills leukemia cells and overcomes chemoresistance. Cancer Res. 2008, 68, 6059–6064. [Google Scholar] [CrossRef]
- Friesen, C.; Roscher, M.; Hormann, I.; Fichtner, I.; Alt, A.; Hilger, R.A.; Debatin, K.M.; Miltner, E. Cell death sensitization of leukemia cells by opioid receptor activation. Oncotarget 2013, 4, 677–690. [Google Scholar] [CrossRef]
- Onken, J.; Friesen, C.; Vajkoczy, P.; Misch, M. Safety and tolerance of d, l-methadone in combination with chemotherapy in patients with glioma. Anticancer Res. 2017, 37, 1227–1235. [Google Scholar]
- Deutsche-Gesellschaft-für-Hämatologie-und-Medizinische-Onkologie. DGHO Stellungnahme Methadon. Available online: https://www.dgho.de/publikationen/stellungnahmen/gute-aerztliche-praxis/methadon/DGHO_Stellungnahme_Methadon 20170426_.pdf/view (accessed on 11 May 2020).
- Deutsche-Krebshilfe. Stellungnahme der Deutschen Krebshilfe zum Thema ‘Methadon in der Krebstherapie’. Available online: https://www.krebshilfe.de/fileadmin/Downloads/PDFs/Stellungnahmen/Deutsche_Krebshilfe_Stellungnahme_Methadon_10_2019.pdf (accessed on 11 May 2020).
- Ferrari, A.; Coccia, C.P.; Bertolini, A.; Sternieri, E. Methadone—metabolism, pharmacokinetics and interactions. Pharmacol. Res. 2004, 50, 551–559. [Google Scholar] [CrossRef]
- Eap, C.B.; Buclin, T.; Baumann, P. Interindividual variability of the clinical pharmacokinetics of methadone: Implications for the treatment of opioid dependence. Clin. Pharmacokinet. 2002, 41, 1153–1193. [Google Scholar] [CrossRef]
- Meresaar, U.; Nilsson, M.I.; Holmstrand, J.; Anggard, E. Single dose pharmacokinetics and bioavailability of methadone in man studied with a stable isotope method. Eur. J. Clin. Pharmacol. 1981, 20, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, K.; Blemmer, T.; Angelo, H.R.; Christrup, L.L.; Drenck, N.E.; Rasmussen, S.N.; Sjogren, P. Stereoselective pharmacokinetics of methadone in chronic pain patients. Ther. Drug Monit. 1996, 18, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Plummer, J.L.; Gourlay, G.K.; Cherry, D.A.; Cousins, M.J. Estimation of methadone clearance: Application in the management of cancer pain. Pain 1988, 33, 313–322. [Google Scholar] [CrossRef]
- Eap, C.B.; Cuendet, C.; Baumann, P. Binding of d-methadone, l-methadone, and dl-methadone to proteins in plasma of healthy volunteers: Role of the variants of alpha 1-acid glycoprotein. Clin. Pharmacol. Ther. 1990, 47, 338–346. [Google Scholar] [CrossRef]
- Garrido, M.J.; Jiminez, R.; Gomez, E.; Calvo, R. Influence of plasma-protein binding on analgesic effect of methadone in rats with spontaneous withdrawal. J. Pharm. Pharmacol. 1996, 48, 281–284. [Google Scholar] [CrossRef]
- Abramson, F.P. Methadone plasma protein binding: Alterations in cancer and displacement from alpha 1-acid glycoprotein. Clin. Pharmacol. Ther. 1982, 32, 652–658. [Google Scholar] [CrossRef]
- Volpe, D.A.; Xu, Y.; Sahajwalla, C.G.; Younis, I.R.; Patel, V. Methadone Metabolism and Drug-Drug Interactions: In Vitro and In Vivo Literature Review. J. Pharm. Sci. 2018, 107, 2983–2991. [Google Scholar] [CrossRef]
- Anggard, E.; Gunne, L.M.; Homstrand, J.; McMahon, R.E.; Sandberg, C.G.; Sullivan, H.R. Disposition of methadone in methadone maintenance. Clin. Pharmacol. Ther. 1975, 17, 258–266. [Google Scholar] [CrossRef]
