The Arg/N-Degron Pathway—A Potential Running Back in Fine-Tuning the Inflammatory Response?



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
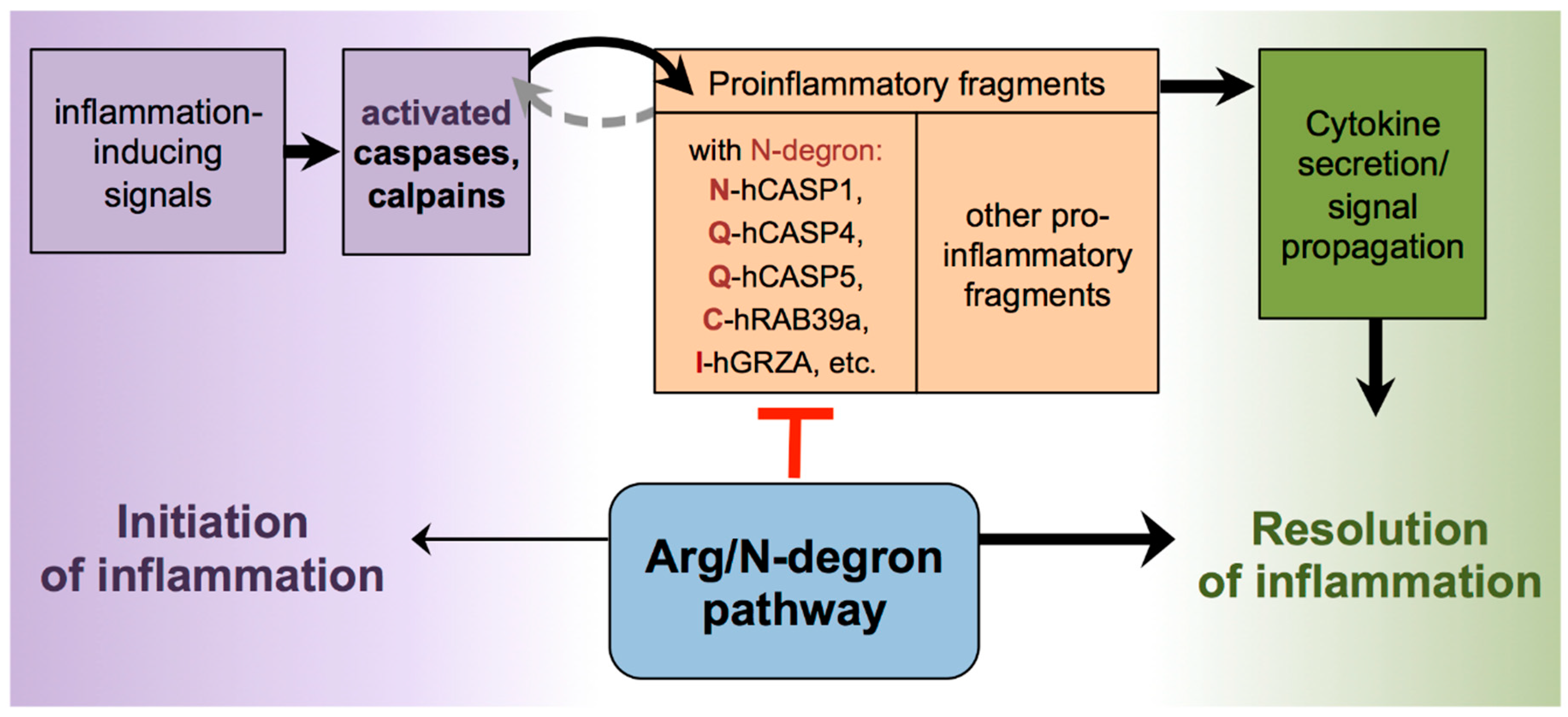
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis of Caspase Substrates
2.2. Cell Culture, Transfections, and Stimulations
2.3. Plasmids, cDNAs, and Primers
2.4. In Vitro Transcription–Translation–Degradation Assay
2.5. siRNA Description and Selection
2.6. cDNA Synthesis and qPCR
2.7. Cell Extracts and Western Blot
2.8. Caspase-1 Activity Assay
2.9. Cytokine Bead Assay (CBA)
2.10. Statistical Analysis
3. Results
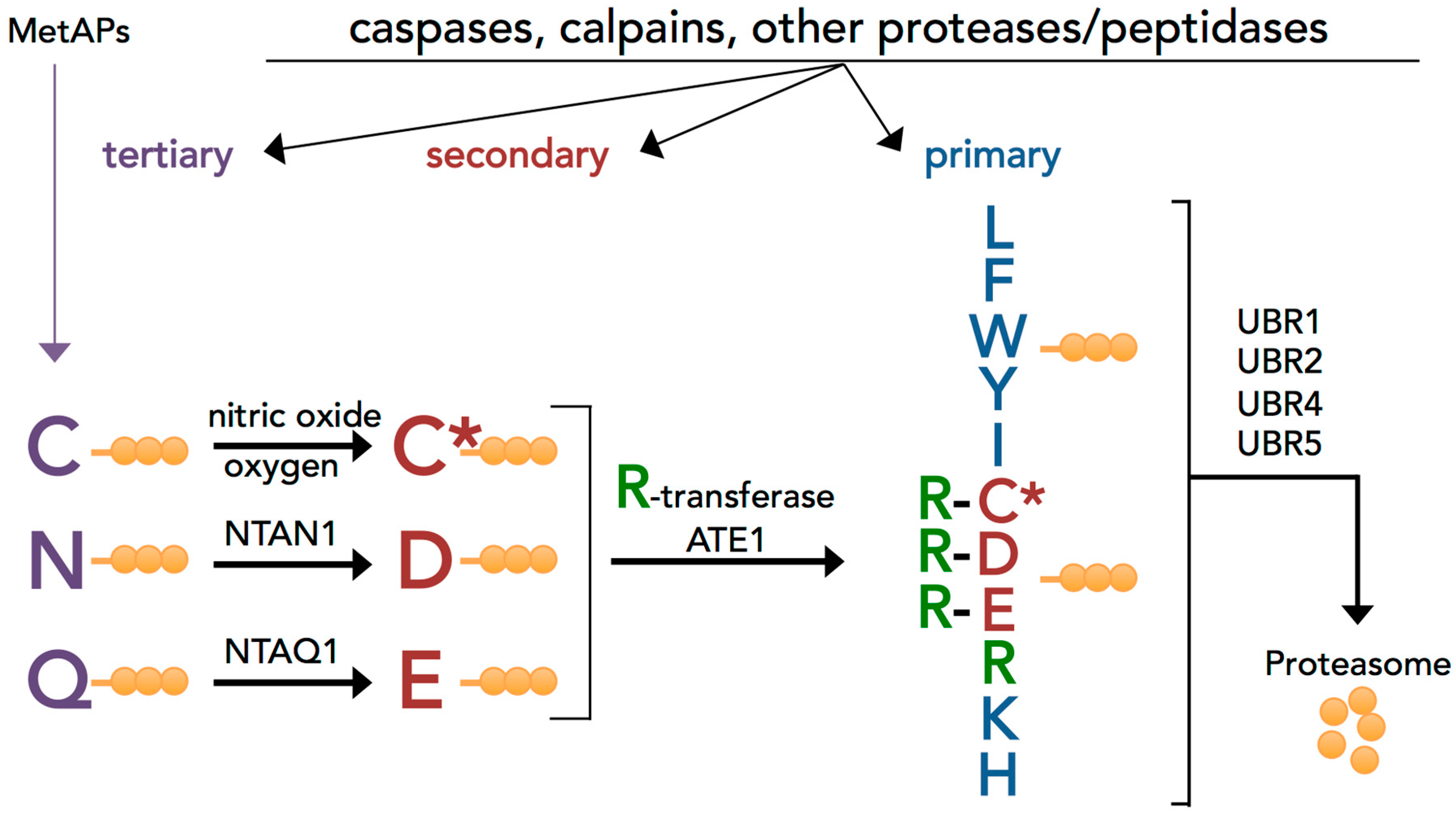
3.1. Evolutionary Conserved Proinflammatory Fragments Contain Destabilizing Residues at Their N-Terminus
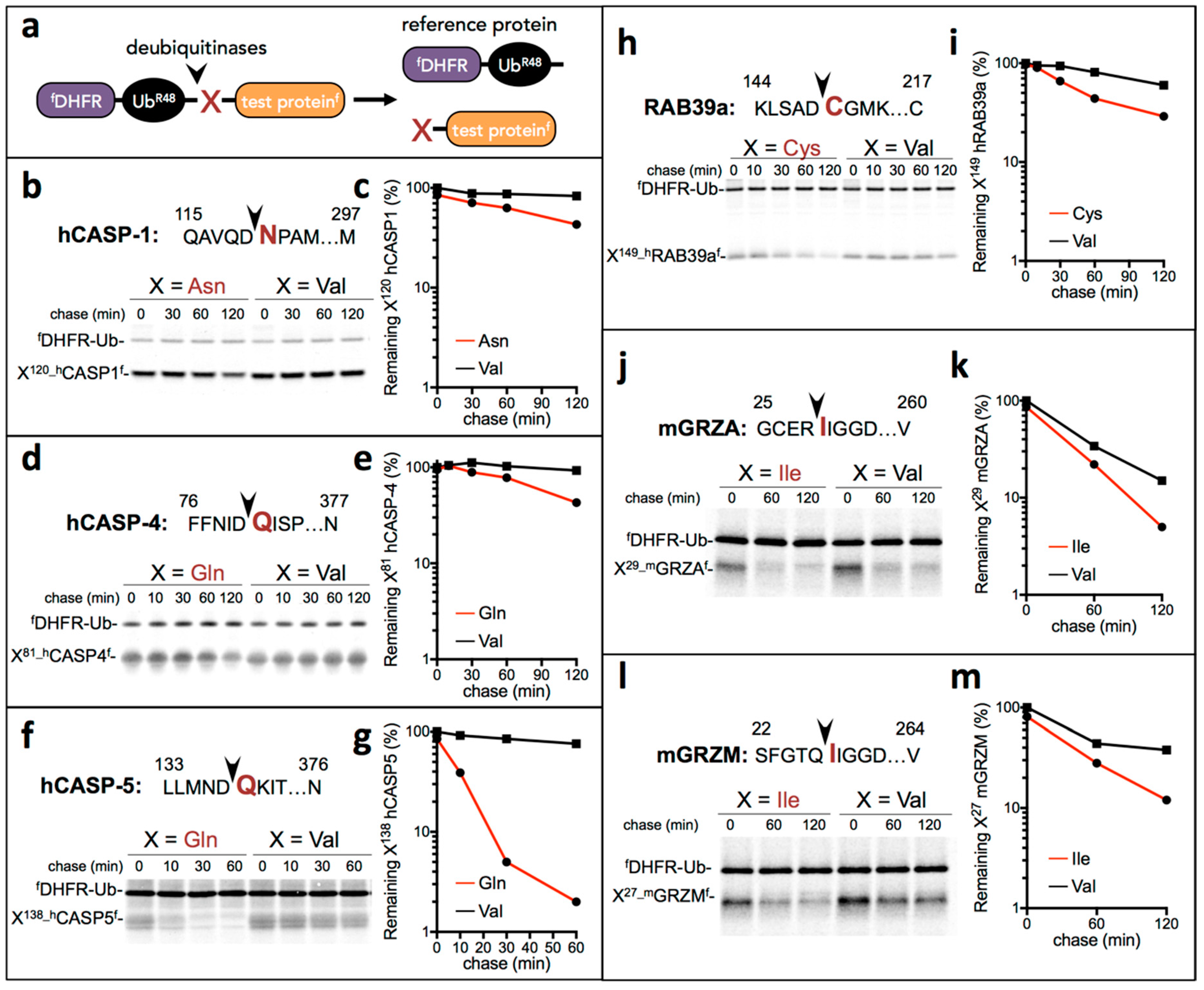
3.2. Proinflammatory Fragments Generated by Proteases are Targeted for Degradation by the Arg/N-Degron Pathway
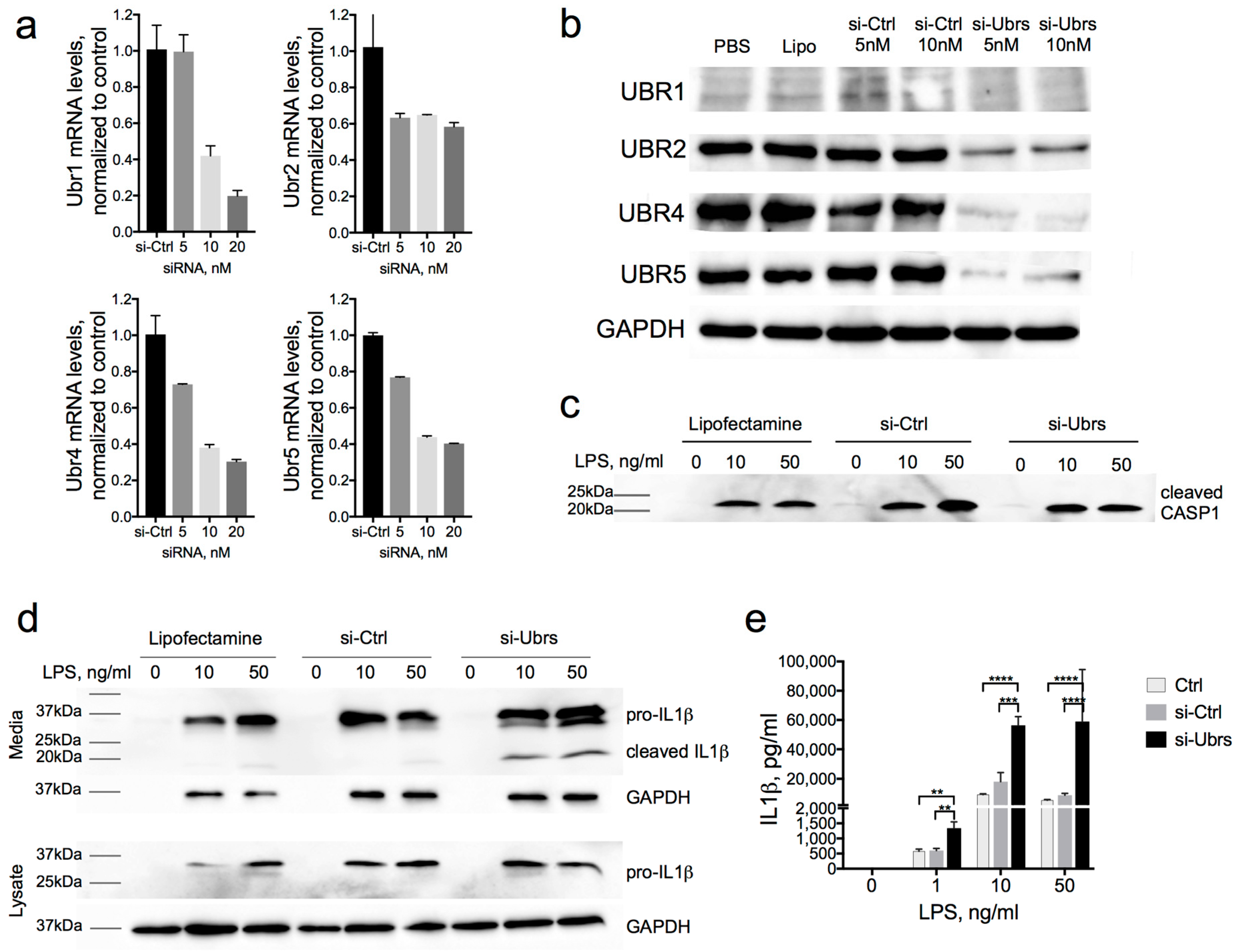
3.3. Partial Downregulation of the Arg/N-Degron Pathway Leads to an Enhanced Inflammatory Response
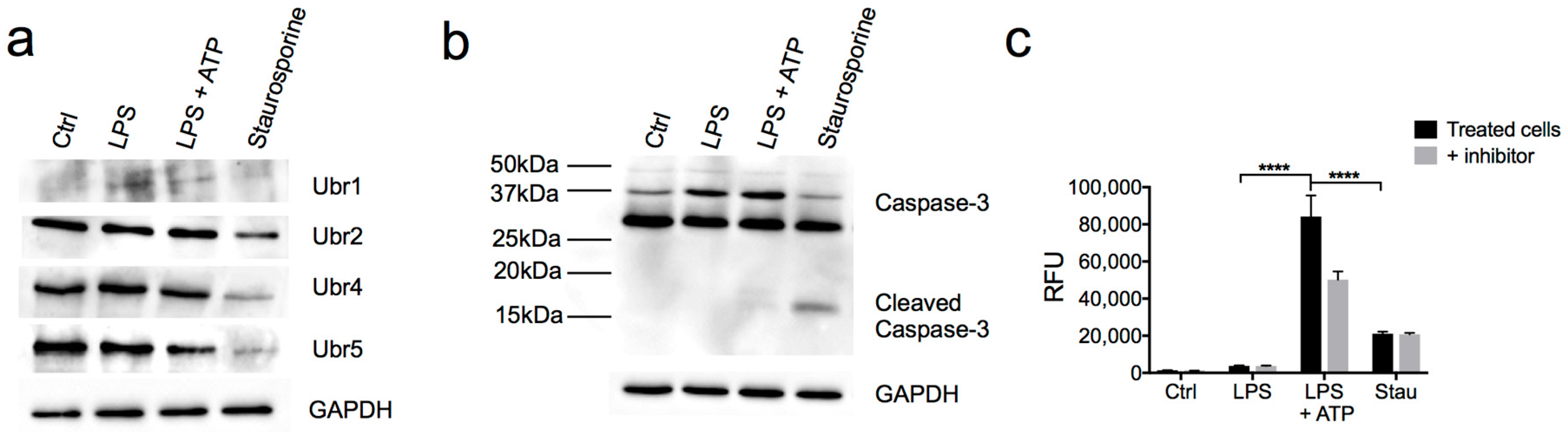
3.4. N-Recognins are not Degraded During the Inflammatory Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of Proil-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Jimenez Fernandez, D.; Lamkanfi, M. Inflammatory Caspases: Key Regulators of Inflammation and Cell Death. Biol. Chem. 2015, 396, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Lopez-Castejon, G.; Brough, D. Caspase-1: Is Il-1 Just the Tip of the Iceberg? Cell Death Dis. 2012, 3, e338. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, H.; Nanayakkara, G.; Li, Y.; Shao, Y.; Johnson, C.; Cheng, J.; Yang, W.Y.; Yang, F.; Lavallee, M.; et al. Novel Extracellular and Nuclear Caspase-1 and Inflammasomes Propagate Inflammation and Regulate Gene Expression: A Comprehensive Database Mining Study. J. Hematol. Oncol. 2016, 9, 122–140. [Google Scholar] [CrossRef]
