Emerging Roles of RNA 3′-end Cleavage and Polyadenylation in Pathogenesis, Diagnosis and Therapy of Human Disorders

Abstract
1. Introduction
1.1. Cleavage and Polyadenylation Factors Interacting with Poorly Conserved Sequence Motifs Control mRNA 3′ end Formation
1.2. Alternative 3′ end Formation Expands Transcriptional Complexity
1.3. Cleavage and Polyadenylation Signal Motifs Determine Alternative Polyadenylation
1.4. Cleavage and Polyadenylation Factor Abundance Regulates Alternative Polyadenylation
2. The Role of Cleavage and Polyadenylation in Disease
2.1. Defects in Cis Resulting in Altered Cleavage and Polyadenylation
2.1.1. Loss of Function Alterations in the Hexamer of a Single PAS Gene Can Affect Gene Expression and Result in Disease
2.1.2. Loss of Function Alterations in a Hexamer of a Multi PAS Gene Resulting in Alternative Polyadenylation and Disease
2.1.3. Gain of Function Alterations Can also Affect Cleavage and Polyadenylation and Result in Disease
2.1.4. Alterations in Non-Hexamer Elements Affecting Cleavage and Polyadenylation and Resulting in Disease
2.1.5. Alterations outside Characterised Elements Affecting Cleavage and Polyadenylation and Resulting in Disease
2.2. Defects in Trans Resulting in Altered Cleavage and Polyadenylation
2.2.1. Alterations in Trans Factors in Alternative Polyadenylation and Disease
2.2.2. Alterations of the CFI Complex in Alternative Polyadenylation and Disease
2.2.3. Alterations of the CSTF Complex in Alternative Polyadenylation and Disease
2.2.4. Alterations of the CFII Complex in Alternative Polyadenylation and Disease
2.2.5. Alterations of the CPSF Complex in Alternative Polyadenylation and Disease
2.2.6. Alterations of Poly(A)Polymerases and Poly(A) Binding Proteins in Alternative Polyadenylation and Disease
2.2.7. Combined Perturbations Affecting Alternative Polyadenylation and Resulting in Disease
3. APA in Molecular Diagnostics
4. Targeting mRNA 3′ end Formation as a Novel Therapy
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manning, K.S.; Cooper, T.A. The roles of RNA processing in translating genotype to phenotype. Nat. Rev. Mol. Cell. Biol. 2017, 18, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell. Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Natalizio, B.J.; Wente, S.R. Postage for the messenger: Designating routes for nuclear mRNA export. Trends Cell Biol. 2013, 23, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Fuke, H.; Ohno, M. Role of poly (A) tail as an identity element for mRNA nuclear export. Nucleic Acids Res. 2008, 36, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Shyu, A.B. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip. Rev. RNA 2011, 2, 167–183. [Google Scholar] [CrossRef]
- Norbury, C.J. Cytoplasmic RNA: A case of the tail wagging the dog. Nat. Rev. Mol. Cell. Biol. 2013, 14, 643–653. [Google Scholar] [CrossRef]
- Kumar, A.; Clerici, M.; Muckenfuss, L.M.; Passmore, L.A.; Jinek, M. Mechanistic insights into mRNA 3′-end processing. Curr. Opin. Struct. Biol. 2019, 59, 143–150. [Google Scholar] [CrossRef]
- Chan, S.; Choi, E.A.; Shi, Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2011, 2, 321–335. [Google Scholar] [CrossRef]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Means to an end: Mechanisms of alternative polyadenylation of messenger RNA precursors. Wiley Interdiscip. Rev. RNA 2014, 5, 183–196. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Danckwardt, S.; Hentze, M.W.; Kulozik, A.E. 3′ end mRNA processing: Molecular mechanisms and implications for health and disease. EMBO J. 2008, 27, 482–498. [Google Scholar] [CrossRef] [PubMed]
- Ogorodnikov, A.; Levin, M.; Tattikota, S.; Tokalov, S.; Hoque, M.; Scherzinger, D.; Marini, F.; Poetsch, A.; Binder, H.; Macher-Goppinger, S.; et al. Transcriptome 3′end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 2018, 9, 5331. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Di Giammartino, D.C.; Taylor, D.; Sarkeshik, A.; Rice, W.J.; Yates, J.R., 3rd; Frank, J.; Manley, J.L. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 2009, 33, 365–376. [Google Scholar] [CrossRef]
- Gruber, A.J.; Schmidt, R.; Gruber, A.R.; Martin, G.; Ghosh, S.; Belmadani, M.; Keller, W.; Zavolan, M. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016, 26, 1145–1159. [Google Scholar] [CrossRef]
- Beaudoing, E.; Freier, S.; Wyatt, J.R.; Claverie, J.M.; Gautheret, D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000, 10, 1001–1010. [Google Scholar] [CrossRef]
- Zarudnaya, M.I.; Kolomiets, I.M.; Potyahaylo, A.L.; Hovorun, D.M. Downstream elements of mammalian pre-mRNA polyadenylation signals: Primary, secondary and higher-order structures. Nucleic Acids Res. 2003, 31, 1375–1386. [Google Scholar] [CrossRef]
- Dantonel, J.C.; Murthy, K.G.; Manley, J.L.; Tora, L. Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature 1997, 389, 399–402. [Google Scholar] [CrossRef]
- Murthy, K.G.; Manley, J.L. Characterization of the multisubunit cleavage-polyadenylation specificity factor from calf thymus. J. Biol. Chem. 1992, 267, 14804–14811. [Google Scholar]
- Kaufmann, I.; Martin, G.; Friedlein, A.; Langen, H.; Keller, W. Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 2004, 23, 616–626. [Google Scholar] [CrossRef]
- Chan, S.L.; Huppertz, I.; Yao, C.; Weng, L.; Moresco, J.J.; Yates, J.R., 3rd; Ule, J.; Manley, J.L.; Shi, Y. CPSF30 and Wdr33 directly bind to AAUAAA in mammalian mRNA 3′ processing. Genes Dev. 2014, 28, 2370–2380. [Google Scholar] [CrossRef] [PubMed]
- Schonemann, L.; Kuhn, U.; Martin, G.; Schafer, P.; Gruber, A.R.; Keller, W.; Zavolan, M.; Wahle, E. Reconstitution of CPSF active in polyadenylation: Recognition of the polyadenylation signal by WDR33. Genes Dev. 2014, 28, 2381–2393. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Faini, M.; Aebersold, R.; Jinek, M. Structural insights into the assembly and polyA signal recognition mechanism of the human CPSF complex. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, Y.; Hamilton, K.; Manley, J.L.; Shi, Y.; Walz, T.; Tong, L. Molecular basis for the recognition of the human AAUAAA polyadenylation signal. Proc. Natl. Acad. Sci. USA 2018, 115, E1419–E1428. [Google Scholar] [CrossRef]
- Martin, G.; Gruber, A.R.; Keller, W.; Zavolan, M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012, 1, 753–763. [Google Scholar] [CrossRef]
- MacDonald, C.C.; Wilusz, J.; Shenk, T. The 64-kilodalton subunit of the CstF polyadenylation factor binds to pre-mRNAs downstream of the cleavage site and influences cleavage site location. Mol. Cell. Biol. 1994, 14, 6647–6654. [Google Scholar] [CrossRef][Green Version]
- Takagaki, Y.; Manley, J.L. RNA recognition by the human polyadenylation factor CstF. Mol. Cell. Biol. 1997, 17, 3907–3914. [Google Scholar] [CrossRef]
- Chen, F.; Wilusz, J. Auxiliary downstream elements are required for efficient polyadenylation of mammalian pre-mRNAs. Nucleic Acids Res. 1998, 26, 2891–2898. [Google Scholar] [CrossRef]
- Takagaki, Y.; Seipelt, R.L.; Peterson, M.L.; Manley, J.L. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell 1996, 87, 941–952. [Google Scholar] [CrossRef]
- Chou, Z.F.; Chen, F.; Wilusz, J. Sequence and position requirements for uridylate-rich downstream elements of polyadenylation signals. Nucleic Acids Res. 1994, 22, 2525–2531. [Google Scholar] [CrossRef][Green Version]
- Kargapolova, Y.; Levin, M.; Lackner, K.; Danckwardt, S. sCLIP-an integrated platform to study RNA-protein interactomes in biomedical research: Identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Res. 2017, 45, 6074–6086. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, K.; Brown, K.M.; Gilmartin, G.M. Analysis of a noncanonical poly(A) site reveals a tripartite mechanism for vertebrate poly(A) site recognition. Genes Dev. 2005, 19, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Gilmartin, G.M.; Doublie, S. Structural basis of UGUA recognition by the Nudix protein CFI(m)25 and implications for a regulatory role in mRNA 3′ processing. Proc. Natl. Acad. Sci. USA 2010, 107, 10062–10067. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P.; Tuting, C.; Schonemann, L.; Kuhn, U.; Treiber, T.; Treiber, N.; Ihling, C.; Graber, A.; Keller, W.; Meister, G.; et al. Reconstitution of mammalian cleavage factor II involved in 3′ processing of mRNA precursors. RNA 2018, 24, 1721–1737. [Google Scholar] [CrossRef]
- Dominski, Z.; Yang, X.C.; Marzluff, W.F. The polyadenylation factor CPSF-73 is involved in histone-pre-mRNA processing. Cell 2005, 123, 37–48. [Google Scholar] [CrossRef]
- Mandel, C.R.; Kaneko, S.; Zhang, H.; Gebauer, D.; Vethantham, V.; Manley, J.L.; Tong, L. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature 2006, 444, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef]
- Conne, B.; Stutz, A.; Vassalli, J.D. The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat. Med. 2000, 6, 637–641. [Google Scholar] [CrossRef]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3, Reviews0004. [Google Scholar] [CrossRef]
- Chabanon, H.; Mickleburgh, I.; Hesketh, J. Zipcodes and postage stamps: mRNA localisation signals and their trans-acting binding proteins. Brief. Funct. Genomic. Proteomic. 2004, 3, 240–256. [Google Scholar] [CrossRef]
- Nourse, J.; Braun, J.; Lackner, K.; Huttelmaier, S.; Danckwardt, S. Large-scale identification of functional microRNA targeting reveals cooperative regulation of the hemostatic system. J. Thromb. Haemost. 2018, 16, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C. Regulation by 3′-Untranslated Regions. Annu. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Proudfoot, N.J. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 2009, 136, 688–700. [Google Scholar] [CrossRef]
- Maniatis, T.; Reed, R. An extensive network of coupling among gene expression machines. Nature 2002, 416, 499–506. [Google Scholar] [CrossRef]
- Rigo, F.; Martinson, H.G. Functional coupling of last-intron splicing and 3′-end processing to transcription in vitro: The poly(A) signal couples to splicing before committing to cleavage. Mol. Cell. Biol. 2008, 28, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Kyburz, A.; Friedlein, A.; Langen, H.; Keller, W. Direct interactions between subunits of CPSF and the U2 snRNP contribute to the coupling of pre-mRNA 3′ end processing and splicing. Mol. Cell 2006, 23, 195–205. [Google Scholar] [CrossRef]
- Kaida, D. The reciprocal regulation between splicing and 3′-end processing. Wiley Interdiscip. Rev. RNA 2016, 7, 499–511. [Google Scholar] [CrossRef]
- Martins, S.B.; Rino, J.; Carvalho, T.; Carvalho, C.; Yoshida, M.; Klose, J.M.; de Almeida, S.F.; Carmo-Fonseca, M. Spliceosome assembly is coupled to RNA polymerase II dynamics at the 3′ end of human genes. Nat. Struct. Mol. Biol. 2011, 18, 1115–1123. [Google Scholar] [CrossRef]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Park, C.Y.; et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef]
- Lian, Z.; Karpikov, A.; Lian, J.; Mahajan, M.C.; Hartman, S.; Gerstein, M.; Snyder, M.; Weissman, S.M. A genomic analysis of RNA polymerase II modification and chromatin architecture related to 3′ end RNA polyadenylation. Genome Res. 2008, 18, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Spies, N.; Nielsen, C.B.; Padgett, R.A.; Burge, C.B. Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell 2009, 36, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.; Schulz, R.; Woodfine, K.; Koltowska, K.; Beechey, C.V.; Peters, J.; Bourc’his, D.; Oakey, R.J. Regulation of alternative polyadenylation by genomic imprinting. Genes Dev. 2008, 22, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Danckwardt, S.; Gantzert, A.S.; Macher-Goeppinger, S.; Probst, H.C.; Gentzel, M.; Wilm, M.; Grone, H.J.; Schirmacher, P.; Hentze, M.W.; Kulozik, A.E. p38 MAPK controls prothrombin expression by regulated RNA 3′ end processing. Mol. Cell 2011, 41, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Edwalds-Gilbert, G.; Prescott, J.; Falck-Pedersen, E. 3′ RNA processing efficiency plays a primary role in generating termination-competent RNA polymerase II elongation complexes. Mol. Cell. Biol. 1993, 13, 3472–3480. [Google Scholar] [CrossRef]