- Dematteis, M.; Auriacombe, M.; D’Agnone, O.; Somaini, L.; Szerman, N.; Littlewood, R.; Alam, F.; Alho, H.; Benyamina, A.; Bobes, J.; et al. Recommendations for buprenorphine and methadone therapy in opioid use disorder: A European consensus. Expert Opin. Pharmacother. 2017, 18, 1987–1999. [Google Scholar] [CrossRef]
- Dole, V.P. Implications of methadone maintenance for theories of narcotic addiction. JAMA 1988, 260, 3025–3029. [Google Scholar] [CrossRef]
- Mohamad, N.; Salehuddin, R.M.; Ghazali, B.; Bakar, N.H.A.; Musa, N.; Ibrahim, M.A.; Adnan, L.H.M.; Rashidi, A.; Ismail, R. Plasma methadone level monitoring in methadone maintenance therapy: A personalised methadone therapy. New Insights Toxic. Drug Test. 2013. [Google Scholar] [CrossRef]
- Jones, A.W.; Holmgren, A.; Ahlner, J. Blood methadone concentrations in living and deceased persons: Variations over time, subject demographics, and relevance of coingested drugs. J. Anal. Toxicol. 2012, 36, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Vital signs: Risk for overdose from methadone used for pain relief—United States, 1999–2010. MMWR Morb. Mortal. Wkly. Rep. 2012, 61, 493–497. [Google Scholar]
- Jantos, R.; Skopp, G. Postmortem blood and tissue concentrations of R-and S-enantiomers of methadone and its metabolite EDDP. Forensic Sci. Int. 2013, 226, 254–260. [Google Scholar] [CrossRef]
- Irey, N.S.; Froede, R.C. Evaluation of deaths from drug overdose: A clinicopathologic study. Am. J. Clin. Pathol. 1974, 61, 778–784. [Google Scholar] [CrossRef]
- Corkery, J.M.; Schifano, F.; Ghodse, A.H.; Oyefeso, A. The effects of methadone and its role in fatalities. Hum. Psychopharmacol. 2004, 19, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, F.; Torrens, M. Pharmacogenetics of Methadone Response. Mol. Diag. Ther. 2018, 22, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, R.A.; Sekine, R.; Homel, P.; Lussier, D.; Yap, Y.; Suzuki, Y.; Schweitzer, P.; Yancovitz, S.R.; Lapin, J.A.; Shaiova, L.; et al. Measurement of QTc in patients receiving chronic methadone therapy. J. Pain Sympt. Manag. 2005, 29, 385–391. [Google Scholar] [CrossRef]
- Kornick, C.A.; Kilborn, M.J.; Santiago-Palma, J.; Schulman, G.; Thaler, H.T.; Keefe, D.L.; Katchman, A.N.; Pezzullo, J.C.; Ebert, S.N.; Woosley, R.L.; et al. QTc interval prolongation associated with intravenous methadone. Pain 2003, 105, 499–506. [Google Scholar] [CrossRef]
- Krantz, M.J.; Lewkowiez, L.; Hays, H.; Woodroffe, M.A.; Robertson, A.D.; Mehler, P.S. Torsade de pointes associated with very-high-dose methadone. Ann. Intern. Med. 2002, 137, 501–504. [Google Scholar] [CrossRef]
- Stringer, J.; Welsh, C.; Tommasello, A. Methadone-associated Q-T interval prolongation and torsades de pointes. Am. J. Health Syst. Pharm. 2009, 66, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro. Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Loimer, N.; Schmid, R. The use of plasma levels to optimize methadone maintenance treatment. Drug Alcohol Depend. 1992, 30, 241–246. [Google Scholar] [CrossRef]
- Seymour, A.; Black, M.; Jay, J.; Cooper, G.; Weir, C.; Oliver, J. The role of methadone in drug-related deaths in the west of Scotland. Addiction 2003, 98, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Albion, C.; Shkrum, M.; Cairns, J. Contributing factors to methadone-related deaths in Ontario. Am. J. Forensic Med. Pathol. 2010, 31, 313–319. [Google Scholar] [CrossRef]