- Hildebrand, D.; Bode, K.A.; Riess, D.; Cerny, D.; Waldhuber, A.; Rommler, F.; Strack, J.; Korten, S.; Orth, J.H.; Miethke, T.; et al. Granzyme A Produces Bioactive Il-1beta through a Nonapoptotic Inflammasome-Independent Pathway. Cell Rep. 2014, 9, 910–917. [Google Scholar] [CrossRef]
- Anthony, D.A.; Andrews, D.M.; Chow, M.; Watt, S.V.; House, C.; Akira, S.; Bird, P.I.; Trapani, J.A.; Smyth, M.J. A Role for Granzyme M in Tlr4-Driven Inflammation and Endotoxicosis. J. Immunol. 2010, 185, 1794–1803. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kB Signaling in Inflammation. Signal. Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef]
- Corn, J.E.; Vucic, D. Ubiquitin in Inflammation: The Right Linkage Makes All the Difference. Nat. Struct. Mol. Biol. 2014, 21, 297–300. [Google Scholar] [CrossRef]
- Kattah, M.G.; Malynn, B.A.; Ma, A. Ubiquitin-Modifying Enzymes and Regulation of the Inflammasome. J. Mol. Biol. 2017, 429, 3471–3485. [Google Scholar] [CrossRef]
- Wu, H. Assembly of Post-Receptor Signaling Complexes for the Tumor Necrosis Factor Receptor Superfamily. Adv. Protein Chem. 2004, 68, 225–279. [Google Scholar]
- Wertz, I.E. Tnfr1-Activated NF-kB Signal Transduction: Regulation by the Ubiquitin/Proteasome System. Curr. Opin. Chem. Biol. 2014, 23, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Luo, R.; Chen, R.; Song, L.; Zhang, S.; Hua, W.; Chen, H. Cleavage of Ikappabalpha by Calpain Induces Myocardial Nf-Kappab Activation, Tnf-Alpha Expression, and Cardiac Dysfunction in Septic Mice. Am. J. Physiol. Heart Circ. Physiol 2014, 306, H833–H843. [Google Scholar] [CrossRef] [PubMed]
- Piatkov, K.I.; Oh, J.H.; Liu, Y.; Varshavsky, A. Calpain-Generated Natural Protein Fragments as Short-Lived Substrates of the N-End Rule Pathway. Proc. Natl. Acad. Sci. USA 2014, 111, E817–E826. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Sun, S.C. Ubiquitin Signaling in Immune Responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef]