- Millevoi, S.; Vagner, S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010, 38, 2757–2774. [Google Scholar] [CrossRef]
- Sheets, M.D.; Ogg, S.C.; Wickens, M.P. Point mutations in AAUAAA and the poly (A) addition site: Effects on the accuracy and efficiency of cleavage and polyadenylation in vitro. Nucleic Acids Res. 1990, 18, 5799–5805. [Google Scholar] [CrossRef]
- Cheng, Y.; Miura, R.M.; Tian, B. Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics 2006, 22, 2320–2325. [Google Scholar] [CrossRef]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef]
- Lutz, C.S. Alternative polyadenylation: A twist on mRNA 3′ end formation. ACS Chem. Biol. 2008, 3, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.; Goncalves, V.; Fernandes, S.; Marques, T.; Pereira, M.; Gama-Carvalho, M. Networks of mRNA Processing and Alternative Splicing Regulation in Health and Disease. Adv. Exp. Med. Biol. 2019, 1157, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.J.; Zavolan, M. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 2019, 20, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Derti, A.; Garrett-Engele, P.; Macisaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- Hoque, M.; Li, W.; Tian, B. Accurate mapping of cleavage and polyadenylation sites by 3′ region extraction and deep sequencing. Methods Mol. Biol. 2014, 1125, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bagga, P.S.; Ford, L.P.; Chen, F.; Wilusz, J. The G-rich auxiliary downstream element has distinct sequence and position requirements and mediates efficient 3′ end pre-mRNA processing through a trans-acting factor. Nucleic Acids Res. 1995, 23, 1625–1631. [Google Scholar] [CrossRef]
- Yonaha, M.; Proudfoot, N.J. Transcriptional termination and coupled polyadenylation in vitro. EMBO J. 2000, 19, 3770–3777. [Google Scholar] [CrossRef]
- Nag, A.; Narsinh, K.; Kazerouninia, A.; Martinson, H.G. The conserved AAUAAA hexamer of the poly(A) signal can act alone to trigger a stable decrease in RNA polymerase II transcription velocity. RNA 2006, 12, 1534–1544. [Google Scholar] [CrossRef][Green Version]
- Fusby, B.; Kim, S.; Erickson, B.; Kim, H.; Peterson, M.L.; Bentley, D.L. Coordination of RNA Polymerase II Pausing and 3′ End Processing Factor Recruitment with Alternative Polyadenylation. Mol. Cell. Biol. 2016, 36, 295–303. [Google Scholar] [CrossRef]
- Pinto, P.A.; Henriques, T.; Freitas, M.O.; Martins, T.; Domingues, R.G.; Wyrzykowska, P.S.; Coelho, P.A.; Carmo, A.M.; Sunkel, C.E.; Proudfoot, N.J.; et al. RNA polymerase II kinetics in polo polyadenylation signal selection. EMBO J. 2011, 30, 2431–2444. [Google Scholar] [CrossRef]
- Millevoi, S.; Decorsiere, A.; Loulergue, C.; Iacovoni, J.; Bernat, S.; Antoniou, M.; Vagner, S. A physical and functional link between splicing factors promotes pre-mRNA 3′ end processing. Nucleic Acids Res. 2009, 37, 4672–4683. [Google Scholar] [CrossRef] [PubMed]
- Neve, J.; Burger, K.; Li, W.; Hoque, M.; Patel, R.; Tian, B.; Gullerova, M.; Furger, A. Subcellular RNA profiling links splicing and nuclear DICER1 to alternative cleavage and polyadenylation. Genome Res. 2016, 26, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Park, J.Y.; Zheng, D.; Hoque, M.; Yehia, G.; Tian, B. Alternative cleavage and polyadenylation in spermatogenesis connects chromatin regulation with post-transcriptional control. BMC Biol. 2016, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, J.; Di Stefano, B.; Wang, X.; Borkent, M.; Forouzmand, E.; Clowers, K.J.; Ji, F.; Schwarz, B.A.; Kalocsay, M.; Elledge, S.J.; et al. Nudt21 Controls Cell Fate by Connecting Alternative Polyadenylation to Chromatin Signaling. Cell 2018, 172, 106–120. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m(6)A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef]
- Parker, M.T.; Knop, K.; Sherwood, A.V.; Schurch, N.J.; Mackinnon, K.; Gould, P.D.; Hall, A.J.; Barton, G.J.; Simpson, G.G. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m(6)A modification. Elife 2020, 9. [Google Scholar] [CrossRef]
- Kubo, T.; Wada, T.; Yamaguchi, Y.; Shimizu, A.; Handa, H. Knock-down of 25 kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res. 2006, 34, 6264–6271. [Google Scholar] [CrossRef]
- Masamha, C.P.; Xia, Z.; Yang, J.; Albrecht, T.R.; Li, M.; Shyu, A.B.; Li, W.; Wagner, E.J. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 2014, 510, 412–416. [Google Scholar] [CrossRef]
- Li, W.; You, B.; Hoque, M.; Zheng, D.; Luo, W.; Ji, Z.; Park, J.Y.; Gunderson, S.I.; Kalsotra, A.; Manley, J.L.; et al. Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015, 11, e1005166. [Google Scholar] [CrossRef]
- Gruber, A.R.; Martin, G.; Keller, W.; Zavolan, M. Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol. 2012, 9, 1405–1412. [Google Scholar] [CrossRef]
- Yao, C.; Biesinger, J.; Wan, J.; Weng, L.; Xing, Y.; Xie, X.; Shi, Y. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2012, 109, 18773–18778. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.W.; Park, C.Y.; Goodarzi, H.; Fak, J.J.; Mele, A.; Moore, M.J.; Saito, Y.; Darnell, R.B. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Rep. 2016, 15, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Dichtl, B.; Blank, D.; Sadowski, M.; Hubner, W.; Weiser, S.; Keller, W. Yhh1p/Cft1p directly links poly(A) site recognition and RNA polymerase II transcription termination. EMBO J. 2002, 21, 4125–4135. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, E.K.; Schnitzler, C.E.; Havlak, P.; Putnam, N.H.; Nguyen, A.D.; Moreland, R.T.; Baxevanis, A.D. Evolutionary profiling reveals the heterogeneous origins of classes of human disease genes: Implications for modeling disease genetics in animals. BMC Evol. Biol. 2014, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Zhou, Q.; Hong, Y.; Li, Q.Q. Role of alternative polyadenylation dynamics in acute myeloid leukaemia at single-cell resolution. RNA Biol. 2019, 16, 785–797. [Google Scholar] [CrossRef]
- Singh, P.; Alley, T.L.; Wright, S.M.; Kamdar, S.; Schott, W.; Wilpan, R.Y.; Mills, K.D.; Graber, J.H. Global changes in processing of mRNA 3′ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009, 69, 9422–9430. [Google Scholar] [CrossRef]
- Xiong, M.; Chen, L.; Zhou, L.; Ding, Y.; Kazobinka, G.; Chen, Z.; Hou, T. NUDT21 inhibits bladder cancer progression through ANXA2 and LIMK2 by alternative polyadenylation. Theranostics 2019, 9, 7156–7167. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, J.X.; Luo, J.H.; Wu, S.; Yuan, G.J.; Ma, N.F.; Feng, Y.; Cai, M.Y.; Chen, R.X.; Lu, J.; et al. CSTF2-Induced Shortening of the RAC1 3′UTR Promotes the Pathogenesis of Urothelial Carcinoma of the Bladder. Cancer Res. 2018, 78, 5848–5862. [Google Scholar] [CrossRef]
- Zhong, J.; Cao, R.X.; Hong, T.; Yang, J.; Zu, X.Y.; Xiao, X.H.; Liu, J.H.; Wen, G.B. Identification and expression analysis of a novel transcript of the human PRMT2 gene resulted from alternative polyadenylation in breast cancer. Gene 2011, 487, 1–9. [Google Scholar] [CrossRef]
- Ni, T.K.; Kuperwasser, C. Premature polyadenylation of MAGI3 produces a dominantly-acting oncogene in human breast cancer. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Miles, W.O.; Lembo, A.; Volorio, A.; Brachtel, E.; Tian, B.; Sgroi, D.; Provero, P.; Dyson, N. Alternative Polyadenylation in Triple-Negative Breast Tumors Allows NRAS and c-JUN to Bypass PUMILIO Posttranscriptional Regulation. Cancer Res. 2016, 76, 7231–7241. [Google Scholar] [CrossRef]
- Yan, H.; Tian, R.; Wang, W.; Zhang, M.; Wu, J.; He, J. Aberrant Ki-67 expression through 3′UTR alternative polyadenylation in breast cancers. FEBS Open Bio 2018, 8, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Ji, P.; Kim, S.; Xia, Z.; Rodriguez, B.; Li, L.; Su, J.; Chen, K.; Masamha, C.P.; Baillat, D.; et al. 3′ UTR shortening represses tumor-suppressor genes in trans by disrupting ceRNA crosstalk. Nat. Genet. 2018, 50, 783–789. [Google Scholar] [CrossRef]
- Gillen, A.E.; Brechbuhl, H.M.; Yamamoto, T.M.; Kline, E.; Pillai, M.M.; Hesselberth, J.R.; Kabos, P. Alternative Polyadenylation of PRELID1 Regulates Mitochondrial ROS Signaling and Cancer Outcomes. Mol. Cancer Res. 2017, 15, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Liaw, H.H.; Lin, C.C.; Juan, H.F.; Huang, H.C. Differential microRNA regulation correlates with alternative polyadenylation pattern between breast cancer and normal cells. PLoS ONE 2013, 8, e56958. [Google Scholar] [CrossRef]
- Akman, H.B.; Oyken, M.; Tuncer, T.; Can, T.; Erson-Bensan, A.E. 3′UTR shortening and EGF signaling: Implications for breast cancer. Hum. Mol. Genet. 2015, 24, 6910–6920. [Google Scholar] [CrossRef]
- Akman, B.H.; Can, T.; Erson-Bensan, A.E. Estrogen-induced upregulation and 3′-UTR shortening of CDC6. Nucleic Acids Res. 2012, 40, 10679–10688. [Google Scholar] [CrossRef]
- Lin, Y.; Li, Z.; Ozsolak, F.; Kim, S.W.; Arango-Argoty, G.; Liu, T.T.; Tenenbaum, S.A.; Bailey, T.; Monaghan, A.P.; Milos, P.M.; et al. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 2012, 40, 8460–8471. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, Y.; Li, Y.; Li, J.; Rao, X.; Chen, C.; Xu, A. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011, 21, 741–747. [Google Scholar] [CrossRef]
- Kim, N.; Chung, W.; Eum, H.H.; Lee, H.O.; Park, W.Y. Alternative polyadenylation of single cells delineates cell types and serves as a prognostic marker in early stage breast cancer. PLoS ONE 2019, 14, e0217196. [Google Scholar] [CrossRef] [PubMed]
- Lembo, A.; Di Cunto, F.; Provero, P. Shortening of 3′UTRs correlates with poor prognosis in breast and lung cancer. PLoS ONE 2012, 7, e31129. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Singh, I.; Tisdale, S.; Abdel-Wahab, O.; Leslie, C.S.; Mayr, C. Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 2018, 561, 127–131. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Robey, R.W.; Knutsen, T.; Zhan, Z.; Ried, T.; Bates, S.E. Escape from hsa-miR-519c enables drug-resistant cells to maintain high expression of ABCG2. Mol. Cancer Ther. 2009, 8, 2959–2968. [Google Scholar] [CrossRef]
- To, K.K.; Zhan, Z.; Litman, T.; Bates, S.E. Regulation of ABCG2 expression at the 3′ untranslated region of its mRNA through modulation of transcript stability and protein translation by a putative microRNA in the S1 colon cancer cell line. Mol. Cell. Biol. 2008, 28, 5147–5161. [Google Scholar] [CrossRef]
- Yang, Q.; Fan, W.; Zheng, Z.; Lin, S.; Liu, C.; Wang, R.; Li, W.; Zuo, Y.; Sun, Y.; Hu, S.; et al. Cleavage and polyadenylation specific factor 4 promotes colon cancer progression by transcriptionally activating hTERT. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1533–1543. [Google Scholar] [CrossRef]
- Mao, Z.; Zhao, H.; Qin, Y.; Wei, J.; Sun, J.; Zhang, W.; Kang, Y. Post-Transcriptional Dysregulation of microRNA and Alternative Polyadenylation in Colorectal Cancer. Front. Genet. 2020, 11, 64. [Google Scholar] [CrossRef]
- Morris, A.R.; Bos, A.; Diosdado, B.; Rooijers, K.; Elkon, R.; Bolijn, A.S.; Carvalho, B.; Meijer, G.A.; Agami, R. Alternative cleavage and polyadenylation during colorectal cancer development. Clin. Cancer Res. 2012, 18, 5256–5266. [Google Scholar] [CrossRef]
- Andres, S.F.; Williams, K.N.; Plesset, J.B.; Headd, J.J.; Mizuno, R.; Chatterji, P.; Lento, A.A.; Klein-Szanto, A.J.; Mick, R.; Hamilton, K.E.; et al. IMP1 3′ UTR shortening enhances metastatic burden in colorectal cancer. Carcinogenesis 2019, 40, 569–579. [Google Scholar] [CrossRef]