- Oldendorf, W.; Hyman, S.; Braun, L.; Oldendorf, S. Blood-brain barrier: Penetration of morphine, codeine, heroin, and methadone after carotid injection. Science 1972, 178, 984–986. [Google Scholar] [CrossRef]
- Peters, M.A. Development of a "blood-brain barrier" to methadone in the newborn rat. J. Pharmacol. Exp. Ther. 1975, 192, 513–520. [Google Scholar]
- Bauer, B.; Yang, X.; Hartz, A.M.; Olson, E.R.; Zhao, R.; Kalvass, J.C.; Pollack, G.M.; Miller, D.S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006, 70, 1212–1219. [Google Scholar] [CrossRef]
- Wang, J.-S.; Ruan, Y.; Taylor, R.M.; Donovan, J.L.; Markowitz, J.S.; DeVane, C.L. Brain penetration of methadone (R)-and (S)-enantiomers is greatly increased by P-glycoprotein deficiency in the blood–brain barrier of Abcb1a gene knockout mice. Psychopharmacology 2004, 173, 132–138. [Google Scholar] [CrossRef]
- Dagenais, C.; Graff, C.L.; Pollack, G.M. Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochem. Pharmacol. 2004, 67, 269–276. [Google Scholar] [CrossRef]
- Coller, J.K.; Barratt, D.T.; Dahlen, K.; Loennechen, M.H.; Somogyi, A.A. ABCB1 genetic variability and methadone dosage requirements in opioid-dependent individuals. Clin. Pharmacol. Ther. 2006, 80, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Crettol, S.; Déglon, J.-J.; Besson, J.; Croquette-Krokar, M.; Hämmig, R.; Gothuey, I.; Monnat, M.; Eap, C.B. ABCB1 and cytochrome P450 genotypes and phenotypes: Influence on methadone plasma levels and response to treatment. Clin. Pharmacol. Ther. 2006, 80, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Crettol, S.; Déglon, J.-J.; Besson, J.; Croquette-Krokar, M.; Hämmig, R.; Gothuey, I.; Monnat, M.; Eap, C. No Influence of ABCB1 Haplotypes on Methadone Dosage Requirement. Clin. Pharmacol. Ther. 2008, 83, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Kreye, G.; Masel, E.-K.; Hackner, K.; Stich, B.; Nauck, F. Methadone as anticancer treatment: Hype, hope, or hazard? Wien. Med. Wochenschr. 2018, 168, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Theile, D.; Mikus, G. Methadone against cancer: Lost in translation. Int. J. Cancer 2018, 143, 1840–1848. [Google Scholar] [CrossRef]
- Kristensen, K.; Christensen, C.B.; Christrup, L.L. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995, 56, PL45–PL50. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef]
- Moreno, E.; Quiroz, C.; Rea, W.; Cai, N.S.; Mallol, J.; Cortes, A.; Lluis, C.; Canela, E.I.; Casado, V.; Ferre, S. Functional mu-Opioid-Galanin Receptor Heteromers in the Ventral Tegmental Area. J. Neurosci. 2017, 37, 1176–1186. [Google Scholar] [CrossRef]
- Cai, N.S.; Quiroz, C.; Bonaventura, J.; Bonifazi, A.; Cole, T.O.; Purks, J.; Billing, A.S.; Massey, E.; Wagner, M.; Wish, E.D.; et al. Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. J. Clin. Investig. 2019, 129, 2730–2744. [Google Scholar] [CrossRef]
- Callahan, R.J.; Au, J.D.; Paul, M.; Liu, C.; Yost, C.S. Functional inhibition by methadone of N-methyl-d-aspartate receptors expressed in Xenopus oocytes: Stereospecific and subunit effects. Anesth. Analg. 2004, 98, 653–659. [Google Scholar] [CrossRef]