- Guillamot, M.; Ouazia, D.; Dolgalev, I.; Yeung, S.T.; Kourtis, N.; Dai, Y.; Corrigan, K.; Zea-Redondo, L.; Saraf, A.; Florens, L.; et al. The E3 Ubiquitin Ligase Spop Controls Resolution of Systemic Inflammation by Triggering Myd88 Degradation. Nat. Immunol. 2019, 20, 1196–1207. [Google Scholar] [CrossRef]
- Aki, D.; Zhang, W.; Liu, Y.C. The E3 Ligase Itch in Immune Regulation and Beyond. Immunol. Rev. 2015, 266, 6–26. [Google Scholar] [CrossRef]
- Elton, L.; Carpentier, I.; Verhelst, K.; Staal, J.; Beyaert, R. The Multifaceted Role of the E3 Ubiquitin Ligase Hoil-1: Beyond Linear Ubiquitination. Immunol. Rev. 2015, 266, 208–221. [Google Scholar] [CrossRef]
- Williams, J.J.; Munro, K.M.; Palmer, T.M. Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (Socs3) Function and Expression. Cells 2014, 3, 546–562. [Google Scholar] [CrossRef]
- Varshavsky, A. N-Degron and C-Degron Pathways of Protein Degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef]
- Rao, H.; Uhlmann, F.; Nasmyth, K.; Varshavsky, A. Degradation of a Cohesin Subunit by the N-End Rule Pathway Is Essential for Chromosome Stability. Nature 2001, 410, 955–959. [Google Scholar] [CrossRef]
- Piatkov, K.I.; Brower, C.S.; Varshavsky, A. The N-End Rule Pathway Counteracts Cell Death by Destroying Proapoptotic Protein Fragments. Proc. Natl. Acad. Sci. USA 2012, 109, E1839–E1847. [Google Scholar] [CrossRef] [PubMed]
- Justa-Schuch, D.; Silva-Garcia, M.; Pilla, E.; Engelke, M.; Kilisch, M.; Lenz, C.; Moller, U.; Nakamura, F.; Urlaub, H.; Geiss-Friedlander, R. Dpp9 Is a Novel Component of the N-End Rule Pathway Targeting the Tyrosine Kinase Syk. Elife 2016, 5, e16370. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.M.; Kwon, Y.T. The Molecular Principles of N-End Rule Recognition. Nat. Struct. Mol. Biol. 2010, 17, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Zakrzewska, A.; Dudgeon, D.D.; Jiang, Y.; Lazo, J.S.; Kwon, Y.T. The Substrate Recognition Domains of the N-End Rule Pathway. J. Biol. Chem. 2009, 284, 1884–1895. [Google Scholar] [CrossRef]