- Yang, X.; Wu, J.; Xu, W.; Tan, S.; Chen, C.; Wang, X.; Sun, J.; Kang, Y. Genome-wide profiling reveals cancer-related genes with switched alternative polyadenylation sites in colorectal cancer. Onco Targets Ther. 2018, 11, 5349–5357. [Google Scholar] [CrossRef]
- Fischl, H.; Neve, J.; Wang, Z.; Patel, R.; Louey, A.; Tian, B.; Furger, A. hnRNPC regulates cancer-specific alternative cleavage and polyadenylation profiles. Nucleic Acids Res. 2019, 47, 7580–7591. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yuan, Q.; Yang, M. A functional germline variant in the P53 polyadenylation signal and risk of esophageal squamous cell carcinoma. Gene 2012, 506, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, Z.; Nepal, M.; Hokutan, K.; Zhang, J.; Yu, H.; Fei, P. DNA methylation at the vicinity of the proximal polyadenylation site in FANCD2 gene involves human malignancy. Cell Cycle 2018, 17, 2204–2206. [Google Scholar] [CrossRef]
- Han, B.; Shen, Y.; Zhang, P.; Jayabal, P.; Che, R.; Zhang, J.; Yu, H.; Fei, P. Overlooked FANCD2 variant encodes a promising, portent tumor suppressor, and alternative polyadenylation contributes to its expression. Oncotarget 2017, 8, 22490–22500. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.P.; Tan, S.; Kang, Y.N.; Wu, J.; Ooi, H.S.; Chen, J.; Shen, T.T.; Qi, Y.; Zhang, X.; Guo, Y.; et al. Genome-wide profiling of polyadenylation sites reveals a link between selective polyadenylation and cancer metastasis. Hum. Mol. Genet. 2015, 24, 3410–3417. [Google Scholar] [CrossRef]
- Kreth, S.; Limbeck, E.; Hinske, L.C.; Schutz, S.V.; Thon, N.; Hoefig, K.; Egensperger, R.; Kreth, F.W. In human glioblastomas transcript elongation by alternative polyadenylation and miRNA targeting is a potent mechanism of MGMT silencing. Acta Neuropathol. 2013, 125, 671–681. [Google Scholar] [CrossRef]
- Chu, Y.; Elrod, N.; Wang, C.; Li, L.; Chen, T.; Routh, A.; Xia, Z.; Li, W.; Wagner, E.J.; Ji, P. Nudt21 regulates the alternative polyadenylation of Pak1 and is predictive in the prognosis of glioblastoma patients. Oncogene 2019, 38, 4154–4168. [Google Scholar] [CrossRef]
- Shao, J.; Zhang, J.; Zhang, Z.; Jiang, H.; Lou, X.; Huang, B.; Foltz, G.; Lan, Q.; Huang, Q.; Lin, B. Alternative polyadenylation in glioblastoma multiforme and changes in predicted RNA binding protein profiles. Omics 2013, 17, 136–149. [Google Scholar] [CrossRef]
- Gruber, A.J.; Schmidt, R.; Ghosh, S.; Martin, G.; Gruber, A.R.; van Nimwegen, E.; Zavolan, M. Discovery of physiological and cancer-related regulators of 3′ UTR processing with KAPAC. Genome Biol. 2018, 19, 44. [Google Scholar] [CrossRef]
- Han, T.; Kim, J.K. Driving glioblastoma growth by alternative polyadenylation. Cell Res. 2014, 24, 1023–1024. [Google Scholar] [CrossRef][Green Version]
- Sun, M.; Ding, J.; Li, D.; Yang, G.; Cheng, Z.; Zhu, Q. NUDT21 regulates 3′-UTR length and microRNA-mediated gene silencing in hepatocellular carcinoma. Cancer Lett. 2017, 410, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, H.; Zhang, W.; Shao, Y.; Liu, Y.; Guan, H.; Wu, J.; Kang, Y.; Zhao, J.; Yu, Q.; et al. NUDT21 negatively regulates PSMB2 and CXXC5 by alternative polyadenylation and contributes to hepatocellular carcinoma suppression. Oncogene 2018, 37, 4887–4900. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ding, J.; Wang, X.; Cheng, Z.; Zhu, Q. NUDT21 regulates circRNA cyclization and ceRNA crosstalk in hepatocellular carcinoma. Oncogene 2020, 39, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Wang, Y.; Zhang, C.; Xuan, Y.; Zhao, S.; Liu, T.; Li, W.; Liao, Y.; Feng, X.; Hao, J.; et al. Cleavage and polyadenylation specific factor 4 targets NF-kappaB/cyclooxygenase-2 signaling to promote lung cancer growth and progression. Cancer Lett. 2016, 381, 1–13. [Google Scholar] [CrossRef]
- Chen, W.; Guo, W.; Li, M.; Shi, D.; Tian, Y.; Li, Z.; Wang, J.; Fu, L.; Xiao, X.; Liu, Q.Q.; et al. Upregulation of cleavage and polyadenylation specific factor 4 in lung adenocarcinoma and its critical role for cancer cell survival and proliferation. PLoS ONE 2013, 8, e82728. [Google Scholar] [CrossRef]
- Ichinose, J.; Watanabe, K.; Sano, A.; Nagase, T.; Nakajima, J.; Fukayama, M.; Yatomi, Y.; Ohishi, N.; Takai, D. Alternative polyadenylation is associated with lower expression of PABPN1 and poor prognosis in non-small cell lung cancer. Cancer Sci. 2014, 105, 1135–1141. [Google Scholar] [CrossRef]
- Huang, J.; Weng, T.; Ko, J.; Chen, N.Y.; Xiang, Y.; Volcik, K.; Han, L.; Blackburn, M.R.; Lu, X. Suppression of cleavage factor Im 25 promotes the proliferation of lung cancer cells through alternative polyadenylation. Biochem. Biophys. Res. Commun. 2018, 503, 856–862. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, X.; Lei, W.; Liang, J.; Xu, Y.; Liu, H.; Ma, S. Genome-wide profiling reveals alternative polyadenylation of mRNA in human non-small cell lung cancer. J. Transl. Med. 2019, 17, 257. [Google Scholar] [CrossRef]
- Aragaki, M.; Takahashi, K.; Akiyama, H.; Tsuchiya, E.; Kondo, S.; Nakamura, Y.; Daigo, Y. Characterization of a cleavage stimulation factor, 3′ pre-RNA, subunit 2, 64 kDa (CSTF2) as a therapeutic target for lung cancer. Clin. Cancer Res. 2011, 17, 5889–5900. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, W.; Ren, C.; Kong, X.; Yan, W.; Chen, X. A PolH Transcript with a Short 3′UTR Enhances PolH Expression and Mediates Cisplatin Resistance. Cancer Res. 2019, 79, 3714–3724. [Google Scholar] [CrossRef]
- Shulman, E.D.; Elkon, R. Cell-type-specific analysis of alternative polyadenylation using single-cell transcriptomics data. Nucleic Acids Res. 2019, 47, 10027–10039. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Toulas, C.; Fouque, F.; Tilkin-Mariame, A.F.; Selves, J.; Guimbaud, R.; Chipoulet, E.; Delmas, C.; Rey, J.M.; Pujol, P.; et al. Decreased efficiency of MSH6 mRNA polyadenylation linked to a 20-base-pair duplication in Lynch syndrome families. Cell Cycle 2012, 11, 2578–2580. [Google Scholar] [CrossRef][Green Version]
- Chen, R.W.; Bemis, L.T.; Amato, C.M.; Myint, H.; Tran, H.; Birks, D.K.; Eckhardt, S.G.; Robinson, W.A. Truncation in CCND1 mRNA alters miR-16-1 regulation in mantle cell lymphoma. Blood 2008, 112, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Wiestner, A.; Tehrani, M.; Chiorazzi, M.; Wright, G.; Gibellini, F.; Nakayama, K.; Liu, H.; Rosenwald, A.; Muller-Hermelink, H.K.; Ott, G.; et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007, 109, 4599–4606. [Google Scholar] [CrossRef] [PubMed]
- Perez-Guijarro, E.; Karras, P.; Cifdaloz, M.; Martinez-Herranz, R.; Canon, E.; Grana, O.; Horcajada-Reales, C.; Alonso-Curbelo, D.; Calvo, T.G.; Gomez-Lopez, G.; et al. Lineage-specific roles of the cytoplasmic polyadenylation factor CPEB4 in the regulation of melanoma drivers. Nat. Commun. 2016, 7, 13418. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Q.; Miao, Y.R.; Yang, J.; Yang, W.; Yu, F.; Wang, D.; Guo, A.Y.; Gong, J. SNP2APA: A database for evaluating effects of genetic variants on alternative polyadenylation in human cancers. Nucleic Acids Res. 2020, 48, D226–D232. [Google Scholar] [CrossRef]
- Gartner, W.; Mineva, I.; Daneva, T.; Baumgartner-Parzer, S.; Niederle, B.; Vierhapper, H.; Weissel, M.; Wagner, L. A newly identified RET proto-oncogene polymorphism is found in a high number of endocrine tumor patients. Hum. Genet. 2005, 117, 143–153. [Google Scholar] [CrossRef]
- Li, W.; Li, W.; Laishram, R.S.; Hoque, M.; Ji, Z.; Tian, B.; Anderson, R.A. Distinct regulation of alternative polyadenylation and gene expression by nuclear poly(A) polymerases. Nucleic Acids Res. 2017, 45, 8930–8942. [Google Scholar] [CrossRef]
- Yang, S.W.; Li, L.; Connelly, J.P.; Porter, S.N.; Kodali, K.; Gan, H.; Park, J.M.; Tacer, K.F.; Tillman, H.; Peng, J.; et al. A Cancer-Specific Ubiquitin Ligase Drives mRNA Alternative Polyadenylation by Ubiquitinating the mRNA 3′ End Processing Complex. Mol. Cell 2020, 77, 1206–1221. [Google Scholar] [CrossRef]
- Zhang, S.; Han, J.; Zhong, D.; Liu, R.; Zheng, J. Genome-wide identification and predictive modeling of lincRNAs polyadenylation in cancer genome. Comput. Biol. Chem. 2014, 52, 1–8. [Google Scholar] [CrossRef]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Warren, R.L.; Gibb, E.A.; MacMillan, D.; Wong, J.; Chiu, R.; Hammond, S.A.; Yang, C.; Nip, K.M.; Ennis, C.A.; et al. Recurrent tumor-specific regulation of alternative polyadenylation of cancer-related genes. BMC Genom. 2018, 19, 536. [Google Scholar] [CrossRef] [PubMed]
- Thivierge, C.; Tseng, H.W.; Mayya, V.K.; Lussier, C.; Gravel, S.P.; Duchaine, T.F. Alternative polyadenylation confers Pten mRNAs stability and resistance to microRNAs. Nucleic Acids Res. 2018, 46, 10340–10352. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, Y.; Lou, Y.; Yang, Y.; Cai, C.; Zhang, Z.; Mills, T.; Chen, N.Y.; Kim, Y.; Muge Ozguc, F.; et al. Comprehensive Characterization of Alternative Polyadenylation in Human Cancer. J. Natl. Cancer Inst. 2018, 110, 379–389. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, X.S.; He, J.; Ma, T.; Lei, W.; Shen, Z.Y. A novel TP53 variant (rs78378222 A > C) in the polyadenylation signal is associated with increased cancer susceptibility: Evidence from a meta-analysis. Oncotarget 2016, 7, 32854–32865. [Google Scholar] [CrossRef]
- Stacey, S.N.; Sulem, P.; Jonasdottir, A.; Masson, G.; Gudmundsson, J.; Gudbjartsson, D.F.; Magnusson, O.T.; Gudjonsson, S.A.; Sigurgeirsson, B.; Thorisdottir, K.; et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 2011, 43, 1098–1103. [Google Scholar] [CrossRef]
- Xu, Y.F.; Li, Y.Q.; Liu, N.; He, Q.M.; Tang, X.R.; Wen, X.; Yang, X.J.; Sun, Y.; Ma, J.; Tang, L.L. Differential genome-wide profiling of alternative polyadenylation sites in nasopharyngeal carcinoma by high-throughput sequencing. J. Biomed. Sci. 2018, 25, 74. [Google Scholar] [CrossRef]
- He, X.; Yang, J.; Zhang, Q.; Cui, H.; Zhang, Y. Shortening of the 3′ untranslated region: An important mechanism leading to overexpression of HMGA2 in serous ovarian cancer. Chin. Med. J. 2014, 127, 494–499. [Google Scholar]
- He, X.J.; Zhang, Q.; Ma, L.P.; Li, N.; Chang, X.H.; Zhang, Y.J. Aberrant Alternative Polyadenylation is Responsible for Survivin Up-regulation in Ovarian Cancer. Chin. Med. J. 2016, 129, 1140–1146. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Y.; Liu, D.; Yang, L. Targeting cleavage and polyadenylation specific factor 1 via shRNA inhibits cell proliferation in human ovarian cancer. J. Biosci. 2017, 42, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, Z.; Yong, L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol. Oncol. 2018, 148, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Venkat, S.; Tisdale, A.A.; Schwarz, J.R.; Alahmari, A.A.; Maurer, H.C.; Olive, K.P.; Eng, K.H.; Feigin, M.E. Alternative polyadenylation drives oncogenic gene expression in pancreatic ductal adenocarcinoma. Genome Res. 2020, 30, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Chang, J.W.; Zhang, W.; Yeh, H.S.; de Jong, E.P.; Jun, S.; Kim, K.H.; Bae, S.S.; Beckman, K.; Hwang, T.H.; Kim, K.S.; et al. mRNA 3′-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015, 6, 7218. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef]
- Rehfeld, A.; Plass, M.; Dossing, K.; Knigge, U.; Kjaer, A.; Krogh, A.; Friis-Hansen, L. Alternative polyadenylation of tumor suppressor genes in small intestinal neuroendocrine tumors. Front. Endocrinol. 2014, 5, 46. [Google Scholar] [CrossRef]
- Weber, M.; Hagedorn, C.H.; Harrison, D.G.; Searles, C.D. Laminar shear stress and 3′ polyadenylation of eNOS mRNA. Circ. Res. 2005, 96, 1161–1168. [Google Scholar] [CrossRef]