- Ebert, B.; Andersen, S.; Krogsgaard-Larsen, P. Ketobemidone, methadone and pethidine are non-competitive N-methyl-d-aspartate (NMDA) antagonists in the rat cortex and spinal cord. Neurosci. Lett. 1995, 187, 165–168. [Google Scholar] [CrossRef]
- Gorman, A.L.; Elliott, K.J.; Inturrisi, C.E. The d- and l-isomers of methadone bind to the non-competitive site on the N-methyl-d-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neurosci. Lett. 1997, 223, 5–8. [Google Scholar] [CrossRef]
- Yang, J.C.; Shan, J.; Ng, K.F.; Pang, P. Morphine and methadone have different effects on calcium channel currents in neuroblastoma cells. Brain Res. 2000, 870, 199–203. [Google Scholar] [CrossRef]
- Xiao, Y.; Smith, R.D.; Caruso, F.S.; Kellar, K.J. Blockade of rat alpha3beta4 nicotinic receptor function by methadone, its metabolites, and structural analogs. J. Pharmacol. Exp. Ther. 2001, 299, 366–371. [Google Scholar]
- Heppe, D.B.; Haigney, M.C.; Krantz, M.J. The effect of oral methadone on the QTc interval in advanced cancer patients: A prospective pilot study. J. Palliat. Med. 2010, 13, 638–639. [Google Scholar] [CrossRef]
- Eap, C.B.; Crettol, S.; Rougier, J.S.; Schlapfer, J.; Sintra Grilo, L.; Deglon, J.J.; Besson, J.; Croquette-Krokar, M.; Carrupt, P.A.; Abriel, H. Stereoselective block of hERG channel by (S)-methadone and QT interval prolongation in CYP2B6 slow metabolizers. Clin. Pharmacol. Ther. 2007, 81, 719–728. [Google Scholar] [CrossRef]
- Ulens, C.; Daenens, P.; Tytgat, J. The dual modulation of GIRK1/GIRK2 channels by opioid receptor ligands. Eur. J. Pharmacol. 1999, 385, 239–245. [Google Scholar] [CrossRef]
- Stoetzer, C.; Kistner, K.; Stüber, T.; Wirths, M.; Schulze, V.; Doll, T.; Foadi, N.; Wegner, F.; Ahrens, J.; Leffler, A. Methadone is a local anaesthetic-like inhibitor of neuronal Na+ channels and blocks excitability of mouse peripheral nerves. Br. J. Anaesth. 2014, 114, 110–120. [Google Scholar] [CrossRef]
- Cuppoletti, J.; Chakrabarti, J.; Tewari, K.; Malinowska, D.H. Methadone but not morphine inhibits lubiprostone-stimulated Cl− currents in T84 intestinal cells and recombinant human ClC-2, but not CFTR Cl− currents. Cell Biochem. Biophys. 2013, 66, 53–63. [Google Scholar] [CrossRef]
- Tournier, N.; Chevillard, L.; Megarbane, B.; Pirnay, S.; Scherrmann, J.M.; Decleves, X. Interaction of drugs of abuse and maintenance treatments with human P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2). Int. J. Neuropsychopharmacol. 2010, 13, 905–915. [Google Scholar] [CrossRef]
- Hemauer, S.J.; Patrikeeva, S.L.; Nanovskaya, T.N.; Hankins, G.D.; Ahmed, M.S. Opiates inhibit paclitaxel uptake by P-glycoprotein in preparations of human placental inside-out vesicles. Biochem. Pharmacol. 2009, 78, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquird cardiac arrthytmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef]
- Huang, M.H.; Shen, A.Y.; Wang, T.S.; Wu, H.M.; Kang, Y.F.; Chen, C.T.; Hsu, T.I.; Chen, B.S.; Wu, S.N. Inhibitory action of methadone and its metabolites on erg-mediated K+ current in GH(3) pituitary tumor cells. Toxicology 2011, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kua, V.M.D.; Rasul, A.; Sreenivasan, S.; Rasool, B.; Younis, T.; Lai, N.S. Methadone hydrochloride and leukemia cells: Effects on cell viability, DNA fragmentation and apoptotic proteins expression level. Pak. J. Pharm. Sci. 2019, 32, 1797–1803. [Google Scholar] [PubMed]
