NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases


 ,
, 
 and
and 
Abstract
1. Neurodegenerative Diseases and Oxidative Stress as Common Pathological Pathway
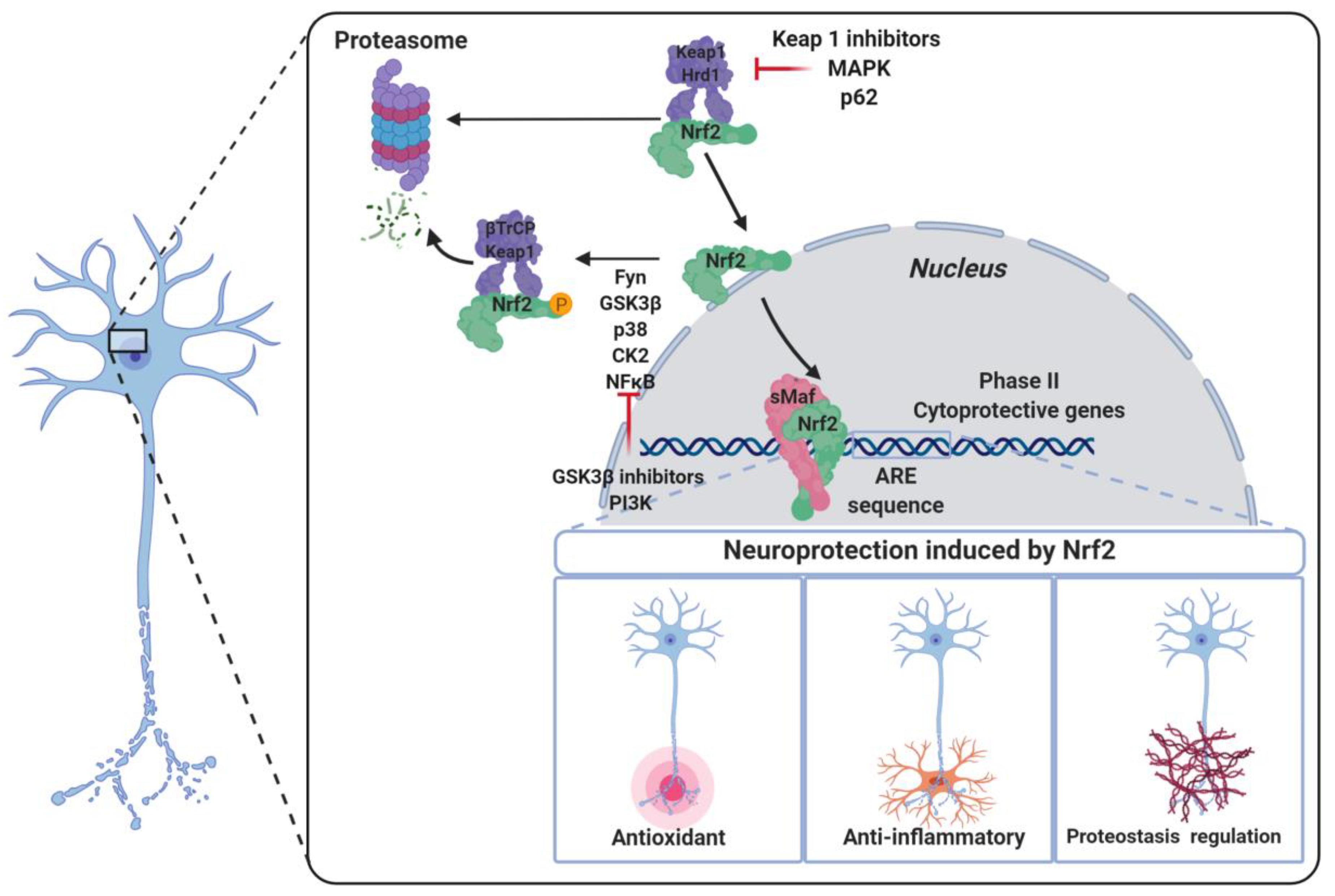
2. The NRF2–ARE Pathway
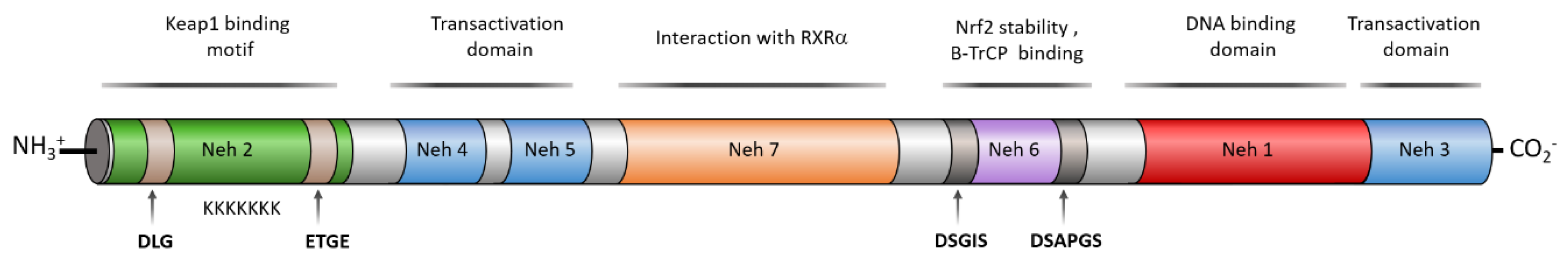
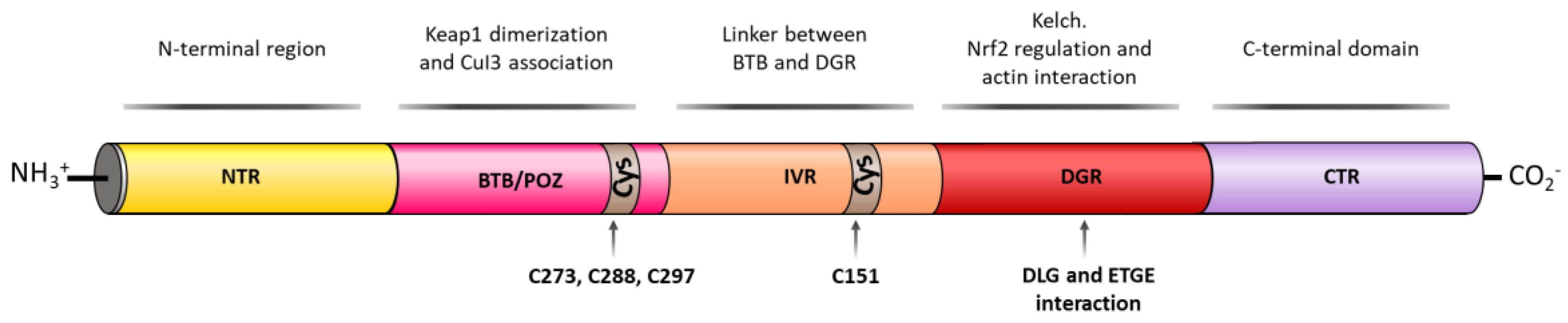
3. Structure of NRF2
4. Neurodegenerative Diseases Hallmarks and Their Crosstalk with NRF2
4.1. Oxidative Stress
4.2. Neuroinflammation
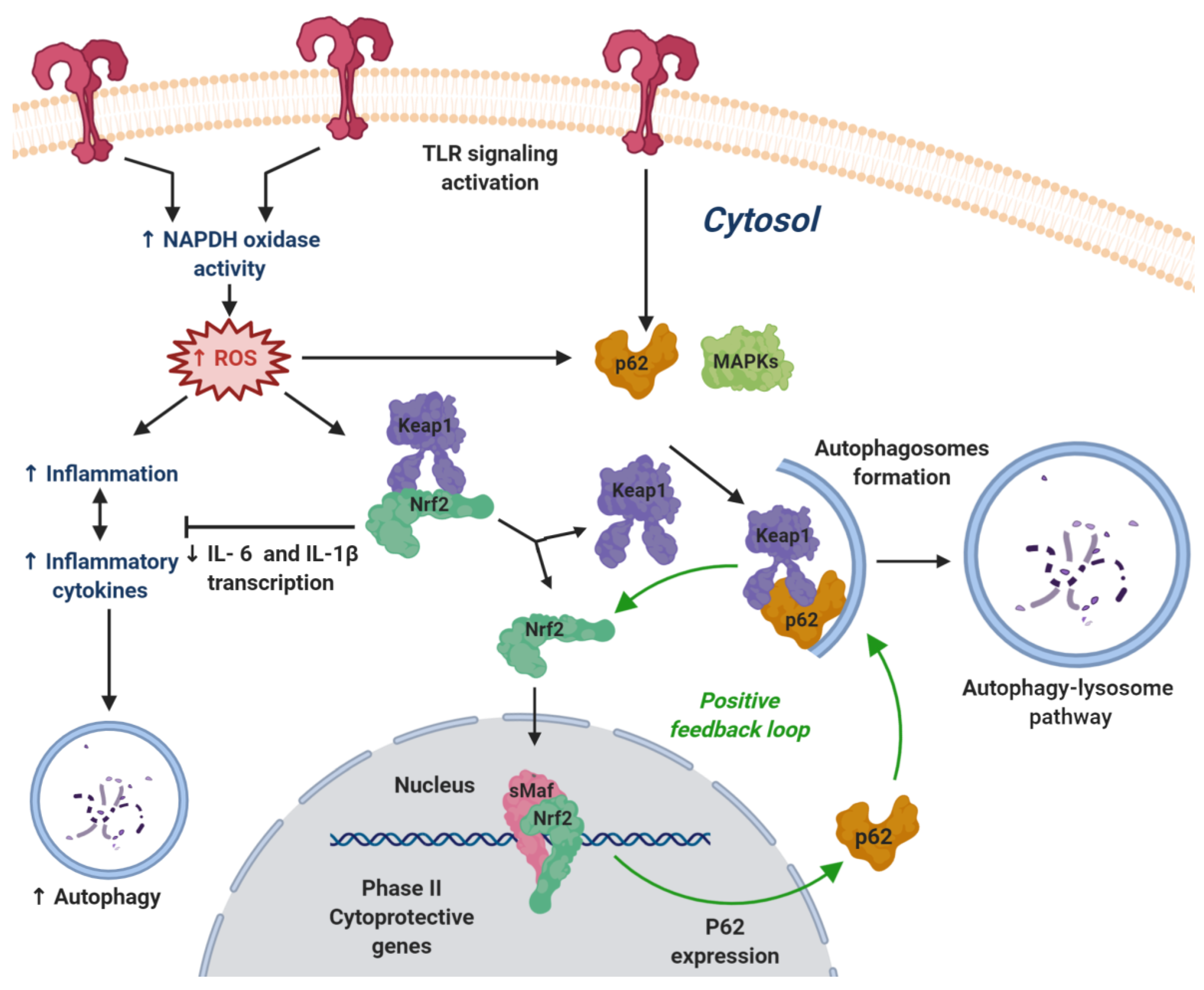
4.2.1. NRF2 and Neuroinflammation: General Aspects
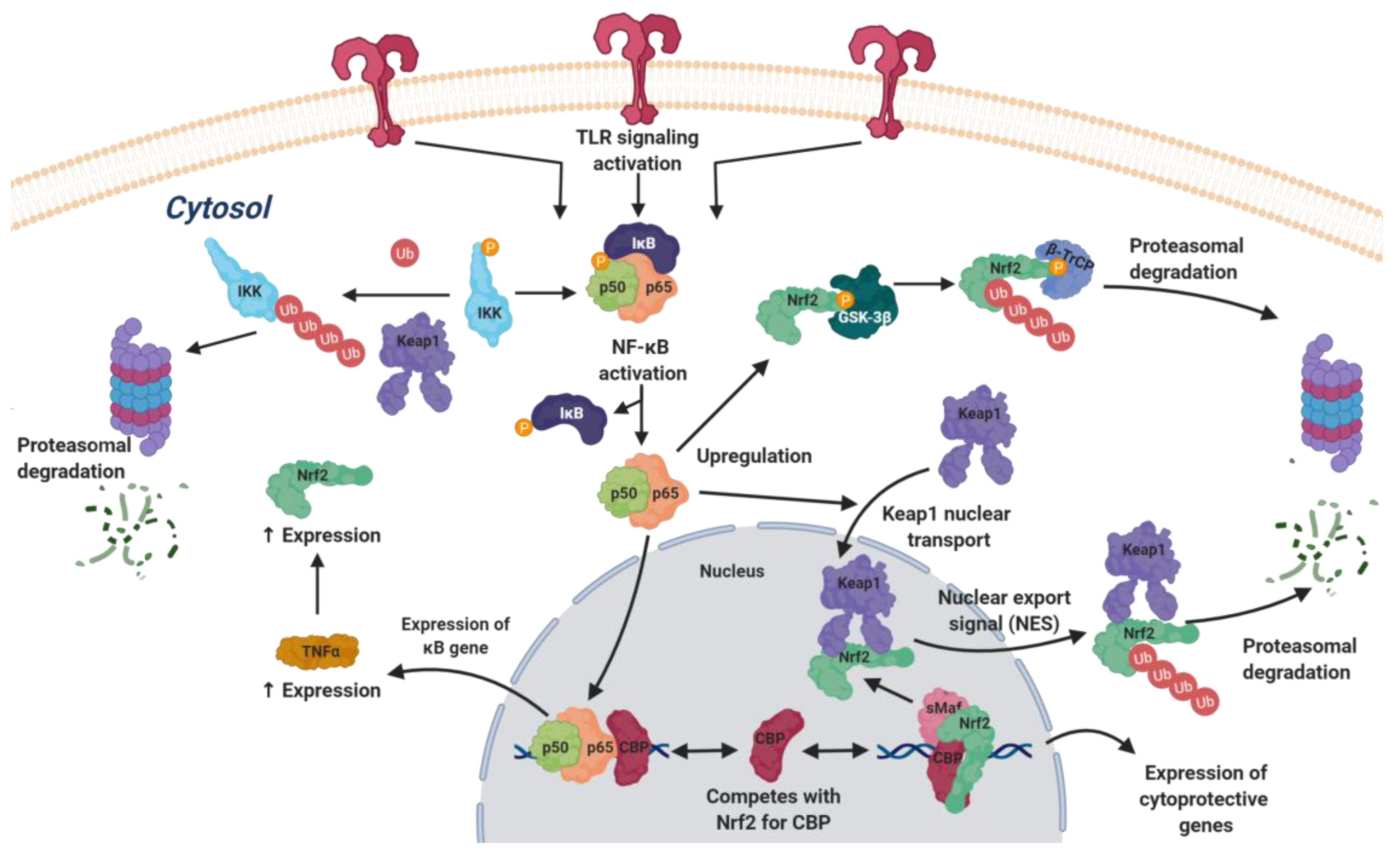
4.2.2. Nrf2 and Toll-Like Receptor Signaling
4.2.3. NRF2 and NFκB
4.3. Proteostasis
5. NRF2 Activation as a Useful Approach to Neurodegenerative Disease Therapy
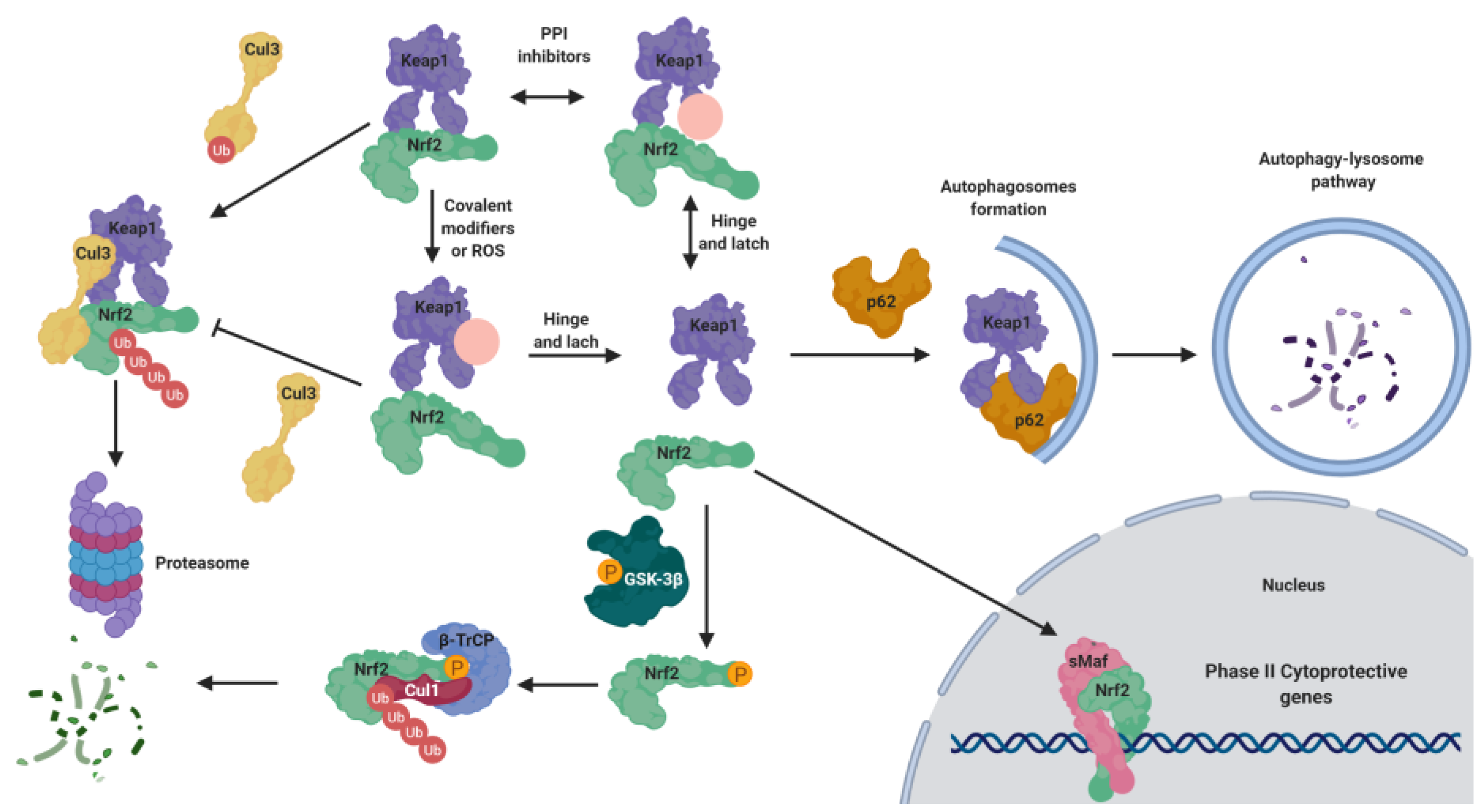
5.1. KEAP1-Dependent Regulation
5.1.1. KEAP1 as an Oxidative Stress Sensor
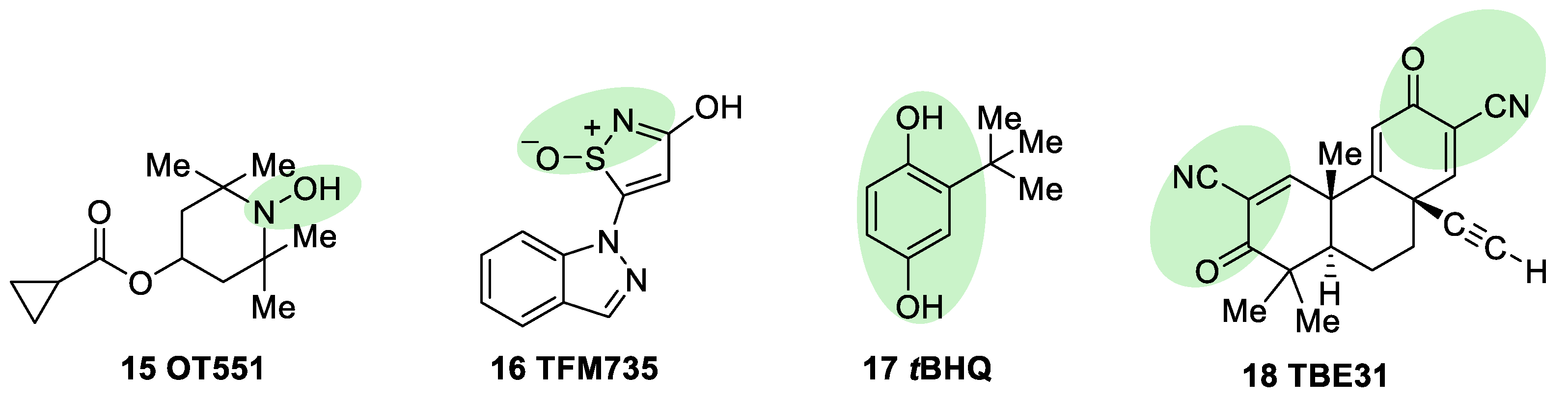
5.1.2. KEAP1 Covalent Modifiers
Natural and Semisynthetic NRF2 Activators and Their Analogues
Synthetic NRF2 Activators
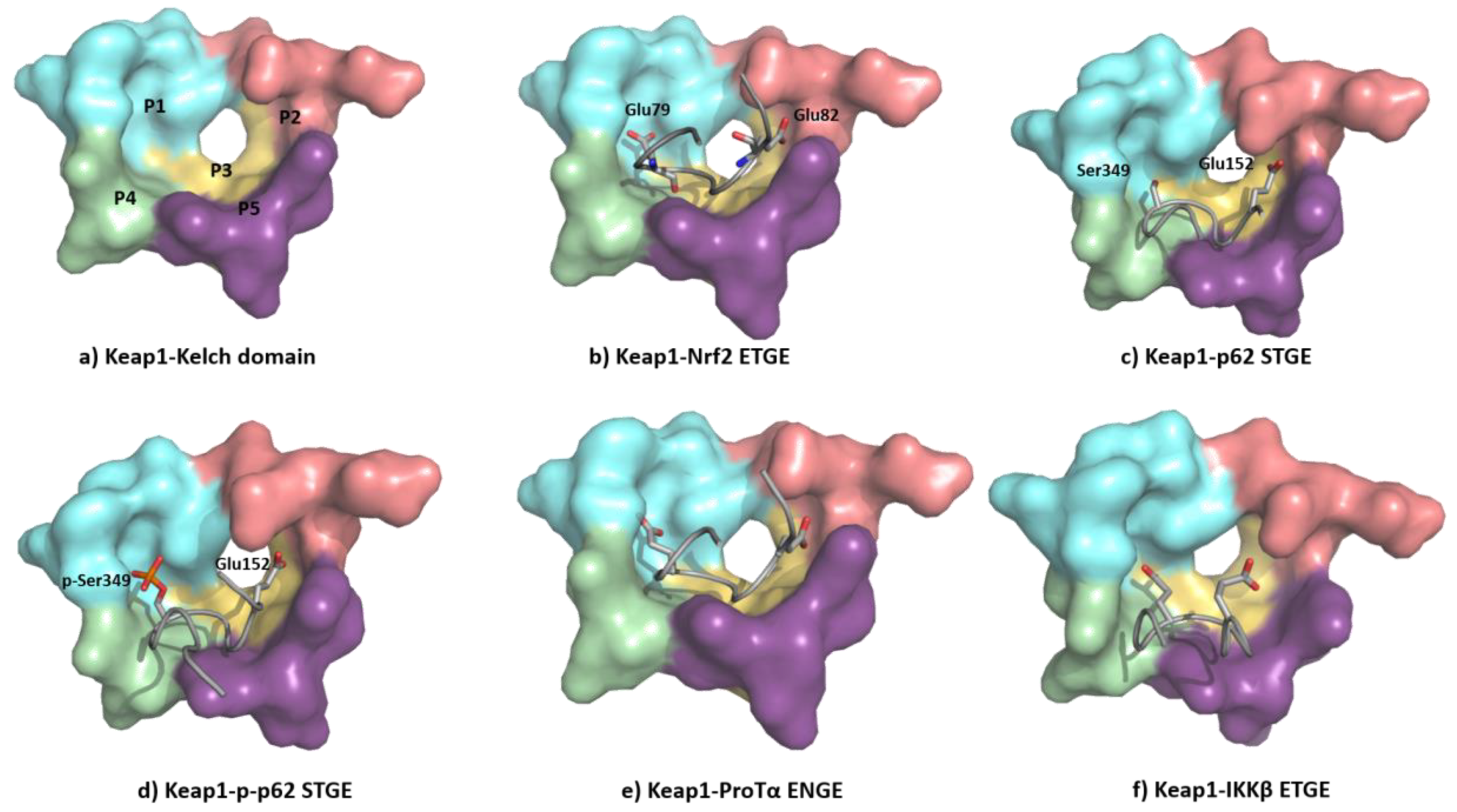
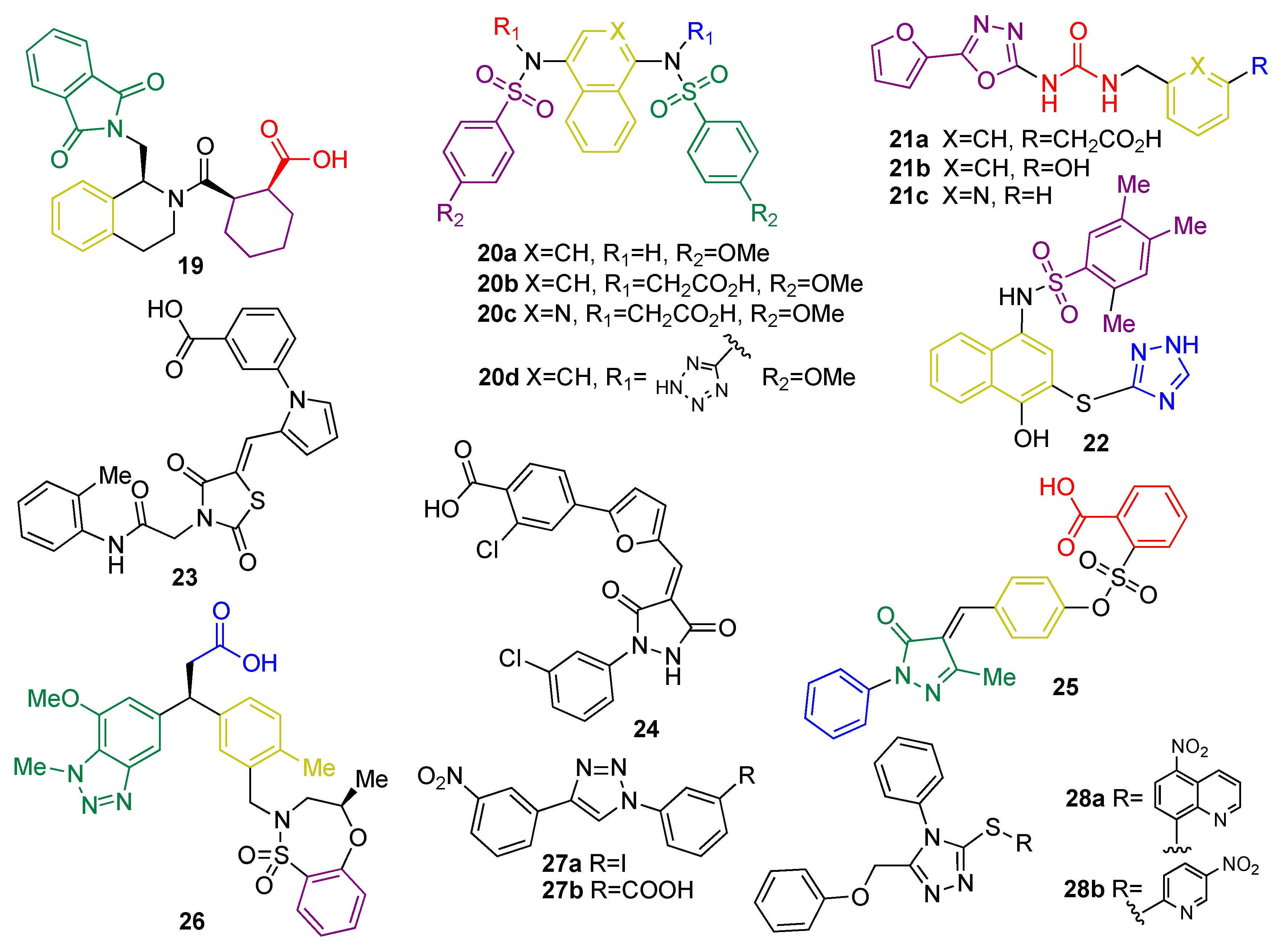
5.1.3. KEAP1 Protein–Protein Interaction Inhibitors
5.1.4. Epigenetic Control of KEAP 1 Expression
5.2. KEAP1-Independent Regulation
5.2.1. NRF2 Regulation by GSK-3β
5.2.2. KEAP1-Independent Regulation of NRF2 Stability
5.2.3. Miscellaneous ARE Transcription Regulators Connected to NRF2
5.2.4. Miscellaneous Pathways Regulating the Post-Translational Phosphorylation of NRF2
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharm. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, D.; Vidal, M.; Barabási, A.L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jia, Z.; Zhu, H. Regulation of NRF2 signaling. React. Oxyg. Species 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (NRF2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An NRF2/small maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The NRF2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. KEAP1 represses nuclear activation of antioxidant responsive elements by NRF2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The KEAP1-BTB protein is an adaptor that bridges NRF2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-KEAP1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of NRF2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by NRF2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef] [PubMed]
- Fuse, Y.; Kobayashi, M. Conservation of the KEAP1-NRF2 system: An evolutionary journey through stressful space and time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. KEAP1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of NRF2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of NRF2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the NRF2 transcription factor in a KEAP1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded NRF2 activation on phase-I and-II drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 2012, 7, e39006. [Google Scholar] [CrossRef]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor NRF2. J. Biol. Chem. 1999, 274, 33627–33636. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jiménez-Moreno, N.; García-Yague, A.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of NRF2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. NRF2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Floyd, R.A. Neuroinflammatory processes are important in neurodegenerative diseases: An hypothesis to explain the increased formation of reactive oxygen and nitrogen species as major factors involved in neurodegenerative disease development. Free Radic. Biol. Med. 1999, 26, 1346–1355. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L.; Close, D.R.; Stern, S.E. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 383–388. [Google Scholar] [CrossRef]
- Arlt, S.; Beisiegel, U.; Kontush, A. Lipid peroxidation in neurodegeneration: New insights into Alzheimer’s disease. Curr. Opin. Lipidol. 2002, 13, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in Substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging: Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef]
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive oxygen species: Physiological and physiopathological effects on synaptic plasticity. J. Exp. Neurosci. 2016, 10 (Suppl 1), 23–48. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; López, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Koppula, S.; Kim, I.S.; More, S.V.; Kim, B.W.; Choi, D.K. Nuclear factor erythroid 2-related factor 2 signaling in Parkinson disease: A promising multi therapeutic target against oxidative stress, neuroinflammation and cell death. CNS Neurol. Disord. Drug Targets 2012, 11, 1015–1029. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Guzmán-Martínez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G. Microglial M1/M2 polarization and metabolic states. Br. J. Pharm. 2016, 173, 649–665. [Google Scholar] [CrossRef]
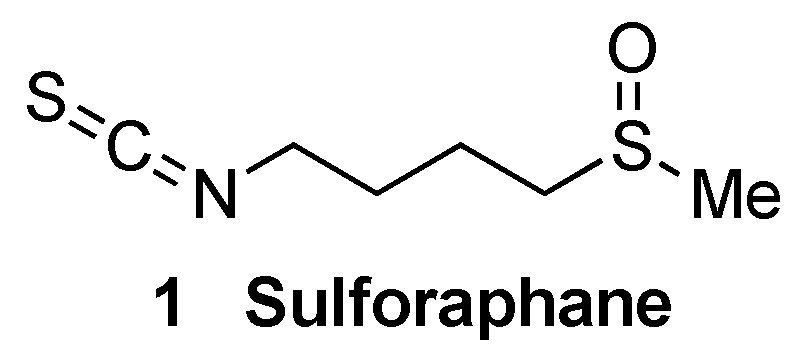
- Townsend, B.E.; Johnson, R.W. Sulforaphane induces NRF2 target genes and attenuates inflammatory gene expression in microglia from brain of young adult and aged mice. Exp. Gerontol. 2016, 73, 42–48. [Google Scholar] [CrossRef]
- Fragoulis, A.; Siegl, S.; Fendt, M.; Jansen, S.; Soppa, U.; Brandenburg, L.O.; Pufe, T.; Weis, J.; Wruck, C.J. Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol. 2017, 12, 843–853. [Google Scholar] [CrossRef]
- Serini, S.; Calviello, G. Reduction of oxidative/nitrosative stress in brain and its involvement in the neuroprotective effect of n-3 PUFA in Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 123–134. [Google Scholar] [CrossRef]