- Sudheesh, A.P.; Mohan, N.; Francis, N.; Laishram, R.S.; Anderson, R.A. Star-PAP controlled alternative polyadenylation coupled poly(A) tail length regulates protein expression in hypertrophic heart. Nucleic Acids Res. 2019, 47, 10771–10787. [Google Scholar] [CrossRef]
- Mohan, N.; Kumar, V.; Kandala, D.T.; Kartha, C.C.; Laishram, R.S. A Splicing-Independent Function of RBM10 Controls Specific 3′ UTR Processing to Regulate Cardiac Hypertrophy. Cell Rep. 2018, 24, 3539–3553. [Google Scholar] [CrossRef]
- Park, J.Y.; Li, W.; Zheng, D.; Zhai, P.; Zhao, Y.; Matsuda, T.; Vatner, S.F.; Sadoshima, J.; Tian, B. Comparative analysis of mRNA isoform expression in cardiac hypertrophy and development reveals multiple post-transcriptional regulatory modules. PLoS ONE 2011, 6, e22391. [Google Scholar] [CrossRef] [PubMed]
- Soetanto, R.; Hynes, C.J.; Patel, H.R.; Humphreys, D.T.; Evers, M.; Duan, G.; Parker, B.J.; Archer, S.K.; Clancy, J.L.; Graham, R.M.; et al. Role of miRNAs and alternative mRNA 3′-end cleavage and polyadenylation of their mRNA targets in cardiomyocyte hypertrophy. Biochim. Biophys. Acta 2016, 1859, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Chorghade, S.; Seimetz, J.; Emmons, R.; Yang, J.; Bresson, S.M.; Lisio, M.; Parise, G.; Conrad, N.K.; Kalsotra, A. Poly(A) tail length regulates PABPC1 expression to tune translation in the heart. Elife 2017, 6. [Google Scholar] [CrossRef]
- Creemers, E.E.; Bawazeer, A.; Ugalde, A.P.; van Deutekom, H.W.; van der Made, I.; de Groot, N.E.; Adriaens, M.E.; Cook, S.A.; Bezzina, C.R.; Hubner, N.; et al. Genome-Wide Polyadenylation Maps Reveal Dynamic mRNA 3′-End Formation in the Failing Human Heart. Circ. Res. 2016, 118, 433–438. [Google Scholar] [CrossRef]
- Yang, Z.; Kaye, D.M. Mechanistic insights into the link between a polymorphism of the 3′UTR of the SLC7A1 gene and hypertension. Hum. Mutat. 2009, 30, 328–333. [Google Scholar] [CrossRef]
- Yang, Z.; Venardos, K.; Jones, E.; Morris, B.J.; Chin-Dusting, J.; Kaye, D.M. Identification of a novel polymorphism in the 3′UTR of the L-arginine transporter gene SLC7A1: Contribution to hypertension and endothelial dysfunction. Circulation 2007, 115, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.K.; Bhalla, K.; Pan, Z.H.; O’Connell, J.R.; Weder, A.B.; Chakravarti, A.; Tian, B.; Chang, Y.P. A polymorphic 3′UTR element in ATP1B1 regulates alternative polyadenylation and is associated with blood pressure. PLoS ONE 2013, 8, e76290. [Google Scholar] [CrossRef]
- Tranter, M.; Helsley, R.N.; Paulding, W.R.; McGuinness, M.; Brokamp, C.; Haar, L.; Liu, Y.; Ren, X.; Jones, W.K. Coordinated post-transcriptional regulation of Hsp70.3 gene expression by microRNA and alternative polyadenylation. J. Biol. Chem. 2011, 286, 29828–29837. [Google Scholar] [CrossRef] [PubMed]
- Uitte de Willige, S.; Rietveld, I.M.; De Visser, M.C.; Vos, H.L.; Bertina, R.M. Polymorphism 10034C>T is located in a region regulating polyadenylation of FGG transcripts and influences the fibrinogen gamma’/gammaA mRNA ratio. J. Thromb. Haemost. 2007, 5, 1243–1249. [Google Scholar] [CrossRef]
- Uitte de Willige, S.; de Visser, M.C.; Houwing-Duistermaat, J.J.; Rosendaal, F.R.; Vos, H.L.; Bertina, R.M. Genetic variation in the fibrinogen gamma gene increases the risk for deep venous thrombosis by reducing plasma fibrinogen gamma’ levels. Blood 2005, 106, 4176–4183. [Google Scholar] [CrossRef]
- Gehring, N.H.; Frede, U.; Neu-Yilik, G.; Hundsdoerfer, P.; Vetter, B.; Hentze, M.W.; Kulozik, A.E. Increased efficiency of mRNA 3′ end formation: A new genetic mechanism contributing to hereditary thrombophilia. Nat. Genet. 2001, 28, 389–392. [Google Scholar] [CrossRef]
- Ceelie, H.; Spaargaren-van Riel, C.C.; Bertina, R.M.; Vos, H.L. G20210A is a functional mutation in the prothrombin gene; effect on protein levels and 3′-end formation. J. Thromb. Haemost. 2004, 2, 119–127. [Google Scholar] [CrossRef]
- Danckwardt, S.; Hartmann, K.; Katz, B.; Hentze, M.W.; Levy, Y.; Eichele, R.; Deutsch, V.; Kulozik, A.E.; Ben-Tal, O. The prothrombin 20209 C-->T mutation in Jewish-Moroccan Caucasians: Molecular analysis of gain-of-function of 3′ end processing. J. Thromb. Haemost. 2006, 4, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Danckwardt, S.; Gehring, N.H.; Neu-Yilik, G.; Hundsdoerfer, P.; Pforsich, M.; Frede, U.; Hentze, M.W.; Kulozik, A.E. The prothrombin 3′end formation signal reveals a unique architecture that is sensitive to thrombophilic gain-of-function mutations. Blood 2004, 104, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Williamson, K.A.; Chou, C.M.; Sapp, J.C.; Ansari, M.; Chapman, H.M.; Cooper, D.N.; Dabir, T.; Dudley, J.N.; Holt, R.J.; et al. NAA10 polyadenylation signal variants cause syndromic microphthalmia. J. Med. Genet. 2019, 56, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Dresser, D.W.; Hacker, A.; Lovell-Badge, R.; Guerrier, D. The genes for a spliceosome protein (SAP62) and the anti-Mullerian hormone (AMH) are contiguous. Hum. Mol. Genet. 1995, 4, 1613–1618. [Google Scholar] [CrossRef]
- Fahiminiya, S.; Al-Jallad, H.; Majewski, J.; Palomo, T.; Moffatt, P.; Roschger, P.; Klaushofer, K.; Glorieux, F.H.; Rauch, F. A polyadenylation site variant causes transcript-specific BMP1 deficiency and frequent fractures in children. Hum. Mol. Genet. 2015, 24, 516–524. [Google Scholar] [CrossRef]
- Ichikawa, S.; Traxler, E.A.; Estwick, S.A.; Curry, L.R.; Johnson, M.L.; Sorenson, A.H.; Imel, E.A.; Econs, M.J. Mutational survey of the PHEX gene in patients with X-linked hypophosphatemic rickets. Bone 2008, 43, 663–666. [Google Scholar] [CrossRef][Green Version]
- Garin, I.; Edghill, E.L.; Akerman, I.; Rubio-Cabezas, O.; Rica, I.; Locke, J.M.; Maestro, M.A.; Alshaikh, A.; Bundak, R.; del Castillo, G.; et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3105–3110. [Google Scholar] [CrossRef]
- Locke, J.M.; Da Silva Xavier, G.; Rutter, G.A.; Harries, L.W. An alternative polyadenylation signal in TCF7L2 generates isoforms that inhibit T cell factor/lymphoid-enhancer factor (TCF/LEF)-dependent target genes. Diabetologia 2011, 54, 3078–3082. [Google Scholar] [CrossRef]
- Nurden, A.T.; Pillois, X.; Fiore, M.; Alessi, M.C.; Bonduel, M.; Dreyfus, M.; Goudemand, J.; Gruel, Y.; Benabdallah-Guerida, S.; Latger-Cannard, V.; et al. Expanding the Mutation Spectrum Affecting alphaIIbbeta3 Integrin in Glanzmann Thrombasthenia: Screening of the ITGA2B and ITGB3 Genes in a Large International Cohort. Hum. Mutat. 2015, 36, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Higgs, D.R.; Goodbourn, S.E.; Lamb, J.; Clegg, J.B.; Weatherall, D.J.; Proudfoot, N.J. Alpha-thalassaemia caused by a polyadenylation signal mutation. Nature 1983, 306, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Cheng, T.C.; Antonarakis, S.E.; Kazazian, H.H., Jr. Thalassemia due to a mutation in the cleavage-polyadenylation signal of the human beta-globin gene. EMBO J. 1985, 4, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Rund, D.; Dowling, C.; Najjar, K.; Rachmilewitz, E.A.; Kazazian, H.H., Jr.; Oppenheim, A. Two mutations in the beta-globin polyadenylylation signal reveal extended transcripts and new RNA polyadenylylation sites. Proc. Natl. Acad. Sci. USA 1992, 89, 4324–4328. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Bennett, C.L.; Brunkow, M.E.; Ramsdell, F.; O’Briant, K.C.; Zhu, Q.; Fuleihan, R.L.; Shigeoka, A.O.; Ochs, H.D.; Chance, P.F. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA-->AAUGAA) leads to the IPEX syndrome. Immunogenetics 2001, 53, 435–439. [Google Scholar] [CrossRef]
- Tian, P.; Li, J.; Liu, X.; Li, Y.; Chen, M.; Ma, Y.; Zheng, Y.Q.; Fu, Y.; Zou, H. Tandem alternative polyadenylation events of genes in non-eosinophilic nasal polyp tissue identified by high-throughput sequencing analysis. Int. J. Mol. Med. 2014, 33, 1423–1430. [Google Scholar] [CrossRef]
- Tian, P.; Sun, Y.; Li, Y.; Liu, X.; Wan, L.; Li, J.; Ma, Y.; Xu, A.; Fu, Y.; Zou, H. A global analysis of tandem 3′UTRs in eosinophilic chronic rhinosinusitis with nasal polyps. PLoS ONE 2012, 7, e48997. [Google Scholar] [CrossRef]
- Hsu, A.P.; Fleisher, T.A.; Niemela, J.E. Mutation analysis in primary immunodeficiency diseases: Case studies. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 517–524. [Google Scholar] [CrossRef]
- Yoon, O.K.; Hsu, T.Y.; Im, J.H.; Brem, R.B. Genetics and regulatory impact of alternative polyadenylation in human B-lymphoblastoid cells. PLoS Genet. 2012, 8, e1002882. [Google Scholar] [CrossRef]
- Graham, R.R.; Kyogoku, C.; Sigurdsson, S.; Vlasova, I.A.; Davies, L.R.; Baechler, E.C.; Plenge, R.M.; Koeuth, T.; Ortmann, W.A.; Hom, G.; et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc. Natl. Acad. Sci. USA 2007, 104, 6758–6763. [Google Scholar] [CrossRef] [PubMed]
- Cunninghame Graham, D.S.; Manku, H.; Wagner, S.; Reid, J.; Timms, K.; Gutin, A.; Lanchbury, J.S.; Vyse, T.J. Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Hum. Mol. Genet. 2007, 16, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Hellquist, A.; Zucchelli, M.; Kivinen, K.; Saarialho-Kere, U.; Koskenmies, S.; Widen, E.; Julkunen, H.; Wong, A.; Karjalainen-Lindsberg, M.L.; Skoog, T.; et al. The human GIMAP5 gene has a common polyadenylation polymorphism increasing risk to systemic lupus erythematosus. J. Med. Genet. 2007, 44, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Andreu, N.; Garcia-Rodriguez, M.; Volpini, V.; Frecha, C.; Molina, I.J.; Fontan, G.; Fillat, C. A novel Wiskott-Aldrich syndrome protein (WASP) complex mutation identified in a WAS patient results in an aberrant product at the C-terminus from two transcripts with unusual polyA signals. J. Hum. Genet. 2006, 51, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Weng, T.; Ko, J.; Masamha, C.P.; Xia, Z.; Xiang, Y.; Chen, N.Y.; Molina, J.G.; Collum, S.; Mertens, T.C.; Luo, F.; et al. Cleavage factor 25 deregulation contributes to pulmonary fibrosis through alternative polyadenylation. J. Clin. Invest. 2019, 129, 1984–1999. [Google Scholar] [CrossRef]
- Mueller, A.A.; van Velthoven, C.T.; Fukumoto, K.D.; Cheung, T.H.; Rando, T.A. Intronic polyadenylation of PDGFRalpha in resident stem cells attenuates muscle fibrosis. Nature 2016, 540, 276–279. [Google Scholar] [CrossRef]
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camano, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef]
- Raz, V.; Dickson, G.; t Hoen, P.A.C. Dysfunctional transcripts are formed by alternative polyadenylation in OPMD. Oncotarget 2017, 8, 73516–73528. [Google Scholar] [CrossRef]
- Abbassi-Daloii, T.; Yousefi, S.; de Klerk, E.; Grossouw, L.; Riaz, M.; t Hoen, P.A.C.; Raz, V. An alanine expanded PABPN1 causes increased utilization of intronic polyadenylation sites. NPJ Aging Mech. Dis. 2017, 3, 6. [Google Scholar] [CrossRef]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef]
- Jenal, M.; Elkon, R.; Loayza-Puch, F.; van Haaften, G.; Kuhn, U.; Menzies, F.M.; Oude Vrielink, J.A.; Bos, A.J.; Drost, J.; Rooijers, K.; et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 2012, 149, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Dickson, J.R.; Kruse, C.; Montagna, D.R.; Finsen, B.; Wolfe, M.S. Alternative polyadenylation and miR-34 family members regulate tau expression. J. Neurochem. 2013, 127, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Barbash, S.; Garfinkel, B.P.; Maoz, R.; Simchovitz, A.; Nadorp, B.; Guffanti, A.; Bennett, E.R.; Nadeau, C.; Turk, A.; Paul, L.; et al. Alzheimer’s brains show inter-related changes in RNA and lipid metabolism. Neurobiol Dis 2017, 106, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Bazan, N.G. Cyclooxygenase 2 RNA message abundance, stability, and hypervariability in sporadic Alzheimer neocortex. J. Neurosci. Res. 1997, 50, 937–945. [Google Scholar] [CrossRef]