- Tasaki, T.; Mulder, L.C.F.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A Family of Mammalian E3 Ubiquitin Ligases That Contain the Ubr Box Motif and Recognize N-Degrons. Mol. Cell. Biol. 2005, 25, 7120–7136. [Google Scholar] [CrossRef]
- Varshavsky, A. The N-End Rule Pathway and Regulation by Proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Kashina, A.S.; Davydov, I.V.; Hu, R.G.; An, J.Y.; Seo, J.W.; Du, F.; Varshavsky, A. An Essential Role of N-Terminal Arginylation in Cardiovascular Development. Science 2002, 297, 96–99. [Google Scholar] [CrossRef]
- Chui, A.J.; Okondo, M.C.; Rao, S.D.; Gai, K.; Griswold, A.R.; Johnson, D.C.; Ball, D.P.; Taabazuing, C.Y.; Orth, E.L.; Vittimberga, B.A.; et al. N-Terminal Degradation Activates the Nlrp1b Inflammasome. Science 2019, 364, 82–85. [Google Scholar] [CrossRef]
- Xu, H.; Shi, J.; Gao, H.; Liu, Y.; Yang, Z.; Shao, F.; Dong, N. The N-End Rule Ubiquitin Ligase Ubr2 Mediates Nlrp1b Inflammasome Activation by Anthrax Lethal Toxin. EMBO J. 2019, 38, e101996. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Finn, R. Twenty Years of the Merops Database of Proteolytic Enzymes, Their Substrates and Inhibitors. Nucleic Acids Res. 2016, 44, D343–D350. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped Blast and Psi-Blast: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- The UniProt, Consortium. Uniprot: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Team, R Core. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Sheng, J.; Kumagai, A.; Dunphy, W.G.; Varshavsky, A. Dissection of C-Mos Degron. EMBO J. 2002, 21, 6061–6071. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. Ubiquitin Fusion Technique and Related Methods. Methods Enzymol. 2005, 399, 777–799. [Google Scholar] [PubMed]
- Leboeuf, D.; Abakumova, T.; Prikazchikova, T.; Rhym, L.; Anderson, D.G.; Zatsepin, T.S.; Piatkov, K.I. Downregulation of the Arg/N-Degron Pathway Sensitizes Cancer Cells to Chemotherapy in Vivo. Mol. Ther. 2020, 28, 1092–1104. [Google Scholar] [CrossRef]