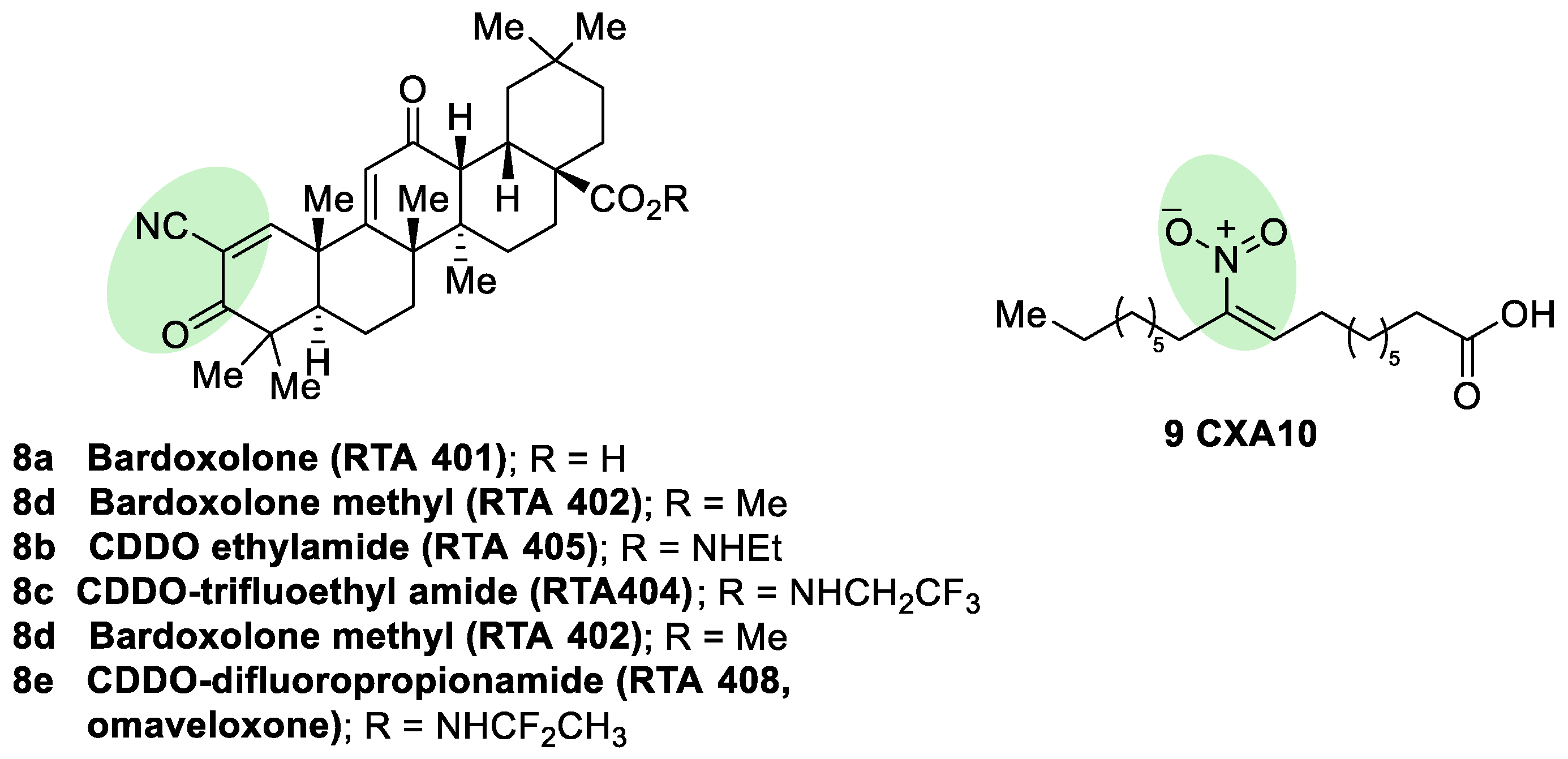
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of NRF2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Quinti, L.; Naidu, D.D.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroinmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Gupta, D. Crosstalk of toll-like receptors signaling and NRF2 pathway for regulation of inflammation. Biomed. Pharmacother. 2018, 108, 1866–1878. [Google Scholar] [CrossRef] [PubMed]
- Cong, Z.X.; Zhou, Y.; Wang, J.W.; Pan, H.; Zhang, D.D.; Zhang, L.; Zhu, L. Temozolomide and irradiation combined treatment-induced NRF2 activation increases chemoradiation sensitivity in human glioblastoma cells. J. Neurooncol. 2014, 116, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Jakaria, M.; Kim, I.-S.; Kim, J.; Ezazul Haque, M.; Choi, D.K. Regulation of toll-like receptor (TLR) signaling pathway by polyphenols in the treatment of age-linked neurodegenerative diseases: Focus on TLR4 signaling. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between NRF2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; Hodgson, R. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and-aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Marsh, A.P. Molecular mechanisms of proteinopathies across neurodegenerative disease: A review. Neurol. Res. Practice 2019, 1, 35. [Google Scholar] [CrossRef]
- Ciccocioppo, F.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Simeone, P.; Lanuti, P.; Pieragostino, D.; Del Boccio, P.; Marchisio, M.; Miscia, S. Neurodegenerative diseases as proteinopathies-driven immune disorders. Neural Regen. Res. 2020, 15, 850–856. [Google Scholar] [PubMed]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox. Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. NRF2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Kim, S.; Choi, K.J.; Cho, S.J.; Yun, S.M.; Jeon, J.P.; Koh, Y.H.; Song, J.; Johnson, G.V.W.; Jo, C. Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and NRF2 transcription factors. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L.J. Expression of Nrf2 in neurodegenerative diseases. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. NRF2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Sarlette, A.; Kramp, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rabano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Moreno-Murciano, P.; Pedraza-Chaverri, J. The transcription factor NRF2 as a new therapeutic target in Parkinson’s disease. Expert Opin. Targets 2009, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Wille, E.; Calingasan, N.Y.; Tampellini, D.; Williams, C.; Gouras, G.K.; Liby, K.; Sporn, M.; Nathan, C.; Beal, M.F.; et al. Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2009, 109, 502–512. [Google Scholar] [CrossRef]
- Kaidery, N.A.; Banerjee, R.; Yang, L.; Smirnova, N.A.; Hushpulian, D.M.; Liby, K.T.; Williams, C.R.; Yamamoto, M.; Kensler, T.W.; Ratan, R.R.; et al. Targeting NRF2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid. Redox Signal. 2013, 18, 139–157. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of NRF2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The NRF2 cell defence pathway: KEAP1-dependent and independent mechanisms of regulation. Biochem. Pharm. 2013, 85, 705–717. [Google Scholar] [CrossRef]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef]
- Snyder, G.H.; Cennerazzo, M.J.; Karalis, A.J.; Field, D. Electrostatic influence of local cysteine environments on disulfide exchange kinetics. Biochemistry 1981, 20, 6509–6519. [Google Scholar] [CrossRef]
- Holland, R.; Fishbein, J.C. Chemistry of the cysteine sensors in Kelch-like ECH-associated protein 1. Antioxid. Redox Signal. 2010, 13, 1749–1761. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of KEAP1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in KEAP1 are required for KEAP1-dependent ubiquitination of NRF2 and for stabilization of NRF2 by chemo-preventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. KEAP1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc and alkenals. Proc. Natl. Acad. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [PubMed]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of NRF2 activation. Pharm. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Gay, K.A.; Mesecar, A.D. Molecular mechanisms of natural products in chemoprevention: Induction of cytoprotective enzymes by NRF2. Mol. Nutr. Food Res. 2008, 52, 84–94. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the KEAP1 sensor modified by inducers. Proc. Natl. Acad. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the NRF2-KEAP1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef]
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y.; You, Q.D. The KEAP1-NRF2-ARE pathway as a potential preventive and therapeutic target: An update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the NRF2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic. Biol. Med. 2016, 99, 544–556. [Google Scholar] [CrossRef]
- Hur, W.; Gray, N.S. Small molecule modulators of antioxidant response pathway. Curr. Opin. Chem. Biol. 2011, 15, 162–173. [Google Scholar] [CrossRef]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. NRF2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent modification at Cys151 dissociates the electrophile sensor KEAP1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of three major cysteine sensors of KEAP1 in stress response. Mol. Cell. Biol. 2015, 36, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent advances of natural polyphenols activators for KEAP1-NRF2 signaling pathway. Chem. Biodivers. 2019, 16, e1900400. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ishii, T.; Akagawa, M.; Nakamura, Y.; Nakayama, T. Covalent binding of tea catechins to protein thiols: The relationship between stability and electrophilic reactivity. Biosci. Biotechnol. Biochem. 2010, 74, 2451–2456. [Google Scholar] [CrossRef]