- Patel, R.; Brophy, C.; Hickling, M.; Neve, J.; Furger, A. Alternative cleavage and polyadenylation of genes associated with protein turnover and mitochondrial function are deregulated in Parkinson’s, Alzheimer’s and ALS disease. BMC Med. Genom. 2019, 12, 60. [Google Scholar] [CrossRef]
- Bai, B.; Hales, C.M.; Chen, P.C.; Gozal, Y.; Dammer, E.B.; Fritz, J.J.; Wang, X.; Xia, Q.; Duong, D.M.; Street, C.; et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16562–16567. [Google Scholar] [CrossRef]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695. [Google Scholar] [CrossRef]
- Masuda, A.; Takeda, J.; Okuno, T.; Okamoto, T.; Ohkawara, B.; Ito, M.; Ishigaki, S.; Sobue, G.; Ohno, K. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev. 2015, 29, 1045–1057. [Google Scholar] [CrossRef]
- Koyama, A.; Sugai, A.; Kato, T.; Ishihara, T.; Shiga, A.; Toyoshima, Y.; Koyama, M.; Konno, T.; Hirokawa, S.; Yokoseki, A.; et al. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016, 44, 5820–5836. [Google Scholar] [CrossRef] [PubMed]
- Flomen, R.; Makoff, A. Increased RNA editing in EAAT2 pre-mRNA from amyotrophic lateral sclerosis patients: Involvement of a cryptic polyadenylation site. Neurosci. Lett. 2011, 497, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Melamed, Z.; Lopez-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; McKenna, M.C.; Rollins, D.A.; Song, M.; Nuriel, T.; Gross, S.S.; Xu, G.; Glatt, C.E. Anxiety-associated alternative polyadenylation of the serotonin transporter mRNA confers translational regulation by hnRNPK. Proc. Natl. Acad. Sci. USA 2013, 110, 11624–11629. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, S.; Subaran, R.; Weissman, M.M.; Hershkowitz, D.; McKenna, M.C.; Talati, A.; Fyer, A.J.; Wickramaratne, P.; Adams, P.B.; Hodge, S.E.; et al. Association of a polyadenylation polymorphism in the serotonin transporter and panic disorder. Biol. Psychiatry 2010, 67, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Hartley, C.A.; McKenna, M.C.; Salman, R.; Holmes, A.; Casey, B.J.; Phelps, E.A.; Glatt, C.E. Serotonin transporter polyadenylation polymorphism modulates the retention of fear extinction memory. Proc. Natl. Acad. Sci. USA 2012, 109, 5493–5498. [Google Scholar] [CrossRef]
- Yasuda, M.; Shabbeer, J.; Osawa, M.; Desnick, R.J. Fabry disease: Novel alpha-galactosidase A 3′-terminal mutations result in multiple transcripts due to aberrant 3′-end formation. Am. J. Hum. Genet. 2003, 73, 162–173. [Google Scholar] [CrossRef]
- Tassone, F.; De Rubeis, S.; Carosi, C.; La Fata, G.; Serpa, G.; Raske, C.; Willemsen, R.; Hagerman, P.J.; Bagni, C. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011, 39, 6172–6185. [Google Scholar] [CrossRef]
- McGinty, R.J.; Puleo, F.; Aksenova, A.Y.; Hisey, J.A.; Shishkin, A.A.; Pearson, E.L.; Wang, E.T.; Housman, D.E.; Moore, C.; Mirkin, S.M. A Defective mRNA Cleavage and Polyadenylation Complex Facilitates Expansions of Transcribed (GAA)n Repeats Associated with Friedreich’s Ataxia. Cell Rep. 2017, 20, 2490–2500. [Google Scholar] [CrossRef]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.; Roos, R.A.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef]
- Gieselmann, V.; Polten, A.; Kreysing, J.; von Figura, K. Arylsulfatase A pseudodeficiency: Loss of a polyadenylylation signal and N-glycosylation site. Proc. Natl. Acad. Sci. USA 1989, 86, 9436–9440. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.S.; Carey, W.F.; Morris, C.P. Importance of the glycosylation and polyadenylation variants in metachromatic leukodystrophy pseudodeficiency phenotype. Hum. Mol. Genet. 1998, 7, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Gennarino, V.A.; Alcott, C.E.; Chen, C.A.; Chaudhury, A.; Gillentine, M.A.; Rosenfeld, J.A.; Parikh, S.; Wheless, J.W.; Roeder, E.R.; Horovitz, D.D.; et al. NUDT21-spanning CNVs lead to neuropsychiatric disease and altered MeCP2 abundance via alternative polyadenylation. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, H.; Qiang, L.; Yamashita, T.; Rhee, D.; Zolin, A.; Vanti, W.; Abeliovich, A. Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 2012, 3, 1084. [Google Scholar] [CrossRef] [PubMed]
- Enoch, M.A.; Hodgkinson, C.A.; Gorodetsky, E.; Goldman, D.; Roy, A. Independent effects of 5′ and 3′ functional variants in the serotonin transporter gene on suicidal behavior in the context of childhood trauma. J. Psychiatr. Res. 2013, 47, 900–907. [Google Scholar] [CrossRef]
- Zheng, D.; Wang, R.; Ding, Q.; Wang, T.; Xie, B.; Wei, L.; Zhong, Z.; Tian, B. Cellular stress alters 3′UTR landscape through alternative polyadenylation and isoform-specific degradation. Nat. Commun. 2018, 9, 2268. [Google Scholar] [CrossRef]
- Falkenberg, K.D.; Braverman, N.E.; Moser, A.B.; Steinberg, S.J.; Klouwer, F.C.C.; Schluter, A.; Ruiz, M.; Pujol, A.; Engvall, M.; Naess, K.; et al. Allelic Expression Imbalance Promoting a Mutant PEX6 Allele Causes Zellweger Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 965–976. [Google Scholar] [CrossRef]
- Nunes, N.M.; Li, W.; Tian, B.; Furger, A. A functional human Poly(A) site requires only a potent DSE and an A-rich upstream sequence. EMBO J. 2010, 29, 1523–1536. [Google Scholar] [CrossRef]
- Pressley, L.; Higgs, D.R.; Clegg, J.B.; Perrine, R.P.; Pembrey, M.E.; Weatherall, D.J. A new genetic basis for hemoglobin-H disease. N Engl. J. Med. 1980, 303, 1383–1388. [Google Scholar] [CrossRef]
- Thein, S.L.; Wallace, R.B.; Pressley, L.; Clegg, J.B.; Weatherall, D.J.; Higgs, D.R. The polyadenylation site mutation in the alpha-globin gene cluster. Blood 1988, 71, 313–319. [Google Scholar] [CrossRef]
- Yuregir, G.T.; Aksoy, K.; Curuk, M.A.; Dikmen, N.; Fei, Y.J.; Baysal, E.; Huisman, T.H. Hb H disease in a Turkish family resulting from the interaction of a deletional alpha-thalassaemia-1 and a newly discovered poly A mutation. Br. J. Haematol. 1992, 80, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.W.; Higgs, D.R.; Murphy, P.; Villegas, A.; de Miguel, A. A mutation in the polyadenylation signal of the alpha 2 globin gene (AATAAA-->AATA--) as a cause of alpha thalassaemia in Asian indians. Br. J. Haematol. 1994, 88, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Harteveld, C.L.; Oosterhuis, W.P.; Schoenmakers, C.H.; Ananta, H.; Kos, S.; Bakker Verweij, M.; van Delft, P.; Arkesteijn, S.G.; Phylipsen, M.; Giordano, P.C. alpha-thalassaemia masked by beta gene defects and a new polyadenylation site mutation on the alpha2-globin gene. Eur. J. Haematol. 2010, 84, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, L.; Efremov, G.D.; Petkov, G.; Kattamis, C.; George, E.; Yang, K.G.; Stoming, T.A.; Huisman, T.H. Two novel polyadenylation mutations leading to beta(+)-thalassemia. Br. J. Haematol. 1990, 75, 122–126. [Google Scholar] [CrossRef]
- Rund, D.; Cohen, T.; Filon, D.; Dowling, C.E.; Warren, T.C.; Barak, I.; Rachmilewitz, E.; Kazazian, H.H., Jr.; Oppenheim, A. Evolution of a genetic disease in an ethnic isolate: Beta-thalassemia in the Jews of Kurdistan. Proc. Natl. Acad. Sci. USA 1991, 88, 310–314. [Google Scholar] [CrossRef]
- El-Kalla, S.; Mathews, A.R. A significant beta-thalassemia heterogeneity in the United Arab Emirates. Hemoglobin 1997, 21, 237–247. [Google Scholar] [CrossRef]
- Waye, J.S.; Eng, B.; Patterson, M.; Reis, M.D.; Macdonald, D.; Chui, D.H. Novel beta-thalassemia mutation in a beta-thalassemia intermedia patient. Hemoglobin 2001, 25, 103–105. [Google Scholar] [CrossRef]
- Ma, S.K.; Lee, A.C.; Chan, A.Y.; Chan, L.C. A novel AATAAA-->CATAAA mutation at the polyadenylation site of the beta-globin gene. Br. J. Haematol. 2001, 115, 230–231. [Google Scholar]
- Jacquette, A.; Le Roux, G.; Lacombe, C.; Goossens, M.; Pissard, S. Compound heterozygosity for two new mutations in the beta-globin gene [codon 9 (+TA) and polyadenylation site (AATAAA-->AAAAAA)] leads to thalassemia intermedia in a Tunisian patient. Hemoglobin 2004, 28, 243–248. [Google Scholar] [CrossRef]
- Giordano, P.C.; Bouva, M.J.; Van Delft, P.; Akkerman, N.; Kappers-Klunne, M.C.; Harteveld, C.L. A new polyadenylation site mutation associated with a mild beta-thalassemia phenotype. Haematologica 2005, 90, 551–552. [Google Scholar]
- Bilgen, T.; Clark, O.A.; Ozturk, Z.; Akif Yesilipek, M.; Keser, I. Two novel mutations in the 3′ untranslated region of the beta-globin gene that are associated with the mild phenotype of beta thalassemia. Int. J. Lab. Hematol. 2013, 35, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Al-Allawi, N.A.; Jalal, S.D.; Mohammad, A.M.; Omer, S.Q.; Markous, R.S. beta -thalassemia intermedia in Northern Iraq: A single center experience. Biomed. Res. Int. 2014, 2014, 262853. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Transcriptional interference and termination between duplicated alpha-globin gene constructs suggests a novel mechanism for gene regulation. Nature 1986, 322, 562–565. [Google Scholar] [CrossRef]
- Whitelaw, E.; Proudfoot, N. Alpha-thalassaemia caused by a poly(A) site mutation reveals that transcriptional termination is linked to 3′ end processing in the human alpha 2 globin gene. EMBO J. 1986, 5, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Battersby, S.; Ogilvie, A.D.; Blackwood, D.H.; Shen, S.; Muqit, M.M.; Muir, W.J.; Teague, P.; Goodwin, G.M.; Harmar, A.J. Presence of multiple functional polyadenylation signals and a single nucleotide polymorphism in the 3′ untranslated region of the human serotonin transporter gene. J. Neurochem. 1999, 72, 1384–1388. [Google Scholar] [CrossRef] [PubMed]
- Heils, A.; Teufel, A.; Petri, S.; Seemann, M.; Bengel, D.; Balling, U.; Riederer, P.; Lesch, K.P. Functional promoter and polyadenylation site mapping of the human serotonin (5-HT) transporter gene. J. Neural Transm. Gen. Sect. 1995, 102, 247–254. [Google Scholar] [CrossRef]
- Bosch, F.; Jares, P.; Campo, E.; Lopez-Guillermo, A.; Piris, M.A.; Villamor, N.; Tassies, D.; Jaffe, E.S.; Montserrat, E.; Rozman, C.; et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: A highly specific marker of mantle cell lymphoma. Blood 1994, 84, 2726–2732. [Google Scholar] [CrossRef]
- Shah, M.; Datson, N.; Srinidhi, L.; Stanton, V.P.; MacDonald, M.; Allard, M.; Youngman, S.; Frischauf, A.M.; Mott, R.; Draths, K.M.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Van Overveld, P.G.; Lemmers, R.J.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.J.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef]
- Zeng, W.; de Greef, J.C.; Chen, Y.Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559. [Google Scholar] [CrossRef]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Matteotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Bristol, L.A.; Rothstein, J.D. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann. Neurol. 1996, 39, 676–679. [Google Scholar] [CrossRef]
- Li, J.B.; Levanon, E.Y.; Yoon, J.K.; Aach, J.; Xie, B.; Leproust, E.; Zhang, K.; Gao, Y.; Church, G.M. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 2009, 324, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef]
- Zhao, J.; Hyman, L.; Moore, C. Formation of mRNA 3′ ends in eukaryotes: Mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev. 1999, 63, 405–445. [Google Scholar] [CrossRef]
- Chen, F.; MacDonald, C.C.; Wilusz, J. Cleavage site determinants in the mammalian polyadenylation signal. Nucleic Acids Res. 1995, 23, 2614–2620. [Google Scholar] [CrossRef]
- Poort, S.R.; Rosendaal, F.R.; Reitsma, P.H.; Bertina, R.M. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996, 88, 3698–3703. [Google Scholar] [CrossRef]
- Balim, Z.; Kosova, B.; Falzon, K.; Bezzina Wettinger, S.; Colak, Y. Budd-Chiari syndrome in a patient heterozygous for the point mutation C20221T of the prothrombin gene. J. Thromb. Haemost. 2003, 1, 852–853. [Google Scholar] [CrossRef]