- Singh, A.; Jayanthan, A.; Farran, A.; Elwi, A.N.; Kim, S.W.; Farran, P.; Narendran, A. Induction of apoptosis in pediatric acute lymphoblastic leukemia (ALL) cells by the therapeutic opioid methadone and effective synergy with Bcl-2 inhibition. Leuk. Res. 2011, 35, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Rosales, J.L.; Meier-Stephenson, V.; Kim, S.; Lee, K.Y.; Narendran, A. Genome-wide loss-of-function genetic screening identifies opioid receptor mu1 as a key regulator of L-asparaginase resistance in pediatric acute lymphoblastic leukemia. Oncogene 2017, 36, 5910–5913. [Google Scholar] [CrossRef]
- Brawanski, K.; Brockhoff, G.; Hau, P.; Vollmann-Zwerenz, A.; Freyschlag, C.; Lohmeier, A.; Riemenschneider, M.J.; Thomé, C.; Brawanski, A.; Proescholdt, M.A. Efficacy of D, L-methadone in the treatment of glioblastoma in vitro. CNS Oncol. 2018. [Google Scholar] [CrossRef]
- Shi, L.; Pohla, H.; Buchner, A.; Zhang, L.; Pongratz, T.; Rühm, A.; Zimmermann, W.; Gederaas, O.A.; Wang, X.; Stepp, H. MOP-dependent enhancement of methadone on the effectiveness of ALA-PDT for A172 cells by upregulating phosphorylated JNK and BCL2. Photodiagnosis Photodyn. Ther. 2020. [Google Scholar] [CrossRef]
- Perez-Alvarez, S.; Cuenca-Lopez, M.D.; de Mera, R.M.M.-F.; Puerta, E.; Karachitos, A.; Bednarczyk, P.; Kmita, H.; Aguirre, N.; Galindo, M.F.; Jordán, J. Methadone induces necrotic-like cell death in SH-SY5Y cells by an impairment of mitochondrial ATP synthesis. Biochim. Biophys. Acta 2010, 1802, 1036–1047. [Google Scholar] [CrossRef]
- Landgraf, V.; Griessmann, M.; Roller, J.; Polednik, C.; Schmidt, M. DL-Methadone as an Enhancer of Chemotherapeutic Drugs in Head and Neck Cancer Cell Lines. Anticancer Res. 2019, 39, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Schultze-Seemann, S.; Kuckuck, I.; Katzenwadel, A.; Wolf, P. Impact of Methadone on Cisplatin Treatment of Bladder Cancer Cells. Anticancer Res. 2018, 38, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; McLaughlin, P.J. Opioids and the apoptotic pathway in human cancer cells. Neuropeptides 2003, 37, 79–88. [Google Scholar] [CrossRef]
- Shi, L.; Buchner, A.; Pohla, H.; Pongratz, T.; Rühm, A.; Zimmermann, W.; Gederaas, O.A.; Zhang, L.; Wang, X.; Stepp, H. Methadone enhances the effectiveness of 5-aminolevulinic acid-based photodynamic therapy for squamous cell carcinoma and glioblastoma in vitro. J. Biophoton. 2019, 12, e201800468. [Google Scholar] [CrossRef]
- Mahmoudi, K.; Garvey, K.L.; Bouras, A.; Cramer, G.; Stepp, H.; Jesu Raj, J.G.; Bozec, D.; Busch, T.M.; Hadjipanayis, C.G. 5-aminolevulinic acid photodynamic therapy for the treatment of high-grade gliomas. J. Neurooncol. 2019, 141, 595–607. [Google Scholar] [CrossRef]
- Ananda, S.; Nowak, A.K.; Cher, L.; Dowling, A.; Brown, C.; Simes, J.; Rosenthal, M.A. Phase 2 trial of temozolomide and pegylated liposomal doxorubicin in the treatment of patients with glioblastoma multiforme following concurrent radiotherapy and chemotherapy. J. Clin. Neurosci. 2011, 18, 1444–1448. [Google Scholar] [CrossRef]
- Hau, P.; Fabel, K.; Baumgart, U.; Rümmele, P.; Grauer, O.; Bock, A.; Dietmaier, C.; Dietmaier, W.; Dietrich, J.; Dudel, C. Pegylated liposomal doxorubicin-efficacy in patients with recurrent high-grade glioma. Cancer 2004, 100, 1199–1207. [Google Scholar] [CrossRef]