- Crawford, E.D.; Wells, J.A. Caspase Substrates and Cellular Remodeling. Annu. Rev. Biochem. 2011, 80, 1055–1087. [Google Scholar] [CrossRef] [PubMed]
- Lévy, F.; JJohnston, A.; Varshavsky, A. Analysis of a Conditional Degradation Signal in Yeast and Mammalian Cells. Eur. J. Biochem. 1999, 259, 244–252. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 Self-Cleavage Is an Intrinsic Mechanism to Terminate Inflammasome Activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Vigano, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat. Commun. 2015, 6, 8761–8774. [Google Scholar] [CrossRef]
- Casson, C.N.; Yu, J.; Reyes, V.M.; Taschuk, F.O.; Yadav, A.; Copenhaver, A.M.; Nguyen, H.T.; Collman, R.G.; Shin, S. Human Caspase-4 Mediates Noncanonical Inflammasome Activation against Gram-Negative Bacterial Pathogens. Proc. Natl. Acad. Sci. USA 2015, 112, 6688–6693. [Google Scholar] [CrossRef]
- Lee, B.L.; Stowe, I.B.; Gupta, A.; Kornfeld, O.S.; Roose-Girma, M.; Anderson, K.; Warming, S.; Zhang, J.; Lee, W.P.; Kayagaki, N. Caspase-11 Auto-Proteolysis Is Crucial for Noncanonical Inflammasome Activation. J. Exp. Med. 2018, 215, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential Requirement for Caspase-1 Autoproteolysis in Pathogen-Induced Cell Death and Cytokine Processing. Cell Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.G.; Sheng, J.; Qi, X.; Xu, Z.; Takahashi, T.T.; Varshavsky, A. The N-End Rule Pathway as a Nitric Oxide Sensor Controlling the Levels of Multiple Regulators. Nature 2005, 437, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.W. Nitric Oxide in Immunity and Inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Becker, C.E.; Creagh, E.M.; O’Neill, L.A. Rab39a Binds Caspase-1 and Is Required for Caspase-1-Dependent Interleukin-1beta Secretion. J. Biol. Chem. 2009, 284, 34531–34537. [Google Scholar] [CrossRef]