- Arya, R. Detection of prothrombin gene polymorphism at position 20209 (PT20209C/T): Pilot study in a black population in the United Kingdom. Thromb. Haemost. 2005, 93, 179–180. [Google Scholar]
- Danckwardt, S.; Kaufmann, I.; Gentzel, M.; Foerstner, K.U.; Gantzert, A.S.; Gehring, N.H.; Neu-Yilik, G.; Bork, P.; Keller, W.; Wilm, M.; et al. Splicing factors stimulate polyadenylation via USEs at non-canonical 3′ end formation signals. EMBO J. 2007, 26, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.W.; Davie, E.W. gamma and gamma’ chains of human fibrinogen are produced by alternative mRNA processing. Biochemistry 1984, 23, 4232–4236. [Google Scholar] [CrossRef] [PubMed]
- Fornace, A.J., Jr.; Cummings, D.E.; Comeau, C.M.; Kant, J.A.; Crabtree, G.R. Structure of the human gamma-fibrinogen gene. Alternate mRNA splicing near the 3′ end of the gene produces gamma A and gamma B forms of gamma-fibrinogen. J. Biol. Chem. 1984, 259, 12826–12830. [Google Scholar]
- Lane, L.K.; Shull, M.M.; Whitmer, K.R.; Lingrel, J.B. Characterization of two genes for the human Na,K-ATPase beta subunit. Genomics 1989, 5, 445–453. [Google Scholar] [CrossRef]
- Young, R.M.; Shull, G.E.; Lingrel, J.B. Multiple mRNAs from rat kidney and brain encode a single Na+,K+-ATPase beta subunit protein. J. Biol. Chem. 1987, 262, 4905–4910. [Google Scholar] [PubMed]
- Montes, C.; Amador, M.; Cuevas, D.; Cordoba, F. Subunit structure of karatasin, the proteinase isolated from Bromelia plumieri (karatas). Agric. Biol. Chem. 1990, 54, 17–24. [Google Scholar] [CrossRef]
- Shao, Y.; Pressley, T.A.; Ismail-Beigi, F. Na,K-ATPase mRNA beta 1 expression in rat myocardium--effect of thyroid status. Eur. J. Biochem. 1999, 260, 1–8. [Google Scholar] [CrossRef]
- Kawakami, K.; Nojima, H.; Ohta, T.; Nagano, K. Molecular cloning and sequence analysis of human Na,K-ATPase beta-subunit. Nucleic Acids Res. 1986, 14, 2833–2844. [Google Scholar] [CrossRef]
- Chang, Y.P.; Liu, X.; Kim, J.D.; Ikeda, M.A.; Layton, M.R.; Weder, A.B.; Cooper, R.S.; Kardia, S.L.; Rao, D.C.; Hunt, S.C.; et al. Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am. J. Hum. Genet. 2007, 80, 253–264. [Google Scholar] [CrossRef]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of alpha-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef]
- Sotiriou, S.; Gibney, G.; Baxevanis, A.D.; Nussbaum, R.L. A single nucleotide polymorphism in the 3′UTR of the SNCA gene encoding alpha-synuclein is a new potential susceptibility locus for Parkinson disease. Neurosci. Lett. 2009, 461, 196–201. [Google Scholar] [CrossRef]
- Baglietto, L.; Lindor, N.M.; Dowty, J.G.; White, D.M.; Wagner, A.; Gomez Garcia, E.B.; Vriends, A.H.; Cartwright, N.R.; Barnetson, R.A.; Farrington, S.M.; et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J. Natl. Cancer Inst. 2010, 102, 193–201. [Google Scholar] [CrossRef]
- Myers, S.M.; Eng, C.; Ponder, B.A.; Mulligan, L.M. Characterization of RET proto-oncogene 3′ splicing variants and polyadenylation sites: A novel C-terminus for RET. Oncogene 1995, 11, 2039–2045. [Google Scholar]
- Knoepfler, P.S.; Lu, Q.; Kamps, M.P. Pbx-1 Hox heterodimers bind DNA on inseparable half-sites that permit intrinsic DNA binding specificity of the Hox partner at nucleotides 3′ to a TAAT motif. Nucleic Acids Res. 1996, 24, 2288–2294. [Google Scholar] [CrossRef][Green Version]
- Toniolo, D.; Rizzolio, F. X chromosome and ovarian failure. Semin. Reprod. Med. 2007, 25, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Allingham-Hawkins, D.J.; Babul-Hirji, R.; Chitayat, D.; Holden, J.J.; Yang, K.T.; Lee, C.; Hudson, R.; Gorwill, H.; Nolin, S.L.; Glicksman, A.; et al. Fragile X premutation is a significant risk factor for premature ovarian failure: The International Collaborative POF in Fragile X study—preliminary data. Am. J. Med. Genet. 1999, 83, 322–325. [Google Scholar] [CrossRef]
- Hagerman, P.J.; Hagerman, R.J. The fragile-X premutation: A maturing perspective. Am. J. Hum. Genet. 2004, 74, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.A.; Coffey, S.M.; Rivera, S.M.; Hessl, D.; Gane, L.W.; Tassone, F.; Greco, C.; Finucane, B.; Nelson, L.; Berry-Kravis, E.; et al. A review of fragile X premutation disorders: Expanding the psychiatric perspective. J. Clin. Psychiatry 2009, 70, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Hagerman, P.J.; Leehey, M.A. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: Two faces of FMR1. Lancet Neurol. 2007, 6, 45–55. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef]
- Puente, X.S.; Bea, S.; Valdes-Mas, R.; Villamor, N.; Gutierrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Flavell, S.W.; Kim, T.K.; Gray, J.M.; Harmin, D.A.; Hemberg, M.; Hong, E.J.; Markenscoff-Papadimitriou, E.; Bear, D.M.; Greenberg, M.E. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 2008, 60, 1022–1038. [Google Scholar] [CrossRef]
- Ji, Z.; Tian, B. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE 2009, 4, e8419. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 7028–7033. [Google Scholar] [CrossRef] [PubMed]
- Smibert, P.; Miura, P.; Westholm, J.O.; Shenker, S.; May, G.; Duff, M.O.; Zhang, D.; Eads, B.D.; Carlson, J.; Brown, J.B.; et al. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep. 2012, 1, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Subtelny, A.O.; Koppstein, D.; Bell, G.W.; Sive, H.; Bartel, D.P. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012, 22, 2054–2066. [Google Scholar] [CrossRef]
- Mayr, C. Evolution and Biological Roles of Alternative 3′UTRs. Trends Cell Biol. 2016, 26, 227–237. [Google Scholar] [CrossRef]
- Elkon, R.; Drost, J.; van Haaften, G.; Jenal, M.; Schrier, M.; Oude Vrielink, J.A.; Agami, R. E2F mediates enhanced alternative polyadenylation in proliferation. Genome Biol. 2012, 13, R59. [Google Scholar] [CrossRef]
- Shepard, P.J.; Choi, E.A.; Lu, J.; Flanagan, L.A.; Hertel, K.J.; Shi, Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 2011, 17, 761–772. [Google Scholar] [CrossRef]
- Muller, S.; Rycak, L.; Afonso-Grunz, F.; Winter, P.; Zawada, A.M.; Damrath, E.; Scheider, J.; Schmah, J.; Koch, I.; Kahl, G.; et al. APADB: A database for alternative polyadenylation and microRNA regulation events. Database 2014, 2014. [Google Scholar] [CrossRef]
- Masamha, C.P.; Xia, Z.; Peart, N.; Collum, S.; Li, W.; Wagner, E.J.; Shyu, A.B. CFIm25 regulates glutaminase alternative terminal exon definition to modulate miR-23 function. RNA 2016, 22, 830–838. [Google Scholar] [CrossRef]
- Halees, A.S.; El-Badrawi, R.; Khabar, K.S. ARED Organism: Expansion of ARED reveals AU-rich element cluster variations between human and mouse. Nucleic Acids Res. 2008, 36, D137–D140. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Ruegsegger, U.; Beyer, K.; Keller, W. Purification and characterization of human cleavage factor Im involved in the 3′ end processing of messenger RNA precursors. J. Biol. Chem. 1996, 271, 6107–6113. [Google Scholar] [CrossRef] [PubMed]
- Sartini, B.L.; Wang, H.; Wang, W.; Millette, C.F.; Kilpatrick, D.L. Pre-messenger RNA cleavage factor I (CFIm): Potential role in alternative polyadenylation during spermatogenesis. Biol. Reprod. 2008, 78, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Routh, A.; Ji, P.; Jaworski, E.; Xia, Z.; Li, W.; Wagner, E.J. Poly(A)-ClickSeq: Click-chemistry for next-generation 3-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res. 2017, 45, e112. [Google Scholar] [CrossRef]
- Zhu, Z.J.; Huang, P.; Chong, Y.X.; Kang, L.X.; Huang, X.; Zhu, Z.X.; Nie, L. MicroRNA-181a promotes proliferation and inhibits apoptosis by suppressing CFIm25 in osteosarcoma. Mol. Med. Rep. 2016, 14, 4271–4278. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, W. Knockdown of NUDT21 inhibits proliferation and promotes apoptosis of human K562 leukemia cells through ERK pathway. Cancer Manag. Res. 2018, 10, 4311–4323. [Google Scholar] [CrossRef]
- Shell, S.A.; Hesse, C.; Morris, S.M., Jr.; Milcarek, C. Elevated levels of the 64-kDa cleavage stimulatory factor (CstF-64) in lipopolysaccharide-stimulated macrophages influence gene expression and induce alternative poly(A) site selection. J. Biol. Chem. 2005, 280, 39950–39961. [Google Scholar] [CrossRef]
- Alt, F.W.; Bothwell, A.L.; Knapp, M.; Siden, E.; Mather, E.; Koshland, M.; Baltimore, D. Synthesis of secreted and membrane-bound immunoglobulin mu heavy chains is directed by mRNAs that differ at their 3′ ends. Cell 1980, 20, 293–301. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Parikh, R.R.; Housman, D.; Yang, Q.; Toppmeyer, D.; Wilson, L.D.; Haffty, B.G. Prognostic value of triple-negative phenotype at the time of locally recurrent, conservatively treated breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Baejen, C.; Andreani, J.; Torkler, P.; Battaglia, S.; Schwalb, B.; Lidschreiber, M.; Maier, K.C.; Boltendahl, A.; Rus, P.; Esslinger, S.; et al. Genome-wide Analysis of RNA Polymerase II Termination at Protein-Coding Genes. Mol. Cell 2017, 66, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.E.; Henneken, L.M.; Liem-Weits, M.; Harrison, P.F.; Swaminathan, A.; Vary, R.; Nikolic, I.; Simpson, K.J.; Powell, D.R.; Beilharz, T.; et al. Requirement for cleavage factor IIm in the control of alternative polyadenylation in breast cancer cells. RNA 2020. [Google Scholar] [CrossRef] [PubMed]
- Kamieniarz-Gdula, K.; Gdula, M.R.; Panser, K.; Nojima, T.; Monks, J.; Wisniewski, J.R.; Riepsaame, J.; Brockdorff, N.; Pauli, A.; Proudfoot, N.J. Selective Roles of Vertebrate PCF11 in Premature and Full-Length Transcript Termination. Mol. Cell 2019, 74, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zheng, D.; Wei, L.; Ding, Q.; Tian, B. Regulation of Intronic Polyadenylation by PCF11 Impacts mRNA Expression of Long Genes. Cell Rep. 2019, 26, 2766–2778. [Google Scholar] [CrossRef]
- Su, Z.; Łabaj, P.P.; Li, S.; Thierry-Mieg, J.; Thierry-Mieg, D.; Shi, W.; Wang, C.; Schroth, G.P.; Setterquist, R.; Thompson, J.F.; et al. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar] [CrossRef]
- Hornshoj, H.; Nielsen, M.M.; Sinnott-Armstrong, N.A.; Switnicki, M.P.; Juul, M.; Madsen, T.; Sallari, R.; Kellis, M.; Orntoft, T.; Hobolth, A.; et al. Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. NPJ Genom. Med. 2018, 3, 1. [Google Scholar] [CrossRef]
- Kuipers, J.; Thurnherr, T.; Moffa, G.; Suter, P.; Behr, J.; Goosen, R.; Christofori, G.; Beerenwinkel, N. Mutational interactions define novel cancer subgroups. Nat. Commun. 2018, 9, 4353. [Google Scholar] [CrossRef]
- Bai, S.; He, B.; Wilson, E.M. Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol. Cell. Biol. 2005, 25, 1238–1257. [Google Scholar] [CrossRef]
- Su, S.; Minges, J.T.; Grossman, G.; Blackwelder, A.J.; Mohler, J.L.; Wilson, E.M. Proto-oncogene activity of melanoma antigen-A11 (MAGE-A11) regulates retinoblastoma-related p107 and E2F1 proteins. J. Biol. Chem. 2013, 288, 24809–24824. [Google Scholar] [CrossRef]
- Lian, Y.; Sang, M.; Ding, C.; Zhou, X.; Fan, X.; Xu, Y.; Lu, W.; Shan, B. Expressions of MAGE-A10 and MAGE-A11 in breast cancers and their prognostic significance: A retrospective clinical study. J. Cancer Res. Clin. Oncol. 2012, 138, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.P.; Xu, M.; Chen, Y.; Shao, W.W. Expression of MAGE-A11 in breast cancer tissues and its effects on the proliferation of breast cancer cells. Mol. Med. Rep. 2013, 7, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Karaca, E.; Weitzer, S.; Pehlivan, D.; Shiraishi, H.; Gogakos, T.; Hanada, T.; Jhangiani, S.N.; Wiszniewski, W.; Withers, M.; Campbell, I.M.; et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 2014, 157, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.E.; Eggens, V.R.; Caglayan, A.O.; Reuter, M.S.; Scott, E.; Coufal, N.G.; Silhavy, J.L.; Xue, Y.; Kayserili, H.; Yasuno, K.; et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 2014, 157, 651–663. [Google Scholar] [CrossRef]