- Oppermann, H.; Matusova, M.; Glasow, A.; Dietterle, J.; Baran-Schmidt, R.; Neumann, K.; Meixensberger, J.; Gaunitz, F. D,L-Methadone does not improve radio- and chemotherapy in glioblastoma in vitro. Cancer ChemoTher. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Grinshpoon, A.; Barchana, M.; Lipshitz, I.; Rosca, P.; Weizman, A.; Ponizovsky, A.M. Methadone maintenance and cancer risk: An Israeli case registry study. Drug Alcohol Depend. 2011, 119, 88–92. [Google Scholar] [CrossRef]
- Krebs, E.E.; Becker, W.C.; Zerzan, J.; Bair, M.J.; McCoy, K.; Hui, S. Comparative mortality among Department of Veterans Affairs patients prescribed methadone or long-acting morphine for chronic pain. Pain 2011, 152, 1789–1795. [Google Scholar] [CrossRef]
- Reddy, A.; Schuler, U.S.; de la Cruz, M.; Yennurajalingam, S.; Wu, J.; Liu, D.; Bruera, E. Overall Survival among Cancer Patients Undergoing Opioid Rotation to Methadone Compared to Other Opioids. J. Palliat. Med. 2017, 20, 656–661. [Google Scholar] [CrossRef]
- Palme, D.; Misovic, M.; Ganser, K.; Klumpp, L.; Salih, H.R.; Zips, D.; Huber, S.M. hERG K+ channels promote survival of irradiated leukemia cells. Front. Pharmacol. 2020, 11, 489. [Google Scholar] [CrossRef]
- Lupp, A.; Richter, N.; Doll, C.; Nagel, F.; Schulz, S. UMB-3, a novel rabbit monoclonal antibody, for assessing mu-opioid receptor expression in mouse, rat and human formalin-fixed and paraffin-embedded tissues. Regul. Pept. 2011, 167, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, E.; Zirjacks, L.; Ganser, K.; Klumpp, L.; Schuler, U.; Zips, D.; Eckert, F.; Huber, S.M. Alternating Electric Fields (TTFields) Activate Cav1.2 Channels in Human Glioblastoma Cells. Cancers 2019, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Klumpp, L.; Misovic, M.; Edalat, L.; Eckert, M.; Klumpp, D.; Ruth, P.; Huber, S.M. K(+) channel signaling in irradiated tumor cells. Eur. Biophys. J. 2016, 45, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Heise, N.; Palme, D.; Misovic, M.; Koka, S.; Rudner, J.; Lang, F.; Salih, H.R.; Huber, S.M.; Henke, G. Non-selective cation channel-mediated Ca2+-entry and activation of Ca2+/calmodulin-dependent kinase II contribute to G2/M cell cycle arrest and survival of irradiated leukemia cells. Cell Physiol. Biochem. 2010, 26, 597–608. [Google Scholar] [CrossRef]
- Palme, D.; Misovic, M.; Schmid, E.; Klumpp, D.; Salih, H.R.; Rudner, J.; Huber, S.M. Kv3.4 potassium channel-mediated electrosignaling controls cell cycle and survival of irradiated leukemia cells. Pflug. Arch. 2013, 465, 1209–1221. [Google Scholar] [CrossRef]
- Stegen, B.; Butz, L.; Klumpp, L.; Zips, D.; Dittmann, K.; Ruth, P.; Huber, S.M. Ca2+-Activated IK K+ Channel Blockade Radiosensitizes Glioblastoma Cells. Mol. Cancer Res. 2015, 13, 1283–1295. [Google Scholar] [CrossRef]
- Klumpp, D.; Misovic, M.; Szteyn, K.; Shumilina, E.; Rudner, J.; Huber, S.M. Targeting TRPM2 Channels Impairs Radiation-Induced Cell Cycle Arrest and Fosters Cell Death of T Cell Leukemia Cells in a Bcl-2-Dependent Manner. Oxid. Med. Cell Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.; Frank, S.C.; Klumpp, L.; Sezgin, E.C.; Eckert, M.; Edalat, L.; Bastmeyer, M.; Zips, D.; Ruth, P.; Huber, S.M. TRPM8 is required for survival and radioresistance of glioblastoma cells. Oncotarget 2017, 8, 95896. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Incidence | Daily Oral Dose [mg] | Organ | Methadone [µM] | Patient Number | Ref. |