- Monteith, A.J.; Vincent, H.A.; Kang, S.; Li, P.; Claiborne, T.M.; Rajfur, Z.; Jacobson, K.; Moorman, N.J.; Vilen, B.J. Mtorc2 Activity Disrupts Lysosome Acidification in Systemic Lupus Erythematosus by Impairing Caspase-1 Cleavage of Rab39a. J. Immunol. 2018, 201, 371–382. [Google Scholar] [CrossRef]
- Broering, R.; Real, C.I.; John, M.J.; Jahn-Hofmann, K.; Ickenstein, L.M.; Kleinehr, K.; Paul, A.; Gibbert, K.; Dittmer, U.; Gerken, G.; et al. Chemical Modifications on Sirnas Avoid Toll-Like-Receptor-Mediated Activation of the Hepatic Immune System in Vivo and in Vitro. Int. Immunol. 2014, 26, 35–46. [Google Scholar] [CrossRef]
- Pop, C.; Salvesen, G.S. Human Caspases: Activation, Specificity, and Regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies Within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Fahlman, R.P. The Anti-Apoptotic Form of Tyrosine Kinase Lyn That Is Generated by Proteolysis Is Degraded by the N-End Rule Pathway. Oncotarget 2014, 5, 2714–2722. [Google Scholar] [CrossRef]
- Xu, Z.; Payoe, R.; Fahlman, R.P. The C-Terminal Proteolytic Fragment of the Breast Cancer Susceptibility Type 1 Protein (Brca1) Is Degraded by the N-End Rule Pathway. J. Biol. Chem. 2012, 287, 7495–7502. [Google Scholar] [CrossRef] [PubMed]
- Ditzel, M.; Wilson, R.; Tenev, T.; Zachariou, A.; Paul, A.; Deas, E.; Meier, P. Degradation of Diap1 by the N-End Rule Pathway Is Essential for Regulating Apoptosis. Nat. Cell Biol. 2003, 5, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Maltby, D.; Wells, J.A. Inflammatory Stimuli Regulate Caspase Substrate Profiles. Mol. Cell Proteom. 2010, 9, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Wensink, A.C.; Hack, C.E.; Bovenschen, N. Granzymes Regulate Proinflammatory Cytokine Responses. J. Immunol. 2015, 194, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Fitzgerald, P. Just So Stories About the Evolution of Apoptosis. Curr. Biol. 2016, 26, R620–R627. [Google Scholar] [CrossRef]