- Lackford, B.; Yao, C.; Charles, G.M.; Weng, L.; Zheng, X.; Choi, E.A.; Xie, X.; Wan, J.; Xing, Y.; Freudenberg, J.M.; et al. Fip1 regulates mRNA alternative polyadenylation to promote stem cell self-renewal. EMBO J. 2014, 33, 878–889. [Google Scholar] [CrossRef]
- Martincic, K.; Alkan, S.A.; Cheatle, A.; Borghesi, L.; Milcarek, C. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat. Immunol. 2009, 10, 1102–1109. [Google Scholar] [CrossRef]
- Mellman, D.L.; Gonzales, M.L.; Song, C.; Barlow, C.A.; Wang, P.; Kendziorski, C.; Anderson, R.A. A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 2008, 451, 1013–1017. [Google Scholar] [CrossRef]
- Laishram, R.S.; Anderson, R.A. The poly A polymerase Star-PAP controls 3′-end cleavage by promoting CPSF interaction and specificity toward the pre-mRNA. EMBO J. 2010, 29, 4132–4145. [Google Scholar] [CrossRef]
- Li, W.; Laishram, R.S.; Ji, Z.; Barlow, C.A.; Tian, B.; Anderson, R.A. Star-PAP control of BIK expression and apoptosis is regulated by nuclear PIPKIalpha and PKCdelta signaling. Mol. Cell 2012, 45, 25–37. [Google Scholar] [CrossRef]
- De Klerk, E.; Venema, A.; Anvar, S.Y.; Goeman, J.J.; Hu, O.; Trollet, C.; Dickson, G.; den Dunnen, J.T.; van der Maarel, S.M.; Raz, V.; et al. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res. 2012, 40, 9089–9101. [Google Scholar] [CrossRef]
- Brais, B.; Bouchard, J.P.; Xie, Y.G.; Rochefort, D.L.; Chretien, N.; Tome, F.M.; Lafreniere, R.G.; Rommens, J.M.; Uyama, E.; Nohira, O.; et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998, 18, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Calado, A.; Kutay, U.; Kuhn, U.; Wahle, E.; Carmo-Fonseca, M. Deciphering the cellular pathway for transport of poly(A)-binding protein II. RNA 2000, 6, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Raz, Y.; van Putten, M.; Paniagua-Soriano, G.; Krom, Y.D.; Florea, B.I.; Raz, V. PABPN1-Dependent mRNA Processing Induces Muscle Wasting. PLoS Genet. 2016, 12, e1006031. [Google Scholar] [CrossRef] [PubMed]
- Anvar, S.Y.; Raz, Y.; Verway, N.; van der Sluijs, B.; Venema, A.; Goeman, J.J.; Vissing, J.; van der Maarel, S.M.; t Hoen, P.A.; van Engelen, B.G.; et al. A decline in PABPN1 induces progressive muscle weakness in oculopharyngeal muscle dystrophy and in muscle aging. Aging 2013, 5, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Kapranov, P.; Foissac, S.; Kim, S.W.; Fishilevich, E.; Monaghan, A.P.; John, B.; Milos, P.M. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 2010, 143, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.E.; Pattison, A.D.; Beilharz, T.H. Alternative polyadenylation in the regulation and dysregulation of gene expression. Semin. Cell Dev. Biol. 2018, 75, 61–69. [Google Scholar] [CrossRef]
- Hoque, M.; Ji, Z.; Zheng, D.; Luo, W.; Li, W.; You, B.; Park, J.Y.; Yehia, G.; Tian, B. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 2013, 10, 133–139. [Google Scholar] [CrossRef]
- Ha, K.C.H.; Blencowe, B.J.; Morris, Q. QAPA: A new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol. 2018, 19, 45. [Google Scholar] [CrossRef]
- Ogorodnikov, A.; Kargapolova, Y.; Danckwardt, S. Processing and transcriptome expansion at the mRNA 3′ end in health and disease: Finding the right end. Pflugers Arch. 2016, 468, 993–1012. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, S.; Pelechano, V.; Jarvelin, A.I.; Tekkedil, M.M.; Anders, S.; Benes, V.; Steinmetz, L.M. An efficient method for genome-wide polyadenylation site mapping and RNA quantification. Nucleic Acids Res. 2013, 41, e65. [Google Scholar] [CrossRef]
- Fox-Walsh, K.; Davis-Turak, J.; Zhou, Y.; Li, H.; Fu, X.D. A multiplex RNA-seq strategy to profile poly(A+) RNA: Application to analysis of transcription response and 3′ end formation. Genomics 2011, 98, 266–271. [Google Scholar] [CrossRef]
- Mangone, M.; Manoharan, A.P.; Thierry-Mieg, D.; Thierry-Mieg, J.; Han, T.; Mackowiak, S.D.; Mis, E.; Zegar, C.; Gutwein, M.R.; Khivansara, V.; et al. The landscape of C. elegans 3′UTRs. Science 2010, 329, 432–435. [Google Scholar] [CrossRef]
- Jan, C.H.; Friedman, R.C.; Ruby, J.G.; Bartel, D.P. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 2011, 469, 97–101. [Google Scholar] [CrossRef]
- Harrison, P.F.; Powell, D.R.; Clancy, J.L.; Preiss, T.; Boag, P.R.; Traven, A.; Seemann, T.; Beilharz, T.H. PAT-seq: A method to study the integration of 3′-UTR dynamics with gene expression in the eukaryotic transcriptome. RNA 2015, 21, 1502–1510. [Google Scholar] [CrossRef]
- Zhang, H.; Lee, J.Y.; Tian, B. Biased alternative polyadenylation in human tissues. Genome Biol. 2005, 6, R100. [Google Scholar] [CrossRef]
- Kwan, T.; Benovoy, D.; Dias, C.; Gurd, S.; Provencher, C.; Beaulieu, P.; Hudson, T.J.; Sladek, R.; Majewski, J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008, 40, 225–231. [Google Scholar] [CrossRef]
- MacIsaac, J.L.; Bogutz, A.B.; Morrissy, A.S.; Lefebvre, L. Tissue-specific alternative polyadenylation at the imprinted gene Mest regulates allelic usage at Copg2. Nucleic Acids Res. 2012, 40, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Siva, K.; Covello, G.; Denti, M.A. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid Ther. 2014, 24, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Miller, T.M. Antisense oligonucleotides: Treating neurodegeneration at the level of RNA. Neurotherapeutics 2013, 10, 486–497. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Matos, L.; Santos, J.I.; Alves, S. RNA Therapeutics: How Far Have We Gone? Adv. Exp. Med. Biol. 2019, 1157, 133–177. [Google Scholar] [CrossRef]
- Marsollier, A.C.; Ciszewski, L.; Mariot, V.; Popplewell, L.; Voit, T.; Dickson, G.; Dumonceaux, J. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: A new gene-silencing approach. Hum. Mol. Genet. 2016, 25, 1468–1478. [Google Scholar] [CrossRef]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P., Jr.; Wagner, K.R. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Nyquist, M.; Li, Y.; Yang, R.; Ho, Y.; Johnson, R.; Ondigi, O.; Voytas, D.F.; Henzler, C.; Dehm, S.M. Targeting a Single Alternative Polyadenylation Site Coordinately Blocks Expression of Androgen Receptor mRNA Splice Variants in Prostate Cancer. Cancer Res. 2017, 77, 5228–5235. [Google Scholar] [CrossRef]
- Chu, W.; Presky, D.H.; Swerlick, R.A.; Burns, D.K. Alternatively processed human E-selectin transcripts linked to chronic expression of E-selectin in vivo. J. Immunol. 1994, 153, 4179–4189. [Google Scholar]
- Vickers, T.A.; Wyatt, J.R.; Burckin, T.; Bennett, C.F.; Freier, S.M. Fully modified 2’ MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res. 2001, 29, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J.L. Alternative cleavage and polyadenylation: The long and short of it. Trends Biochem. Sci. 2013, 38, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Martinson, H.G. An active role for splicing in 3′-end formation. Wiley Interdiscip. Rev. RNA 2011, 2, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell. Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Englbrecht, C.; Chari, A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip. Rev. RNA 2011, 2, 718–731. [Google Scholar] [CrossRef]
- Gunderson, S.I.; Polycarpou-Schwarz, M.; Mattaj, I.W. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol. Cell 1998, 1, 255–264. [Google Scholar] [CrossRef]
- Kaida, D.; Berg, M.G.; Younis, I.; Kasim, M.; Singh, L.N.; Wan, L.; Dreyfuss, G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 2010, 468, 664–668. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Vorlova, S.; Rocco, G.; Lefave, C.V.; Jodelka, F.M.; Hess, K.; Hastings, M.L.; Henke, E.; Cartegni, L. Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol. Cell 2011, 43, 927–939. [Google Scholar] [CrossRef]
- Goraczniak, R.; Behlke, M.A.; Gunderson, S.I. Gene silencing by synthetic U1 adaptors. Nat. Biotechnol. 2009, 27, 257–263. [Google Scholar] [CrossRef]
- Beckley, S.A.; Liu, P.; Stover, M.L.; Gunderson, S.I.; Lichtler, A.C.; Rowe, D.W. Reduction of target gene expression by a modified U1 snRNA. Mol. Cell. Biol. 2001, 21, 2815–2825. [Google Scholar] [CrossRef]
- Fortes, P.; Cuevas, Y.; Guan, F.; Liu, P.; Pentlicky, S.; Jung, S.P.; Martinez-Chantar, M.L.; Prieto, J.; Rowe, D.; Gunderson, S.I. Inhibiting expression of specific genes in mammalian cells with 5′ end-mutated U1 small nuclear RNAs targeted to terminal exons of pre-mRNA. Proc. Natl. Acad. Sci. USA 2003, 100, 8264–8269. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.A.; Sabripour, M.; Crooke, S.T. U1 adaptors result in reduction of multiple pre-mRNA species principally by sequestering U1snRNP. Nucleic Acids Res. 2011, 39, e71. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, L.; Gonzalez-Rojas, S.J.; Abad, A.; Razquin, N.; Abad, X.; Fortes, P. Increased in vivo inhibition of gene expression by combining RNA interference and U1 inhibition. Nucleic Acids Res. 2012, 40, e8. [Google Scholar] [CrossRef]
- Vickers, T.A.; Crooke, S.T. siRNAs targeted to certain polyadenylation sites promote specific, RISC-independent degradation of messenger RNAs. Nucleic Acids Res. 2012, 40, 6223–6234. [Google Scholar] [CrossRef]
- Niu, X.; He, W.; Song, B.; Ou, Z.; Fan, D.; Chen, Y.; Fan, Y.; Sun, X. Combining Single Strand Oligodeoxynucleotides and CRISPR/Cas9 to Correct Gene Mutations in beta-Thalassemia-induced Pluripotent Stem Cells. J. Biol. Chem. 2016, 291, 16576–16585. [Google Scholar] [CrossRef]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef]
- Song, B.; Fan, Y.; He, W.; Zhu, D.; Niu, X.; Wang, D.; Ou, Z.; Luo, M.; Sun, X. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015, 24, 1053–1065. [Google Scholar] [CrossRef]
- Gao, Y.; Guo, X.; Santostefano, K.; Wang, Y.; Reid, T.; Zeng, D.; Terada, N.; Ashizawa, T.; Xia, G. Genome Therapy of Myotonic Dystrophy Type 1 iPS Cells for Development of Autologous Stem Cell Therapy. Mol. Ther. 2016, 24, 1378–1387. [Google Scholar] [CrossRef]
- Xia, G.; Gao, Y.; Jin, S.; Subramony, S.H.; Terada, N.; Ranum, L.P.; Swanson, M.S.; Ashizawa, T. Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 2015, 33, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.G.; McGarrity, G.J. Gene therapy progress and prospects: Reprograming gene expression by trans-splicing. Gene Ther. 2005, 12, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Mansfield, S.G.; Bartel, R.C.; Hiriyanna, S.; Mitchell, L.G.; Garcia-Blanco, M.A.; Walsh, C.E. Phenotype correction of hemophilia A mice by spliceosome-mediated RNA trans-splicing. Nat. Med. 2003, 9, 1015–1019. [Google Scholar] [CrossRef]
- Kleiman, F.E.; Manley, J.L. The BARD1-CstF-50 interaction links mRNA 3′ end formation to DNA damage and tumor suppression. Cell 2001, 104, 743–753. [Google Scholar] [CrossRef]
- Rozenblatt-Rosen, O.; Nagaike, T.; Francis, J.M.; Kaneko, S.; Glatt, K.A.; Hughes, C.M.; LaFramboise, T.; Manley, J.L.; Meyerson, M. The tumor suppressor Cdc73 functionally associates with CPSF and CstF 3′ mRNA processing factors. Proc. Natl. Acad. Sci. USA 2009, 106, 755–760. [Google Scholar] [CrossRef]
- Topalian, S.L.; Kaneko, S.; Gonzales, M.I.; Bond, G.L.; Ward, Y.; Manley, J.L. Identification and functional characterization of neo-poly(A) polymerase, an RNA processing enzyme overexpressed in human tumors. Mol. Cell. Biol. 2001, 21, 5614–5623. [Google Scholar] [CrossRef] [PubMed]