- Yang, G.Z.; Wang, Z.J.; Bai, F.; Qin, X.J.; Cao, J.; Lv, J.Y.; Zhang, M.S. Epigallocatechin-3-gallate protects HUVECs from PM2.5-induced oxidative stress injury by activating critical antioxidant pathways. Molecules 2015, 20, 6626–6639. [Google Scholar] [CrossRef]
- Ma, L.; Cao, T.T.; Kandpal, G.; Warren, L.; Fred Hess, J.; Seabrook, G.R.; Ray, W.J. Genome-wide microarray analysis of the differential neuroprotective effects of antioxidants in neuroblastoma cells overexpressing the familial Parkinson’s disease alpha-synuclein A53T mutation. Neurochem. Res. 2010, 35, 130–142. [Google Scholar] [CrossRef]
- Itoh, T.; Tabuchi, M.; Mizuguchi, N.; Imano, M.; Tsubaki, M.; Nishida, S.; Hashimoto, S.; Matsuo, K.; Nakayama, T.; Ito, A.; et al. Neuroprotective effect of (-)-epigallocatechin-3-gallate in rats when administered pre- or post-traumatic brain injury. J. Neural. Transm. 2013, 120, 767–783. [Google Scholar] [CrossRef]
- Xu, Q.; Kanthasamy, A.G.; Reddy, M.B. Epigallocatechin gallate protects against TNFalpha-or H2O2-induced apoptosis by modulating iron related proteins in a cell culture model. Int. J. Vitam. Nutr. Res. 2018, 88, 158–165. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P.; Sharkey, J.; Zhang, Y.; Holtzclaw, W.D.; Wang, X.J.; David, E.; Schiavoni, K.H.; Finlayson, S.; Mierke, D.F.; et al. An exceptionally potent inducer of cytoprotective enzymes: Elucidation of the structural features that determine inducer potency and reactivity with KEAP1. J. Biol. Chem. 2010, 285, 33747–33755. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate NRF2 through inhibition of ubiquitination activity of KEAP1. Mol Cell Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Sashwati, R.; Packer, L. Fas mediated apoptosis of human Jurkat T-cells: Intracellular events and potentiation by redox-active alpha-lipoic acid. Cell Death Differ. 1999, 6, 481–491. [Google Scholar] [CrossRef]
- Chaudhary, P.; Marracci, G.; Galipeau, D.; Pocius, E.; Morris, B.; Bourdette, D. Lipoic acid reduces inflammation in a mouse focal cortical experimental autoimmune encephalomyelitis model. J Neuroimmunol. 2015, 289, 68–74. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, H.; Zhao, X.; Liu, L.; Zhang, H.; Xuan, M.; Guo, Z.; Wang, H.; Liu, C. Neurochemical effects of the R form of alpha-lipoic acid and its neuroprotective mechanism in cellular models of Parkinson’s disease. Int. J. Biochem. Cell. Biol. 2017, 87, 86–94. [Google Scholar] [CrossRef]
- Agarwal, N.B.; Jain, S.; Agarwal, N.K.; Mediratta, P.K.; Sharma, K.K. Modulation of pentylenetetrazole-induced kindling and oxidative stress by curcumin in mice. Phytomedicine 2011, 18, 756–759. [Google Scholar] [CrossRef]
- Dong, W.; Yang, B.; Wang, L.; Li, B.; Guo, X.; Zhang, M.; Jiang, Z.; Fu, J.; Pi, J.; Guan, D.; et al. Curcumin plays neuroprotective roles against traumatic brain injury partly via NRF2 signaling. Toxicol. Appl. Pharm. 2018, 346, 28–36. [Google Scholar] [CrossRef]
- Rao, M.N.A. Nitric oxide scavenging by curcuminoids. J. Pharm. Pharm. 1997, 49, 105–107. [Google Scholar]
- Ren, L.; Zhan, P.; Wang, Q.; Wang, C.; Liu, Y.; Yu, Z.; Zhang, S. Curcumin upregulates the NRF2 system by repressing inflammatory signaling-mediated KEAP1 expression in insulin-resistant conditions. Biochem. Biophys. Res. Commun. 2019, 514, 691–698. [Google Scholar] [CrossRef]
- Wang, B.F.; Cui, Z.W.; Zhong, Z.H.; Sun, Y.H.; Sun, Q.F.; Yang, G.Y.; Bian, L.G. Curcumin attenuates brain edema in mice with intracerebral hemorrhage through inhibition of AQP4 and AQP9 expression. Acta Pharm. Sin. 2015, 36, 939–948. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, C.; Zhao, S.; Yuan, D.; Lian, G.; Wang, X.; Wang, L.; Yang, J. Demethoxycurcumin, a natural derivative of curcumin attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia induced by lipopolysaccharide. Int. Immunopharmacol. 2010, 10, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1091–1100. [Google Scholar]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the KEAP1-NRF2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, Y.; Zhang, G.; Fang, H.; Wang, H.; Zang, H.; Xie, T.; Wang, W. Activation of NRF2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 2014, 1544, 54–61. [Google Scholar] [CrossRef]
- Duran, C.G.; Burbank, A.J.; Mills, K.H.; Duckworth, H.R.; Aleman, M.M.; Kesic, M.J.; Peden, D.B.; Pan, Y.; Zhou, H.; Hernandez, M.L. A proof-of-concept clinical study examining the NRF2 activator sulforaphane against neutrophilic airway inflammation. Respir. Res. 2016, 17, 89. [Google Scholar] [CrossRef]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and other nutrigenomic NRF2 activators: Can the clinician’s expectation be matched by the reality? Oxid. Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef]
- Park, H.M.; Kim, J.A.; Kwak, M.K. Protection against amyloid beta cytotoxicity by sulforaphane: Role of the proteasome. Arch. Pharmacal Res. 2009, 32, 109–115. [Google Scholar] [CrossRef]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y.S. Amelioration of Alzheimer’s disease by neuropro-tective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef]
- Han, J.M.; Lee, Y.J.; Lee, S.Y.; Kim, E.M.; Moon, Y.; Kim, H.W.; Hwang, O. Protective effect of sulfo-raphane against dopaminergic cell death. Pharm. L Exp. 2007, 321, 249–256. [Google Scholar] [CrossRef]