- Munoz-Pinedo, C. Signaling Pathways That Regulate Life and Cell Death: Evolution of Apoptosis in the Context of Self-Defense. Adv. Exp. Med. Biol. 2012, 738, 124–143. [Google Scholar]
- Ameisen, J.C. On the Origin, Evolution, and Nature of Programmed Cell Death: A Timeline of Four Billion Years. Cell Death Differ. 2002, 9, 367–393. [Google Scholar] [CrossRef]
- Kruger, B.; Mayerle, J.; Zenker, M.; Lerch, M.M. Johanson-Blizzard Syndrome: Pathophysiology of Ubiquitin Ligase Deficiency in Pancreatic Acinar Cells. Pathology 2009, 41, 51. [Google Scholar] [CrossRef]
- Zenker, M.; Mayerle, J.; Lerch, M.M.; Tagariello, A.; Zerres, K.; Durie, P.R.; Beier, M.; Hulskamp, G.; Guzman, C.; Rehder, H.; et al. Deficiency of Ubr1, a Ubiquitin Ligase of the N-End Rule Pathway, Causes Pancreatic Dysfunction, Malformations and Mental Retardation (Johanson-Blizzard Syndrome). Nat. Genet. 2005, 37, 1345–1350. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Xia, Z.; Davydov, I.V.; Lecker, S.H.; Varshavsky, A. Construction and Analysis of Mouse Strains Lacking the Ubiquitin Ligase Ubr1 (E3a) of the N-End Rule Pathway. Mol. Cell. Biol. 2001, 21, 8007–8021. [Google Scholar] [CrossRef]
- Zenker, M.; Mayerle, J.; Reis, A.; Lerch, M.M. Genetic Basis and Pancreatic Biology of Johanson-Blizzard Syndrome. Endocrinol. Metab. Clin. 2006, 35, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Brower, C.S.; Varshavsky, A. Ablation of Arginylation in the Mouse N-End Rule Pathway: Loss of Fat, Higher Metabolic Rate, Damaged Spermatogenesis, and Neurological Perturbations. PLoS ONE 2009, 4, e7757. [Google Scholar] [CrossRef] [PubMed]
- de Marchi, R.; Sorel, M.; Mooney, B.; Fudal, I.; Goslin, K.; Kwasniewska, K.; Ryan, P.T.; Pfalz, M.; Kroymann, J.; Pollmann, S.; et al. The N-End Rule Pathway Regulates Pathogen Responses in Plants. Sci. Rep. 2016, 6, 26020. [Google Scholar] [CrossRef] [PubMed]