- Cools, J.; Stover, E.H.; Wlodarska, I.; Marynen, P.; Gilliland, D.G. The FIP1L1-PDGFRalpha kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia. Curr. Opin. Hematol. 2004, 11, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Cools, J.; Malone, J.M., 3rd; Schrier, S.L.; Gilliland, D.G.; Coutre, S.E. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: Implications for diagnosis, classification, and management. Blood 2004, 103, 2879–2891. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Mills, T.; Huang, J.; Chen, N.Y.; Mertens, T.C.J.; Collum, S.D.; Lee, G.; Xiang, Y.; Han, L.; Zhou, Y.; et al. Transforming growth factor beta1 alters the 3′-UTR of mRNA to promote lung fibrosis. J. Biol. Chem. 2019, 294, 15781–15794. [Google Scholar] [CrossRef]
- Collins, I.; Weber, A.; Levens, D. Transcriptional consequences of topoisomerase inhibition. Mol. Cell. Biol. 2001, 21, 8437–8451. [Google Scholar] [CrossRef]
- Hampsey, M.; Reinberg, D. Tails of intrigue: Phosphorylation of RNA polymerase II mediates histone methylation. Cell 2003, 113, 429–432. [Google Scholar] [CrossRef][Green Version]
- Di Giammartino, D.C.; Shi, Y.; Manley, J.L. PARP1 represses PAP and inhibits polyadenylation during heat shock. Mol. Cell 2013, 49, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Shirai, C.L.; White, B.S.; Tripathi, M.; Tapia, R.; Ley, J.N.; Ndonwi, M.; Kim, S.; Shao, J.; Carver, A.; Saez, B.; et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat. Commun. 2017, 8, 14060. [Google Scholar] [CrossRef]
- Penman, S.; Rosbash, M.; Penman, M. Messenger and heterogeneous nuclear RNA in HeLa cells: Differential inhibition by cordycepin. Proc. Natl. Acad. Sci. USA 1970, 67, 1878–1885. [Google Scholar] [CrossRef]
- Rose, K.M.; Bell, L.E.; Jacob, S.T. Specific inhibition of chromatin-associated poly(A) synthesis in vitro by cordycepin 5′-triphosphate. Nature 1977, 267, 178–180. [Google Scholar] [CrossRef]
- Ryner, L.C.; Manley, J.L. Requirements for accurate and efficient mRNA 3′ end cleavage and polyadenylation of a simian virus 40 early pre-RNA in vitro. Mol. Cell. Biol. 1987, 7, 495–503. [Google Scholar] [CrossRef]
- Zarkower, D.; Wickens, M. Specific pre-cleavage and post-cleavage complexes involved in the formation of SV40 late mRNA 3′ termini in vitro. EMBO J. 1987, 6, 4185–4192. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.C.; Hsiao, J.R.; Lian, Y.Y.; Lin, C.Y.; Huang, B.M. The apoptotic effect of cordycepin on human OEC-M1 oral cancer cell line. Cancer Chemother. Pharmacol. 2007, 60, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Thomadaki, H.; Tsiapalis, C.M.; Scorilas, A. Polyadenylate polymerase modulations in human epithelioid cervix and breast cancer cell lines, treated with etoposide or cordycepin, follow cell cycle rather than apoptosis induction. Biol. Chem. 2005, 386, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Koc, Y.; Urbano, A.G.; Sweeney, E.B.; McCaffrey, R. Induction of apoptosis by cordycepin in ADA-inhibited TdT-positive leukemia cells. Leukemia 1996, 10, 1019–1024. [Google Scholar]
- Chen, L.S.; Stellrecht, C.M.; Gandhi, V. RNA-directed agent, cordycepin, induces cell death in multiple myeloma cells. Br. J. Haematol. 2008, 140, 391–682. [Google Scholar] [CrossRef]
- Imesch, P.; Hornung, R.; Fink, D.; Fedier, A. Cordycepin (3′-deoxyadenosine), an inhibitor of mRNA polyadenylation, suppresses proliferation and activates apoptosis in human epithelial endometriotic cells in vitro. Gynecol. Obstet. Invest. 2011, 72, 43–49. [Google Scholar] [CrossRef]
- Ashraf, S.; Radhi, M.; Gowler, P.; Burston, J.J.; Gandhi, R.D.; Thorn, G.J.; Piccinini, A.M.; Walsh, D.A.; Chapman, V.; de Moor, C.H. The polyadenylation inhibitor cordycepin reduces pain, inflammation and joint pathology in rodent models of osteoarthritis. Sci. Rep. 2019, 9, 4696. [Google Scholar] [CrossRef]
- Kondrashov, A.; Meijer, H.A.; Barthet-Barateig, A.; Parker, H.N.; Khurshid, A.; Tessier, S.; Sicard, M.; Knox, A.J.; Pang, L.; De Moor, C.H. Inhibition of polyadenylation reduces inflammatory gene induction. RNA 2012, 18, 2236–2250. [Google Scholar] [CrossRef]
- Cowley, M.; Wood, A.J.; Bohm, S.; Schulz, R.; Oakey, R.J. Epigenetic control of alternative mRNA processing at the imprinted Herc3/Nap1l5 locus. Nucleic Acids Res. 2012, 40, 8917–8926. [Google Scholar] [CrossRef]
- Harries, L.W. RNA Biology Provides New Therapeutic Targets for Human Disease. Front. Genet. 2019, 10, 205. [Google Scholar] [CrossRef]
- Bava, F.A.; Eliscovich, C.; Ferreira, P.G.; Minana, B.; Ben-Dov, C.; Guigo, R.; Valcarcel, J.; Mendez, R. CPEB1 coordinates alternative 3′-UTR formation with translational regulation. Nature 2013, 495, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, V.; Lemke, S.B.; Levine, M. ELAV mediates 3′ UTR extension in the Drosophila nervous system. Genes Dev. 2012, 26, 2259–2264. [Google Scholar] [CrossRef]
- Karpova, N.N.; Pickenhagen, A.; Lindholm, J.; Tiraboschi, E.; Kulesskaya, N.; Agustsdottir, A.; Antila, H.; Popova, D.; Akamine, Y.; Bahi, A.; et al. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 2011, 334, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Nakayama, Y.; Sano, O.; Nakao, S.; Shimizu-Ogasawara, M.; Toyoshiba, H.; Nakanishi, A.; Aparicio, S. Decoding Transcriptome Dynamics of Genome-Encoded Polyadenylation and Autoregulation with Small-Molecule Modulators of Alternative Polyadenylation. Cell Chem. Biol. 2018, 25, 1470–1484. [Google Scholar] [CrossRef] [PubMed]






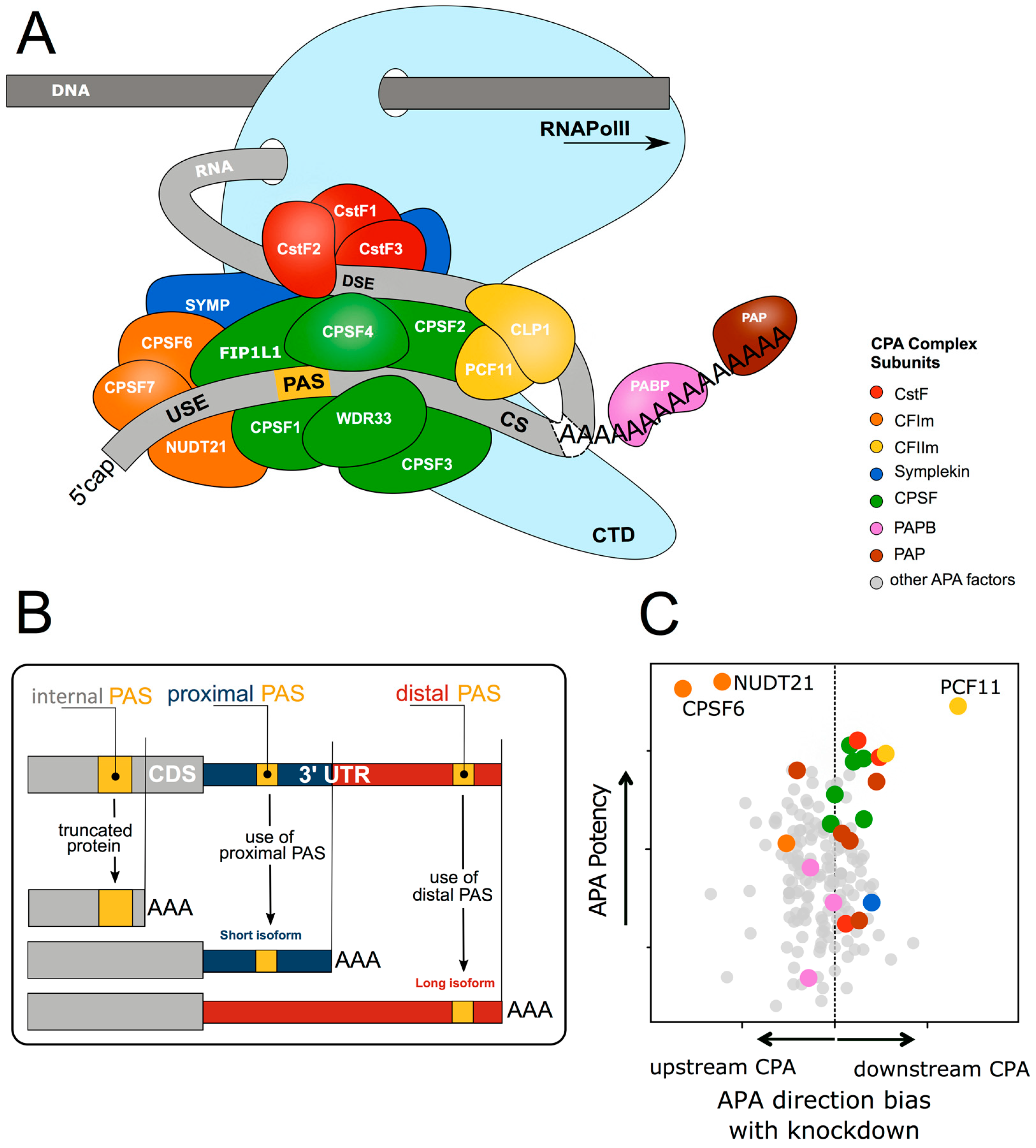{kind=link}
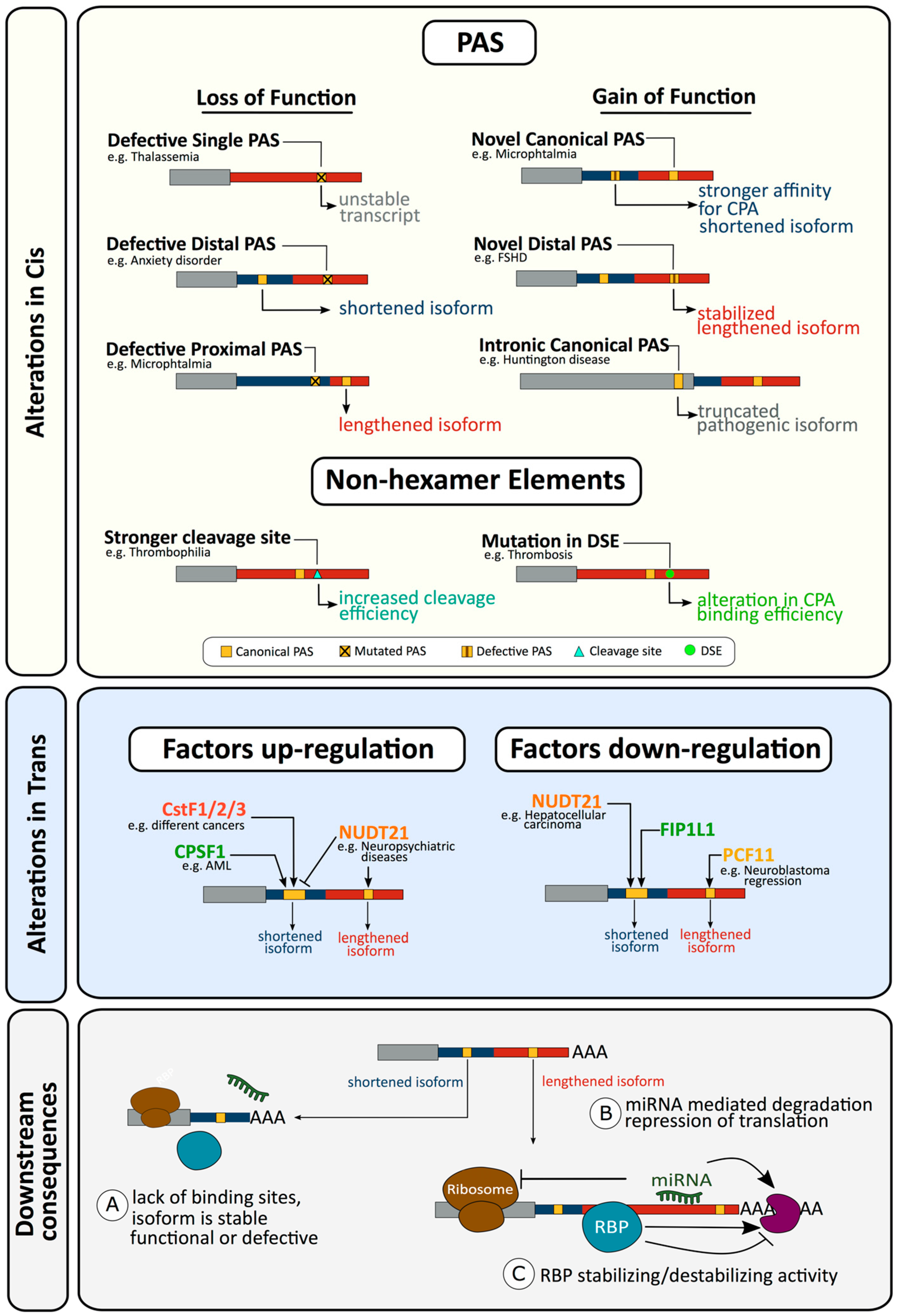{kind=link}
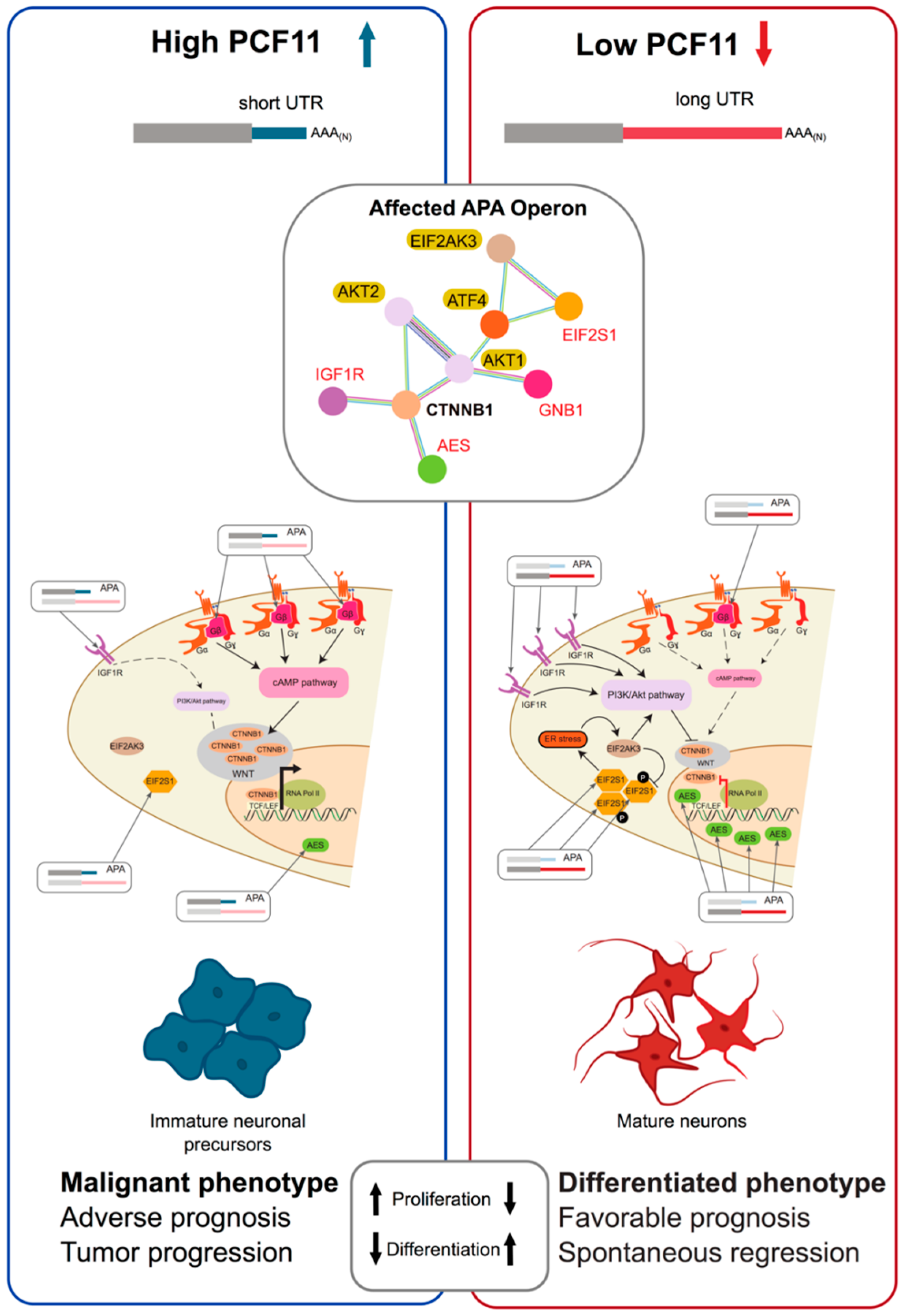{kind=link}
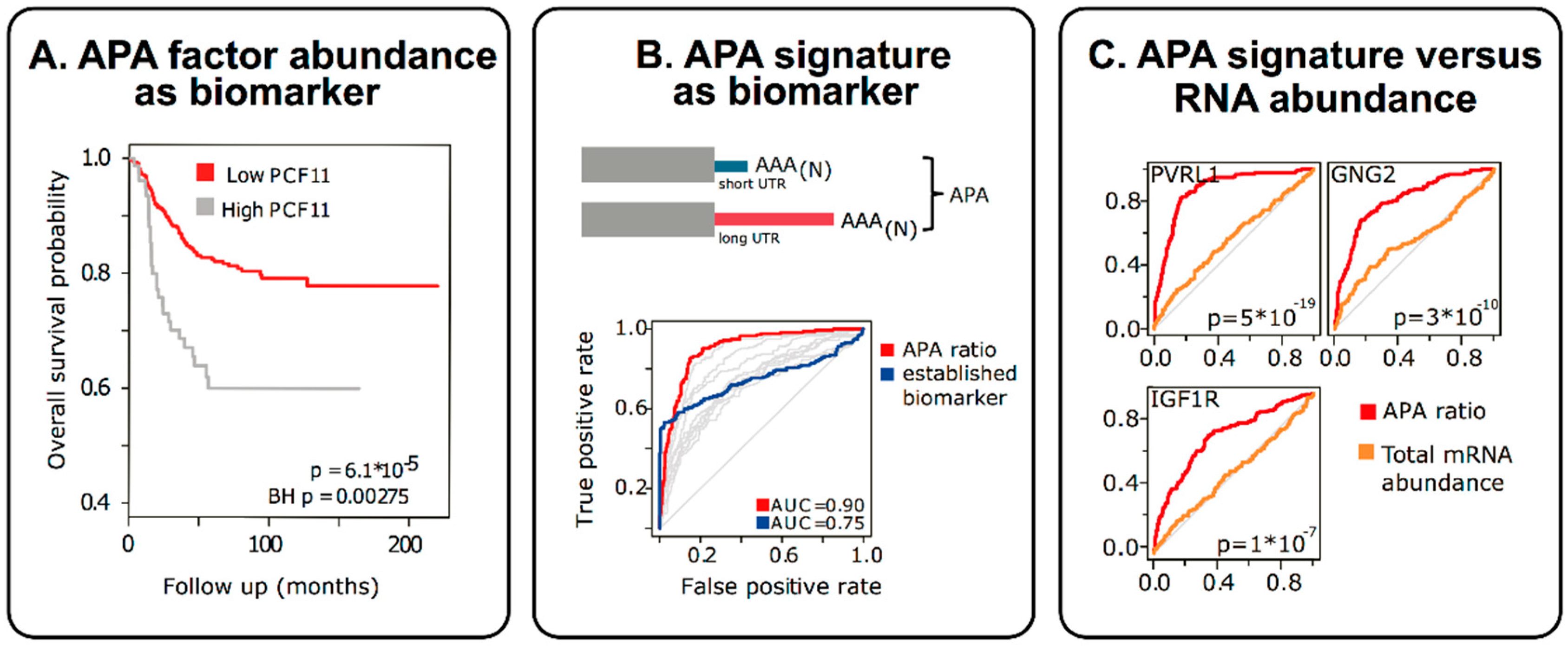{kind=link}
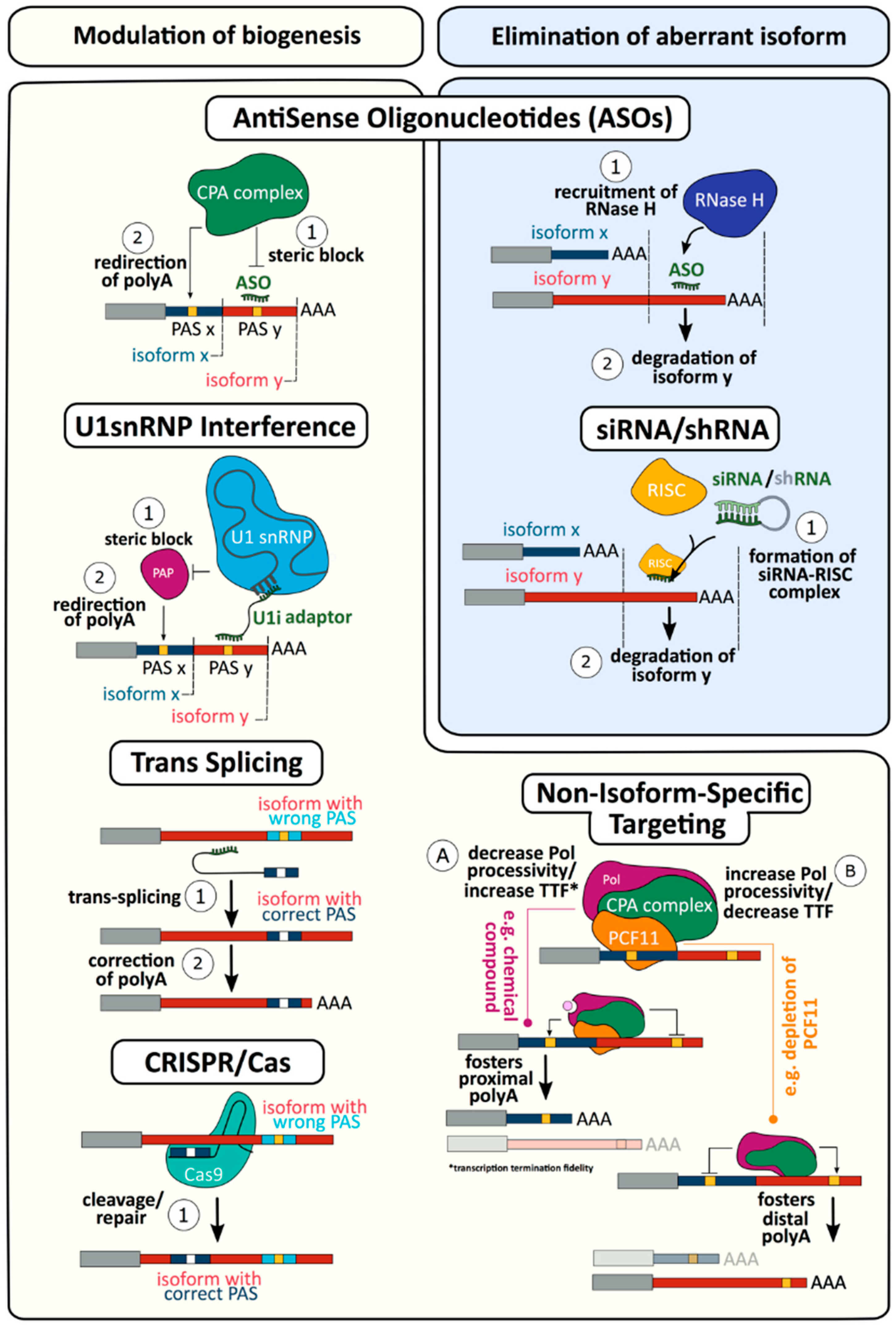{kind=link}
| Disease | Type | Effector Factors | Cis/Trans | Gain/Loss | References |
|---|---|---|---|---|---|
| Cancer | |||||
| Acute myeloid leukaemia | APA | CPSF | Trans | Unknown | [86] |
| B-cell leukemia/lymphoma | APA-S | CFII, CPSF, CstF | Trans | Unknown | [87] |
| Bladder | APA-S | CFI, CstF | Trans | Unknown | [88,89] |
| Breast | APA-I | Estrogen | Trans | Unknown | [90,91] |
| Breast | APA-S | CstF, Estrogen | Trans | Unknown | [92,93,94,95,96,97,98,99,100,101,102] |
| Chronic lymphocytic leukaemia | APA-I | Unknown | Trans | Unknown | [103] |
| Colon | APA-S | Unknown | Trans | Unknown | [104,105] |
| Colorectal | APA-S | CPSF, CFI, hnRNPC | Trans | Unknown | [106,107,108,109,110,111] |
| Esophageal squamous cell carcinoma | CPA | PAS | Cis | Loss | [112] |
| Fanconi Anemia | APA-I | PAS-methylation, SF3A1 | Trans | Gain | [113,114] |
| Gastric | APA-S | Unknown | Trans | Unknown | [115] |
| Glioblastoma | APA-L | Unknown | Trans | Unknown | [116] |
| Glioblastoma | APA-S | CFI, PTBP1 | Trans | Loss | [78,117,118,119,120] |
| Hepatocellular carcinoma | APA-S | CFI | Trans | Unknown | [121,122,123] |
| Lung | APA-S | CFI, CstF | Trans | Unknown | [102,124,125,126,127,128,129,130,131] |
| Lynch syndrome | CPA | PAS | Cis | Loss | [132] |
| Mantle cell lymphoma | APA-S | PAS | Cis | Gain | [133,134] |
| Melanoma | CPA | Unknown | Trans | Unknown | [135] |
| Multiple | APA | UTR3 SNP | Cis | SNP | [136] |
| Multiple | APA-I | PAS-Pbx1 binding | Cis | Gain | [137] |
| Multiple | APA-S | CFI, CstF, PAPs, PABPN1 | Trans | Unknown | [138,139,140,141,142,143,144,145] |
| Multiple | CPA | PAS | Cis | Loss | [146,147] |
| Nasopharyngeal carcinoma | APA | Unknown | Trans | Unknown | [148] |
| Neuroblastoma | APA-L | CFII | Trans | Loss | [13] |
| Ovarian | APA-S | Unknown | Trans | Unknown | [149,150,151,152] |
| Pancreatic ductal adenocarcinoma | APA-S | Unknown | Trans | Unknown | [153] |
| Proliferative conditions | APA-S | Unknown | Trans | Unknown | [154,155] |
| Prostate | CPA | Other | Trans | Loss | [156] |
| Small intestinal neuroendocrine | APA-S | Unknown | Trans | Unknown | [157] |
| Cardiovascular | |||||
| Atherosclerosis | CPA | Unknown | Trans | Unknown | [158] |
| Cardiac hypertrophy | APA | CstF, PAP | Trans | Unknown | [159,160,161,162] |
| Cardiac hypertrophy | CPA | PABPC1 | Trans | Unknown | [163] |
| Cardiomyopathy, Dilated | APA | CFII, CPSF, PABPN1 | Trans | Unknown | [164] |
| Hypertension | APA-L | UTR3 SNP | Cis | Unknown | [165,166] |
| Hypertension | APA-S | DSE | Cis | Gain | [167] |
| Ischemia/reperfusion injury | APA-S | Unknown | Trans | Unknown | [168] |
| Thrombosis (Deep vein) | APA-I | DSE | Cis | Gain | [169,170] |
| Thrombosis (Venous) | CPA | CS, DSE | Cis | Gain | [171,172,173,174] |
| Congenital Abnormalities | |||||
| Microphthalmia | APA-L | PAS | Cis | Loss | [175] |
| Mullerian aplasia | CPA | Unknown | Trans | Unknown | [176] |
| Bone fragility (Osteogenesis imperfecta) | CPA | PAS | Cis | Loss | [177] |
| Rickets | APA | Unknown | Trans | Unknown | [178] |
| Endocrine | |||||
| Diabetes, neonatal | CPA | PAS | Cis | Loss | [179] |
| Diabetes, type 2 | APA | Unknown | Trans | Unknown | [180] |
| Hematological | |||||
| Glanzmann Thrombasthenia | CPA | PAS | Cis | Loss | [181] |
| α-Thalassemia | CPA | PAS | Cis | Loss | [182] |
| β-Thalassemia | CPA | PAS | Cis | Loss | [183,184] |
| Immunological | |||||
| IPEX syndrome | CPA | PAS | Cis | Loss | [185,186] |
| Nasal polyps | APA | Unknown | Trans | Unknown | [187,188] |
| Severe combined immunodeficiency | CPA | PAS | Cis | Loss | [189] |
| Systemic lupus erythematosus | APA-L | PAS | Cis | Gain, Loss | [190,191,192,193] |
| Wiskott-Aldrich syndrome | CPA | PAS | Cis | Loss | [194] |
| Lung Disease | |||||
| Pulmonary fibrosis | APA-S | CFI | Trans | Loss | [195] |
| Musculoskeletal | |||||
| Muscle fibrosis | APA-I | Unknown | Trans | Unknown | [196] |
| Muscular Dystrophy, Facioscapulohumeral | CPA | PAS | Cis | Gain | [197] |
| Muscular dystrophy, Oculopharyngeal | APA-S | PABP | Trans | Loss | [198,199] |
| Myotonic dystrophy | APA-S | CFI | Trans | Loss | [200] |
| Oculopharyngeal muscular dystrophy | APA | PABPN1 | Trans | Loss | [201] |
| Neurological | |||||
| Alzheimer Disease | APA | Unknown | Trans | Unknown | [202,203,204,205,206] |
| Alzheimer Disease | CPA | U1 snRNP | Trans | Unknown | [207] |
| Amyotrophic lateral sclerosis | APA | FUS, TARDBP | Trans | Loss | [206,208,209,210,211] |
| Amyotrophic lateral sclerosis | CPA | PAS | Cis | Gain | [212,213] |
| Anxiety Disorders | APA-S | PAS | Cis | SNP | [214,215,216] |
| Fabry disease | CPA | PAS | Cis | Loss | [217] |
| Fragile X syndrome | APA-S | UTR5 repeats | Cis | Unknown | [218] |
| Friedreich’s Ataxia | APA | CPSF | Trans | Loss | [219] |
| Huntington’s disease | CPA | PAS | Cis | Gain | [220] |
| Metachromatic leukodystrophy pseudodeficiency | APA-L | PAS | Cis | Loss | [221,222] |
| Neuropsychiatric disease | APA-L | CFI | Trans | Gain | [223] |
| Parkinson disease | APA | UTR3 SNP | Cis | Unknown | [224] |
| Parkinson disease | APA-L | PAS dopamine | Trans | Unknown | [206,224] |
| Suicidal behavior | APA-S | PAS | Cis | SNP | [225] |
| Other | |||||
| Stress | APA | Unknown | Trans | Unknown | [226] |
| Zellweger syndrome | APA-S | PAS | Cis | Loss | [227] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nourse, J.; Spada, S.; Danckwardt, S. Emerging Roles of RNA 3′-end Cleavage and Polyadenylation in Pathogenesis, Diagnosis and Therapy of Human Disorders. Biomolecules 2020, 10, 915. https://doi.org/10.3390/biom10060915
Nourse J, Spada S, Danckwardt S. Emerging Roles of RNA 3′-end Cleavage and Polyadenylation in Pathogenesis, Diagnosis and Therapy of Human Disorders. Biomolecules. 2020; 10(6):915. https://doi.org/10.3390/biom10060915
Chicago/Turabian StyleNourse, Jamie, Stefano Spada, and Sven Danckwardt. 2020. "Emerging Roles of RNA 3′-end Cleavage and Polyadenylation in Pathogenesis, Diagnosis and Therapy of Human Disorders" Biomolecules 10, no. 6: 915. https://doi.org/10.3390/biom10060915
APA StyleNourse, J., Spada, S., & Danckwardt, S. (2020). Emerging Roles of RNA 3′-end Cleavage and Polyadenylation in Pathogenesis, Diagnosis and Therapy of Human Disorders. Biomolecules, 10(6), 915. https://doi.org/10.3390/biom10060915