- Buendia, I.; Navarro, E.; Michalska, P.; Gameiro, I.; Egea, J.; Abril, S.; López, A.; González-Lafuente, L.; López, M.G.; León, R. New melatonin-cinnamate hybrids as multi-target drugs for neurodegenerative diseases: NRF2-induction, antioxidant effect and neuroprotection. Future Med. Chem. 2015, 7, 1961–1969. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, J.; Lin, H.; Gu, K.; Feng, F. Recent progress in the development of small molecule NRF2 modulators: A patent review (2012–2016). Expert Opin. Pat. 2017, 27, 763–785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Bardoxolone brings NRF2-based therapies to light. Antioxid. Redox Signal. 2013, 19, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.A.; O’Donnell, V.B.; Schopfer, F.J. The discovery of nitro fatty acids as products of metabolic and inflammatory reactions and mediators of adaptive cell signaling. Nitric Oxide 2018, 77, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Deen, A.J.; Sihvola, V.; Härkönen, J.; Patinen, T.; Adinolfi, S.; Levonen, A.L. Regulation of stress signaling pathways by nitro-fatty acids. Nitric Oxide 2018, 78, 170–175. [Google Scholar] [CrossRef]
- Baker, L.M.; Baker, P.R.; Golin-Bisello, F.; Schopfer, F.J.; Fink, M.; Woodcock, S.R.; Branchaud, B.P.; Radi, R.; Freeman, B.A. Nitro- fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J. Biol. Chem. 2007, 282, 31085–31093. [Google Scholar]
- Schopfer, F.J.; Vitturi, D.A.; Jorkasky, D.K.; Freeman, B.A. Nitro-fatty acids: New drug candidates for chronic inflammatory and fibrotic diseases. Nitric Oxide 2018, 79, 31–37. [Google Scholar] [CrossRef]
- Hoxtermann, S.; Nuchel, C.; Altmeyer, P. Fumaric acid esters suppress peripheral CD4-and CD8-positive lymphocytes in psoriasis. Dermatology 1998, 196, 223–230. [Google Scholar] [CrossRef]
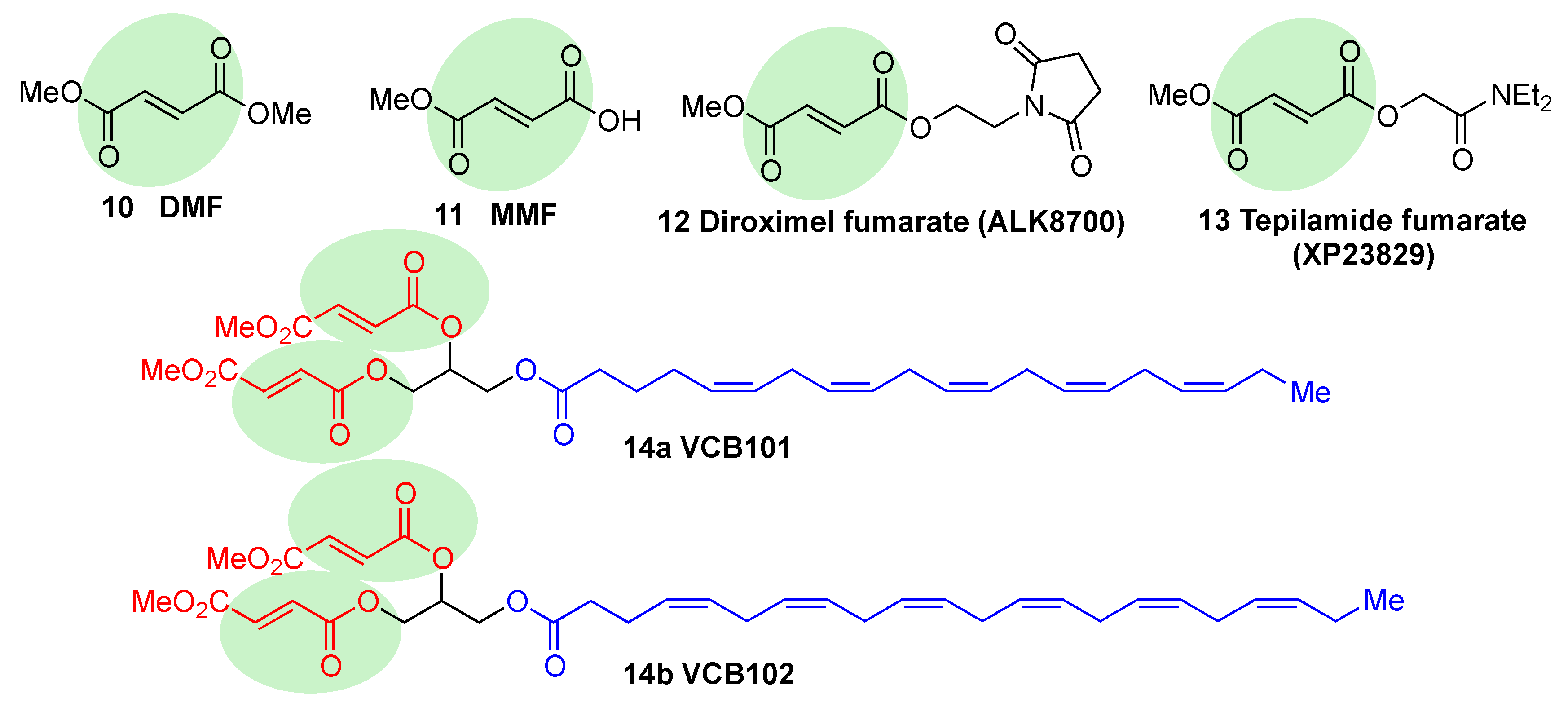
- Deeks, E.D. Dimethyl fumarate: A review in relapsing-remitting MS. Drugs 2016, 76, 243–254. [Google Scholar] [CrossRef]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef]
- Muller, S.; Behnen, M.; Bieber, K.; Moller, S.; Hellberg, L.; Witte, M.; Hansel, M.; Zillikens, D.; Solbach, W.; Laskay, T.; et al. Dimethyl fumarate impairs neutrophil functions. J. Invest. Dermatol. 2016, 136, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Majkutewicz, I.; Kurowska, E.; Podlacha, M.; Myslinska, D.; Grembecka, B.; Rucinski, J.; Pierzynowska, K.; Wrona, D. Age-dependent effects of dimethyl fumarate on cognitive and neuropathological features in the streptozotocin-induced rat model of Alzheimer’s disease. Brain Res. 2018, 1686, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Kunze, R.; Urrutia, A.; Hoffmann, A.; Liu, H.; Helluy, X.; Pham, M.; Reischl, S.; Korff, T.; Marti, H.H. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood-brain barrier integrity. Exp. Neurol. 2015, 266, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [CrossRef]
- Bahn, G.; Jo, D.G. Therapeutic approaches to Alzheimer’s disease through modulation of NRF2. Neuromolecular Med. 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the NRF2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Zeidan, T.A.; Duncan, S.; Hencken, C.P.; Wynn, T.A.; Sanrame, C.N. Prodrugs of Fumarates and Their Use in Treating Various Diseases. US8669281B1, 3 November 2014. [Google Scholar]
- Zarling, J.A.; Brunt, V.E.; Vallerga, A.K.; Li, W.; Tao, A.; Zarling, D.A.; Minson, C.T. Nitroxide pharmaceutical development for age-related degeneration and disease. Front. Genet. 2015, 6, 00325. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Pisano, B.; Dugo, L.; Ianaro, A.; Patel, N.S.; Caputi, A.P.; Thiemermann, C. Tempol reduces the activation of nuclear factor-kappaB in acute inflammation. Free. Radic. Res. 2004, 38, 813–819. [Google Scholar] [CrossRef]
- Greenwald, B.Y.M.; Anzi, S.; Ben Sasson, S.; Bianco-Peled, H.; Kohen, R. Can nitroxides evoke the KEAP1-Nrf2-ARE pathway in skin? Free Rad. Med. Biol. 2014, 77, 258–279. [Google Scholar]
- Hirotsu, Y.; Katsuoka, F.; Itoh, K.; Yamamoto, M. NRF2 degron- fused reporter system: A new tool for specific evaluation of NRF2 inducers. Genes Cells 2011, 16, 406–415. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor NRF2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Sukumari-Ramesh, S.; Alleyne, C.H., Jr. Post-injury administration of tert-butylhydroquinone attenuates acute neurological injury after intracerebral hemorrhage in mice. J. Mol. Neurosci. 2016, 58, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel NRF2-inducer prevents mitochondrial defects and oxidative stress in Friedreich’s ataxia models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Lu, M.C.; You, Q.D. Discovery and development of Kelch-like ECH-associated protein 1. Nuclear factor erythroid 2-related factor 2 (KEAP1: NRF2) protein-protein interaction inhibitors: Achievements, challenges, and future directions. J. Med. Chem. 2016, 59, 10837–10858. [Google Scholar] [CrossRef]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the KEAP1: NRF2 interface provides mechanistic insight into NRF2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, Z.; Zhou, Y.; Xiao, Q.; Wang, H.; Peng, Y. Emerging substrate proteins of Kelch-like ECH associated protein 1 (KEAP1) and potential challenges for the development of small-molecule inhibitors of the KEAP1-nuclear factor erythroid 2-related factor 2 (NRF2) protein-protein interaction. J. Med. Chem. 2020, 63. [Google Scholar] [CrossRef]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, E.; Park, B.K. Physical and functional interaction of sequestosome 1 with KEAP1 regulates the KEAP1-NRF2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the KEAP1-NRF2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Lin, X.; Li, S.; Zhao, Y.; Ma, X.; Zhang, K.; He, X.; Wang, Z. Interaction domains of p62: A bridge between p62 and selective autophagy. DNA Cell Biol. 2013, 32, 220–227. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the KEAP1-NRF2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Nakamura, Y.; Yokoyama, S. Structural analysis of the complex of KEAP1 with a prothymosin α peptide. Acta Cryst. F 2008, 64, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Halder, S.K.; Matsunaga, H.; Sasaki, K.; Maeda, S. Neuroprotective impact of prothymosin alpha-derived hexapeptide against retinal ischemia-reperfusion. Neuroscience 2016, 318, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Karapetian, R.N.; Evstafieva, A.G.; Abaeva, I.S.; Chichkova, N.V.; Filonov, G.S.; Rubtsov, Y.P.; Sukhacheva, E.A.; Melnikov, S.V.; Schneider, U.; Wanker, E.E.; et al. Nuclear oncoprotein prothymosin α is a partner of KEAP1: Implications for expression of oxidative stress-protecting genes. Mol. Cell. Biol. 2005, 25, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa B kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-kappa B signaling by KEAP1 regulation of IKKβ activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Chu, H.X.; Xi, M.Y.; Yang, T.T.; Jia, J.M.; Huang, J.J.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Insight into the intermolecular recognition mechanism between KEAP1 and IKKβ combining homology modelling, protein-protein docking, molecular dynamics simulations and virtual alanine mutation. PLoS ONE 2013, 8, e75076. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-κB signaling by targeting IKKβ. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef]
- Wells, G. Peptide and small molecule inhibitors of the Keap1-Nrf2 protein-protein interaction. Biochem. Soc. Trans. 2015, 43, 674–679. [Google Scholar] [CrossRef]
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of KEAP1-NRF2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043. [Google Scholar] [CrossRef]
- Ontoria, J.M.; Biancofiore, I.; Fezzardi, P.; Ferrigno, F.; Torrente, E.; Colarusso, S.; Bianchi, E.; Andreini, M.; Patsilinakos, A.; Kempf, G.; et al. Combined peptide and small-molecule approach towards non-acidic THIQ inhibitors of the KEAP1/NRF2 interaction. ACS Med. Chem. Lett. 2020. [Google Scholar] [CrossRef]
- Jain, A.D.; Potteti, H.; Richardson, B.G.; Kingsley, L.; Luciano, J.P.; Ryuzoji, A.F.; Lee, H.; Krunic, A.; Mesecar, A.D.; Reddy, S.P.; et al. Probing the structural requirements of non-electrophilic naphthalene-based NRF2 activators. Eur. J. Med. Chem. 2015, 103, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.G.; Jain, A.D.; Potteti, H.R.; Lazzara, P.R.; David, B.P.; Tamatam, C.R.; Choma, E.; Skowron, K.; Dye, K.; Siddiqui, Z.; et al. Replacement of a naphthalene scaffold in Kelch-like ECHAssociated protein 1 (KEAP1)/nuclear factor (erythroid-derived 2)-like 2 (NRF2) inhibitors. J. Med. Chem. 2018, 61, 8029–8047. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Tan, S.J.; Ji, J.A.; Chen, Z.Y.; Yuan, Z.W.; You, Q.D.; Jiang, Z.Y. Polar recognition group study of KEAP1-NRF2 protein-protein interaction inhibitors. ACS Med. Chem. Lett. 2016, 7, 835–840. [Google Scholar] [CrossRef]
- Shimozono, R.; Asaoka, Y.; Yoshizawa, Y.; Aoki, T.; Noda, H.; Yamada, M.; Kaino, M.; Mochizuki, H. NRF2 activators attenuate the progression of nonalcoholic steatohepatitis-related fibrosis in a dietary rat model. Mol. Pharm. 2013, 84, 62–70. [Google Scholar] [CrossRef]
- Zhuang, C.L.; Narayanapillai, S.; Zhang, W.N.; Sham, Y.Y.; Xing, C.G. Rapid identification of KEAP1-NRF2 small-molecule inhibitors through structure based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef]
- Kim, S.; Viswanath, A.N.I.; Park, J.H.; Lee, H.E.; Park, A.Y.; Choi, J.W.; Kim, H.J.; Londhe, A.M.; Jang, B.K.; Lee, J.; et al. NRF2 activator via interference of NRF2-KEAP1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 2020, 167, 107989. [Google Scholar] [CrossRef]
- Davies, T.G.; Wixted, W.E.; Coyle, J.E.; Griffiths-Jones, C.; Hearn, K.; McMenamin, R.; Norton, D.; Rich, S.J.; Richardson, C.; Saxty, G.; et al. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: Nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 2016, 59, 3991–4006. [Google Scholar] [CrossRef]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, synthesis, and evaluation of triazole derivatives that induce NRF2 dependent gene products and inhibit the KEAP1-NRF2. J. Med. Chem. 2015, 58, 7186–7194. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M.; Abagyan, R.; Casale, M. Small Molecule Activators of NRF2 Pathway. WO2014197818 A2, 3 November 2014. [Google Scholar]
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2-and NRF2-targeting thiazole-containing compound with therapeutic activity in Huntington’s disease models. Cell. Chem. Biol. 2016, 23, 849–861. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, F.; Gertz, K.; Kronenberg, G.; Harms, C.; Fink, K.B.; Meisel, A.; Endres, M. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp. Neurol. 2008, 210, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Pancani, T.; Formentini, L.; Mascagni, P.; Fossati, G.; Leoni, F.; Moroni, F.; Chiarugi, A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharm. 2006, 70, 1876–1884. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor NRF2 and protects against cerebral ischemic damage. Free Radic Biol Med. 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. NRF2 pathway in age-related neurological disorders: Insights into microRNAs. Cell. Physiol. Biochem. 2018, 47, 1951–1976. [Google Scholar] [CrossRef]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates NRF2 pathway by targeting KEAP1 expression. Free Radic. Biol. Med. 2015, 89, 548–556. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. GSK-3β acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed]