- Goslin, K.; Eschen-Lippold, L.; Naumann, C.; Linster, E.; Sorel, M.; Klecker, M.; de Marchi, R.; Kind, A.; Wirtz, M.; Lee, J.; et al. Differential N-End Rule Degradation of Rin4/Noi Fragments Generated by the Avrrpt2 Effector Protease. Plant Physiol. 2019, 180, 2272–2289. [Google Scholar] [CrossRef]
- Heck, J.W.; Cheung, S.K.; Hampton, R.Y. Cytoplasmic Protein Quality Control Degradation Mediated by Parallel Actions of the E3 Ubiquitin Ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA 2010, 107, 1106–1111. [Google Scholar] [CrossRef]
- Dubnikov, T.; Ben-Gedalya, T.; Cohen, E. Protein Quality Control in Health and Disease. CHS Perspect. Biol. 2017, 9, a023523. [Google Scholar] [CrossRef]
- Currais, A.; Fischer, W.; Maher, P.; Schubert, D. Intraneuronal Protein Aggregation as a Trigger for Inflammation and Neurodegeneration in the Aging Brain. FASEB J. 2017, 31, 5–10. [Google Scholar] [CrossRef]
- Brower, C.S.; Piatkov, K.I.; Varshavsky, A. Neurodegeneration-Associated Protein Fragments as Short-Lived Substrates of the N-End Rule Pathway. Mol. Cell 2013, 50, 161–171. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leboeuf, D.; Pyatkov, M.; Zatsepin, T.S.; Piatkov, K. The Arg/N-Degron Pathway—A Potential Running Back in Fine-Tuning the Inflammatory Response? Biomolecules 2020, 10, 903. https://doi.org/10.3390/biom10060903
Leboeuf D, Pyatkov M, Zatsepin TS, Piatkov K. The Arg/N-Degron Pathway—A Potential Running Back in Fine-Tuning the Inflammatory Response? Biomolecules. 2020; 10(6):903. https://doi.org/10.3390/biom10060903
Chicago/Turabian StyleLeboeuf, Dominique, Maxim Pyatkov, Timofei S. Zatsepin, and Konstantin Piatkov. 2020. "The Arg/N-Degron Pathway—A Potential Running Back in Fine-Tuning the Inflammatory Response?" Biomolecules 10, no. 6: 903. https://doi.org/10.3390/biom10060903
APA StyleLeboeuf, D., Pyatkov, M., Zatsepin, T. S., & Piatkov, K. (2020). The Arg/N-Degron Pathway—A Potential Running Back in Fine-Tuning the Inflammatory Response? Biomolecules, 10(6), 903. https://doi.org/10.3390/biom10060903