|---|---|---|---|---|---|---|
| anti-glioma therapy | methadone maintenance | 30 | blood | 0.59–0.67 | 1 | [19] |
| drug substitution | methadone maintenance | 83.3 * | blood | 1.5 * 2 # | 104 | [46] |
| drug abuse | methadone-associated death | Blood | 2.6 * 14.5 # | 17 | [38] | |
| brain | 12.0 *,$ | 17 | ||||
| drug substitution/drug abuse | methadone-associated death | 20–80 | Blood | 1.6 ** 10.6 # | 15 | [37] |
| brain | 2.2 **,$ 8.6 #,$ | 15 | ||||
| CSF | 0.7 ** 2.8 # | 8 | ||||
| drug substitution/drug abuse | § methadone-associated death | blood | 2.6 * (2.3 **) 8.5 # | 52 | [47] | |
| drug substitution/drug abuse | § methadone-associated death | blood | 1.3 * (2.3 **) 9.7 # | 11 | [48] |
| Protein | Gene | Cell Model | Cellular Effects | Methadone IC50 | Enantiomer | Refs |
|---|---|---|---|---|---|---|
| N-methyl-d-aspartate receptors | GRIN1 GRIN2A GRIN2B GRIN2C GRIN2D | Heterologous Xenopus laevis Expression system rat cortical membranes spinal cord rat forebrain spinal cord | blockage of NMDA currents | ≥~2 µM ≥~0.1 µM ≥~4 µM ~3.1 µM >~3.1 µM ~18 µM ~4.3 µM | R(−)/S(+) R(−) S(+) R(−)/S(+) R(−)/S(+) R(−)/S(+) R(−)/S(+) | [63,64,65] |
| L type Ca2+ channels T type Ca2+ channels | CACNA1C CACNA1D CACNA1S CACNA1F CACNA1G CACNA1H CACNA1I | mouse neuroblastoma | Blockage of T- and L-type Ca2+ currents | >~10 µM | R(−)/S(+) ? | [66] |
| α3β4 nicotinic receptor | CHRNA3 CHRNB4 | nAChRs-transfected HEK293 cells | blockage of 86Rb+ efflux | 1.9 ± 0.2 μM | R(−)/S(+) | [67] |
| hERG1 | KCNH2 | GH3 pituitary tumor cells HEK293 stably expressing hERG | blockage of hERG currents | ~10 µM 7 µM 2 µM | R(−)/S(+) R(−) S(+) | [68,69] |
| GIRK1/GIRK2 | KCNJ3/KCNJ6 | Xenopus laevis oocytes injected with mRNA | blockage of GIRK1/GIRK2 currents | ~53 μM | R(−)/S(+) | [70] |
| Nav1.2 Nav1.3 Nav1.7 Nav1.8 | SCN2A SCN3A SCN9A SCN10A | mouse peripheral nerves | blockage of excitability | 86–119 µM | R(−)/S(+) | [71] |
| ClC-2 | CLCN2 | T84 intestinal cells HEK293EBNA stably expressing hClC-2 | blockage of hClC-2 currents | 100 Nm 100–230 nM | R(−)/S(+) R(−)/S(+) | [72] |
| p-glyco-protein (hMDR) | ABCB1 | hMDR -transfected HEK293 cells human placental inside-out vesicles | calcein efflux paclitaxel uptake | >25 µM Ki = 18 µM | R(−)/S(+) R(−), S(+) R(−)/S(+) | [73,74] |
| Tumor Model Carcinoma Cells | Methadone | Read-Out | Effects | Ref |
|---|---|---|---|---|
| H187 (SCLC) and H157 (NSCLC) cells | 0.01–0.1 µM | dehydroge- nase activity, trypan blue exclusion, delayed plating growth assay | impaired viability IC50 0.3–10 nM | [14] |
| NCI-N417 (SCLC), NCI-H460 (NSCLC) xenografts | 10 mg/(kg d) | tumor volume | growth delay | [14] |
| MIA PaCa-2 pancreatic and HT-29 colon adeno-carcinoma, CAL-27 (HNSCC) cells | 10 µM | TUNEL, annexin-V binding, trypan blue exclusion | cell viability was not altered | [86] |
| FaDu, HLaC78 and PJ41 (HNSCC) cells | 2–32 µM | dehydroge- nase activity | impaired viability IC50 >> 32 µM cisplatin-, doxorubicin-, 5-FU- and paclitaxel sensitization IC50 ≥ 32 µM | [84] |