- Velichkova, M.; Hasson, T. KEAP1 regulates the oxidation-sensitive shuttling of NRF2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol. Cell. Biol. 2005, 25, 4501–4513. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of NRF2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef]
- Shaw, M.; Cohen, P. Role of protein kinase B and the MAP kinase cascade in mediating the EGF-dependent inhibition of glycogen synthase kinase 3 in Swiss 3T3 cells. Febs. Lett. 1999, 461, 120–124. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704. [Google Scholar] [CrossRef]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK-3β inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Martínez, A. GSK-3 Inhibitors: Preclinical and clinical focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium promotes longevity through GSK-3/NRF2-dependent hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef]
- Hu, B.; Wu, Y.; Liu, J.; Shen, X.; Tong, F.; Xu, G.; Shen, R. GSK-3β inhibitor induces expression ofNRF2/TrxR2 signaling pathway to protect against renal ischemia/reperfusion injury in diabetic rats. Kidney Blood Press. Res. 2016, 41, 937–946. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Armagan, G.; Sevgili, E.; Gurkan, F.T.; Kose, F.A.; Bilgic, T.; Dagci, T.; Saso, L. Regulation of the NRF2 pathway by glycogen synthase kinase-3β in MPP(+)-induced cell damage. Molecules 2019, 24, 1377. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, P.; Qiao, Y.; Ge, Y.; Wang, Y.; Quan, S.; Yao, R.; Zhuang, S.; Wang, L.J.; Du, Y.; et al. Genetic and pharmacologic targeting of glycogen synthase kinase 3β reinforces the NRF2 antioxidant defense against podocytopathy. J. Am. Soc. Nephrol. 2016, 27, 2289–2308. [Google Scholar] [CrossRef]
- Pang, T.; Wang, Y.J.; Gao, Y.X.; Xu, Y.; Li, Q.; Zhou, Y.B.; Xu, L.; Huang, Z.J.; Liao, H.; Zhang, L.Y.; et al. A novel GSK-3β inhibitor YQ138 prevents neuronal injury induced by glutamate and brain ischemia through activation of the NRF2 signaling pathway. Acta Pharm. Sin. 2016, 37, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, T.; Wang, H.; Jiang, Y.; Peng, S. Obacunone attenuates high glucose-induced oxidative damage in NRK-52E cells by inhibiting the activity of GSK-3β. Biochem. Biophys. Res. Commun. 2019, 513, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Hong, L.; Tian, Y.; Yin, C.; Zhu, C.; Feng, H. Corilagin alleviates acetaminophen-induced hepatotoxicity via enhancing the AMPK/GSK-3β-NRF2 signaling pathway. Cell. Commun. Signal. 2019, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Martín, D.; Rojo, A.I.; Salinas, M.; Díaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef]
- Pitha-Rowe, I.; Liby, K.; Royce, D.; Sporn, M. Synthetic triterpenoids attenuate cytotoxic retinal injury: Cross-talk between Nrf2 and PI3K/AKT signaling through inhibition of the lipid phosphatase PTEN. Invest. Ophthalmol. Vis. Sci. 2009, 50, 5339–5347. [Google Scholar] [CrossRef]
- Shearn, C.T.; Smathers, R.L.; Backos, D.S.; Reigan, P.; Orlicky, D.J.; Petersen, D.R. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic. Biol. Med. 2013, 65, 680–692. [Google Scholar] [CrossRef]
- Worby, C.A.; Dixon, J.E. Pten. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Goode, N.; Hughes, K.; Woodgett, J.R.; Parker, P.J. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J. Biol. Chem. 1992, 267, 16878–16882. [Google Scholar]
- Cohen, P.; Frame, S. The renaissance of GSK-3. Nat. Rev. Mol. Cell. Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Bayascas, J.R. PDK1: The major transducer of PI 3-kinase actions. Curr. Top. Microbiol. Immunol. 2010, 346, 9–29. [Google Scholar]
- Hayes, J.D.; Ebisine, K.; Sharma, R.S.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Regulation of the CNC-bZIP transcription factor Nrf2 by KEAP1 and the axis between GSK-3 and β-TrCP. Curr. Opin. Toxicol. 2016, 1, 92–103. [Google Scholar] [CrossRef]
- Fuchs, S.Y.; Spiegelman, V.S.; Suresh Kumar, K.G. The many faces of β-TrCP E3 ubiquitin ligases: Reflections in the magic mirror of cancer. Oncogene 2004, 23, 2028–2036. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Robertson, H.; Hayes, J.D.; Sutherland, C. A partnership with the proteasome; The destructive nature of GSK-3. Biochem. Pharm. 2018, 147, 77–92. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of NRF2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Peters, M.; Jang, Y.; Shi, W.; Pintilie, M.; Fletcher, G.C.; DeLuca, C.; Liepa, J.; Zhou, L.; Snow, B.; et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 2005, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Nakamura, T.; Cho, S.J.; Han, X.; Holland, E.A.; Qu, J.; Petsko, G.A.; Yates, J.R.; Liddington, R.C.; Lipton, S.A. Transnitrosylation from DJ-1 to PTEN attenuates neuronal cell death in Parkinson’s disease models. J. Neurosci. 2014, 34, 15123–15131. [Google Scholar] [CrossRef] [PubMed]
- Juricek, L.; Coumoul, X. The aryl hydrocarbon receptor and the nervous system. Int. J. Mol. Sci. 2018, 19, 2504. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef]
- Dietrich, C. Antioxidants functions of the aryl hydrocarbon receptor. Stem. Cells Int. 2016, 2016, 10. [Google Scholar] [CrossRef]
- Szybińska, A.; Leśniak, W. P53 dysfunction in neurodegenerative diseases-The cause or effect of pathological changes? Aging Dis. 2017, 8, 506–518. [Google Scholar] [CrossRef]
- Hiemstra, S.; Niemeijer, M.; Koedoot, E.; Wink, S.; Klip, J.E.; Vlasveld, M.; de Zeeuw, E.; van Os, B.; White, W.; van de Water, B. Comprehensive landscape of NRF2 and p53 pathway activation dynamics by oxidative stress and DNA damage. Chem. Res. Toxicol. 2017, 30, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between NRF2 and p21(Cip1/WAF1) upregulates the NRF2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jiang, T.; Wang, H.; Tao, S.; Lau, A.; Fang, D.; Zhang, D.D. Does NRF2 contribute to p53-mediated control of cell survival and death? Antioxid. Redox Sign. 2012, 17, 1670–1675. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, S.; Zhang, C.; Kong, A.-N.T. Epigenetic regulation of KEAP1-NRF2 signaling. Free Radical Biol. Med. 2015, 88, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Zawia, N.; Lahiri, D.K.; Cardozo-Peláez, F. Epigenetics, oxidative stress, and Alzheimer’s disease. Free Radical Biol. Med. 2009, 46, 1241–1249. [Google Scholar] [CrossRef]
- Cao, H.; Wang, L.; Chen, B.; Zheng, P.; He, Y.; Ding, Y.; Deng, Y.; Lu, X.; Guo, X.; Zhang, Y.; et al. DNA demethylation upregulated NRF2 expression in Alzheimer’s disease cellular model. Front. Aging Neurosci. 2016, 7, 244. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, J.; Chang, N. Epigenetic modification of NRF2 by sulforaphane increases the antioxidative and anti-inflammatory capacity in a cellular model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 824, 1–10. [Google Scholar] [CrossRef]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of NRF2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef]
- Ba, Q.; Cui, C.; Wen, L.; Feng, S.; Zhou, J.; Yang, K. Schisandrin B shows neuroprotective effect in 6-OHDA-induced Parkinson’s disease via inhibiting the negative modulation of miR-34a on NRF2 pathway. Biomed Pharm. 2015, 75, 165–172. [Google Scholar] [CrossRef]
- Wang, P.; Liang, X.; Lu, Y.; Zhao, X.; Liang, J. MicroRNA-93 downregulation ameliorates cerebral ischemic injury through the NRF2/HO-1 defense pathway. Neurochem. Res. 2016, 41, 2627–2635. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, H.; Wang, R.; Wang, P.; Tao, Z.; Gao, L.; Yan, F.; Liu, X.; Yu, S.; Ji, X.; et al. MicroRNA-424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke 2015, 46, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Zhang, D.D. Non-canonical activation of NRF2: New insights and its relevance to disease. Curr. Pathobiol. Rep. 2017, 5, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Fao, L.; Mota, S.I.; Rego, A.C. Shaping the NRF2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing. Res. Rev. 2019, 54, 100942. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lei, W.; Mandlekar, S.; Weber, M.J.; Der, C.J.; Wu, J.; Kong, A.N. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J. Biol. Chem. 1999, 274, 27545–27552. [Google Scholar] [CrossRef]
- Turjanski, A.G.; Vaque, J.P.; Gutkind, J.S. MAP kinases and the control of nuclear events. Oncogene 2007, 26, 3240–3253. [Google Scholar] [CrossRef]
- Ainbinder, E.; Bergelson, S.; Pinkus, R.; Daniel, V. Regulatory mechanisms involved in activator-protein-1 (AP-1)-mediated activation of glutathione-S-transferase gene expression by chemical agents. Eur. J. Biochem. 1997, 243, 49–57. [Google Scholar] [CrossRef]
- Fao, L.; Mota, S.I.; Rego, A.C. c-Src regulates NRF2 activity through PKCdelta after oxidant stimulus. Biochim. Biophys. Acta. Mol. Cell. Res. 2019, 1866, 686–698. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef]
- Yu, R.; Mandlekar, S.; Lei, W.; Fahl, W.E.; Tan, T.H.; Kong, A.N. P38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J. Biol. Chem. 2000, 275, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of NRF2 at multiple sites by MAP kinases has a limited contribution in modulating the NRF2-dependent antioxidant response. PLoS ONE 2009, 4, e6588. [Google Scholar] [CrossRef]
- Prikas, E.; Poljak, A.; Ittner, A. Mapping p38α mitogen-activated protein kinase signaling by proximity-dependent labeling. Protein Sci. 2020, 29, 1196–1210. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox. Signal 2006, 8, 1775–1789. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef]
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405. [Google Scholar] [CrossRef]
- Keum, Y.S.; Yu, S.; Chang, P.P.; Yuan, X.; Kim, J.H.; Xu, C.; Han, J.; Agarwal, A.; Kong, A.N. Mechanism of action of sulforaphane: Inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006, 66, 8804–8813. [Google Scholar] [CrossRef]
- McNally, S.J.; Harrison, E.M.; Ross, J.A.; Garden, O.J.; Wigmore, S.J. Curcumin induces heme oxygenase 1 through generation of reactive oxygen species, p38 activation and phosphatase inhibition. Int. Int. J. Mol. Med. 2007, 19, 165–172. [Google Scholar]
- Rojo, A.I.; Medina-Campos, O.N.; Rada, P.; Zúñiga-Toala, A.; López-Gazcón, A.; Espada, S.; Pedraza-Chaverri, J.; Cuadrado, A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a KEAP1-independent regulation of NRF2 stability: Role of glycogen synthase kinase-3. Free Radic. Biol. Med. 2012, 52, 473–487. [Google Scholar] [CrossRef]
- Sabapathy, K. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 145–169. [Google Scholar]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell. Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Kawasaki, H.; Nishina, H. Diverse roles of JNK and MKK pathways in the brain. J. Signal Transduct. 2012, 459265. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Wang, X.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Zhu, X. c-Jun phosphorylation in Alzheimer’s disease. J. Neurosci. Res. 2007, 85, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- White, L.R.; Toft, M.; Kvam, S.N.; Farrer, M.J.; Aasly, J.O. MAPK-pathway activity, Lrrk2 G2019S, and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Mitsios, N.; Gaffney, J.; Krupinski, J.; Mathias, R.; Wang, Q.; Hayward, S.; Rubio, F.; Kumar, P.; Kumar, S.; Slevin, M. Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem. Biophys. 2007, 47, 73–86. [Google Scholar] [CrossRef]