| FaDu (HNSCC) cells | 32 µM | annexin-V binding | cell viability was not altered, enhancement of ALA-PDT | [87] |
| T24 and HT-1376 bladder cancer cells | 0.3–32 µM | dehydroge- nase activity, annexin-V binding PI staining | impaired viability IC50 >> 32 µM cisplatin sensitization ~32 µM and >>32 µM | [85] |
| leukemia | ||||
| CEM and HL-60 leukemia cells | 10–30 µM | subG1 population PI-staining | apoptotic cell death IC50 ~15 µM | [17] |
| CCRF-CEM and HL-60 leukemia cells | 60–200 µM | dehydroge-nase activity | impaired viability IC50 ≥ 100 µM | [78] |
| ALL leukemia cells | 1–323 µM | cell number | impaired viability IC50 >20 µM | [79] |
| ALL leukemia cells | 0.3–32 µM | subG1 population PI-staining | apoptotic cell death, IC50 >32 µM, doxorubicin sensitization IC50 <0.3–≥32 µM | [18] |
| ALL leukemia cells | 20 µM | western blot, cell viability assay | OPMR1 knockdown enhanced asparaginase resistance, methadone sensitized to asparaginase treatment | [80] |
| ALL leukemia xenografts | 20–120 mg/(kg d) | tumor volume | growth delay, doxorubicin sensitization | [18] |
| neuroblastoma | ||||
| SH-SY5Y human neuroblastomacell line | 100–1000 µM | LDH activity, caspase activity, cyt-c release, ATP concentration | caspase independent cell death, bioenergetic crisis IC50 ~500µM, | [83] |
| glioblastoma | ||||
| U118MG and A172 glioblastoma cells | 3.2–32 µM | subG1 population PI-staining | cell death IC50 > 32 µM, doxorubicin sensitization IC50 ≤3 - ~10 µM | [16] |
| U87MG glioblastoma xenografts | 60–120 mg/(kg d) | tumor volume | growth delay | [16] |
| U87MG, U251 and primary glioblastoma cells | 1–145 µM | crystal violet staining, annexin-V- - binding | impaired viability/apoptotic cell death IC50 (25) ≥100 µM, TMZ sensitization IC50 (~50) >>145 µM | [81] |
| primary glioblastoma cells | 1–30 µM | ATP concentration, dehydroge- nase activity | impaired viability IC50 between 10 and 30 µM, TMZ sensitization IC50 >> 30 µM, radiosensitization IC50 >> 30 µM | [91] |
| A172 glioblastoma cells | 32 µM | annexin-V binding, 7- 7-AAD exclusion | unaltered viability, enhancement of ALA-PDT | [87] |
| A172 glioblastoma cells | 0.065 µM | annexin-V binding | apoptotic cell death, enhancement of ALA-PDT | [82] |
| A172 glioblastoma cells | 2–32 µM | dehydroge- nase activity | impaired viability IC50 >>32 µM cisplatin-, doxorubicin-, 5-FU- and paclitaxel sensitization IC50 ≤4 - ≥32 µM | [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vatter, T.; Klumpp, L.; Ganser, K.; Stransky, N.; Zips, D.; Eckert, F.; Huber, S.M. Against Repurposing Methadone for Glioblastoma Therapy. Biomolecules 2020, 10, 917. https://doi.org/10.3390/biom10060917
Vatter T, Klumpp L, Ganser K, Stransky N, Zips D, Eckert F, Huber SM. Against Repurposing Methadone for Glioblastoma Therapy. Biomolecules. 2020; 10(6):917. https://doi.org/10.3390/biom10060917
Chicago/Turabian StyleVatter, Tatjana, Lukas Klumpp, Katrin Ganser, Nicolai Stransky, Daniel Zips, Franziska Eckert, and Stephan M. Huber. 2020. "Against Repurposing Methadone for Glioblastoma Therapy" Biomolecules 10, no. 6: 917. https://doi.org/10.3390/biom10060917
APA StyleVatter, T., Klumpp, L., Ganser, K., Stransky, N., Zips, D., Eckert, F., & Huber, S. M. (2020). Against Repurposing Methadone for Glioblastoma Therapy. Biomolecules, 10(6), 917. https://doi.org/10.3390/biom10060917