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, K.; Zhang, J.; Hai, Y.; Wang, P.; Wang, H.; Liu, Q.; Wong, C.C.; Yao, J.; Gao, Y.; et al. JNK phosphorylates the Neh6 domain of NRF2 and downregulates cytoprotective genes in acetaminophen-induced liver injury. Hepatology 2020, 71, 1787–1801. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances NRF2-mediated transcriptional activation of the antioxidant response element. Degradation of NRF2 by the 26 S proteasome. Degradation of NRF2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar]
- Zipper, L.M.; Mulcahy, R.T. Inhibition of ERK and p38 MAP kinases inhibits binding of NRF2 and induction of GCS genes. Biochem. Biophys. Res. Commun. 2000, 278, 484–492. [Google Scholar] [CrossRef]
- Yang, S.Y.; Pyo, M.C.; Nam, M.H.; Lee, K.W. ERK/NRF2 pathway activation by caffeic acid in HepG2 cells alleviates its hepatocellular damage caused by t-butylhydroperoxide-induced oxidative stress. BMC Complement. Altern. Med. 2019, 19, 139. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Kim, K.S.; Ko, W.; Li, B.; Jeong, G.S.; Jang, J.H.; Oh, H.; Kim, Y.C. The cytoprotective effect of sulfuretin against tert-butyl hydroperoxide-induced hepatotoxicity through NRF2/ARE and JNK/ERK MAPK-mediated heme oxygenase-1 expression. Int. J. Mol. Sci. 2014, 15, 8863–8877. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, T.; Makiyama, A.; Samukawa, Y.; Jiang, S.; Yamashita, Y.; Ashida, H. A physiological concentration of luteolin induces phase II drug-metabolizing enzymes through the ERK1/2 signaling pathway in HepG2 cells. Arch. Biochem. Biophys. 2019, 663, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Xu, C.; Pan, Z.; Keum, Y.S.; Kim, J.H.; Shen, G.; Yu, S.; Oo, K.T.; Ma, J.; Kong, A.N. Butylated hydroxyanisole regulates ARE-mediated gene expression via NRF2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850. [Google Scholar] [CrossRef]
- Mitani, T.; Yoshioka, Y.; Furuyashiki, T.; Yamashita, Y.; Shirai, Y.; Ashida, H. Enzymatically synthesized glycogen inhibits colitis through decreasing oxidative stress. Free Radic. Biol. Med. 2017, 106, 355–367. [Google Scholar] [CrossRef]
- Steinberg, S.F. Mechanisms for redox-regulation of protein kinase C. Front. Pharm. 2015, 6, 128. [Google Scholar] [CrossRef]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of NRF2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of NRF2 at Ser40 by protein kinase C in response to antioxidants leads to the release of NRF2 from INRF2, but is not required for NRF2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar]
- Fang, X.; Yu, S.; Tanyi, J.L.; Lu, Y.; Woodgett, J.R.; Mills, G.B. Convergence of multiple signaling cascades at glycogen synthase kinase 3: EDG receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol. Cell. Biol. 2002, 22, 2099–2110. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Pisano, M.R.; Friedman, E. Attenuated protein kinase C activity and translocation in Alzheimer’s disease brain. Neurobiol. Aging 1994, 15, 293–298. [Google Scholar] [CrossRef]
- Talman, V.; Pascale, A.; Jantti, M.; Amadio, M.; Tuominen, R.K. Protein Kinase C Activation as a potential therapeutic strategy in Alzheimer’s disease: Is there a role for embryonic lethal abnormal vision-like proteins? Basic Clin. Pharm. Toxicol. 2016, 119, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human NRF2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic. Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of NRF2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of NRF2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. Phosphorylation of tyrosine 568 controls nuclear export of NRF2. J. Biol. Chem. 2006, 281, 12132–12142. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Disease | Clinical Trial | Ref |
|---|---|---|---|
Pioglitazone  | Type II diabetes | Approved | |
| Several diseases | 300 clinical trials | -- | |
Tideglusib  | AD | Phase II | NCT01350362 |
| Congenital myotonic dystrophy | Phase II/III | NCT03692312 | |
| Autism spectrum disorders | Phase II | NCT02586935 | |
| Progressive supranuclear palsy | - | NCT01049399 | |
TDZD-8 | AD | Preclinical evaluation | |
SB216763 | AD | Preclinical evaluation | |
YQ138 | Brain ischemia | Preclinical evaluation | |
Obacunone  | Natural product positioned toward several diseases | Preclinical evaluation | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cores, Á.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules 2020, 10, 904. https://doi.org/10.3390/biom10060904
Cores Á, Piquero M, Villacampa M, León R, Menéndez JC. NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules. 2020; 10(6):904. https://doi.org/10.3390/biom10060904
Chicago/Turabian StyleCores, Ángel, Marta Piquero, Mercedes Villacampa, Rafael León, and J. Carlos Menéndez. 2020. "NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases" Biomolecules 10, no. 6: 904. https://doi.org/10.3390/biom10060904
APA StyleCores, Á., Piquero, M., Villacampa, M., León, R., & Menéndez, J. C. (2020). NRF2 Regulation Processes as a Source of Potential Drug Targets against Neurodegenerative Diseases. Biomolecules, 10(6), 904. https://doi.org/10.3390/biom10060904