Ubiquitomics: An Overview and Future


Abstract
:1. Ubiquitous and Complex
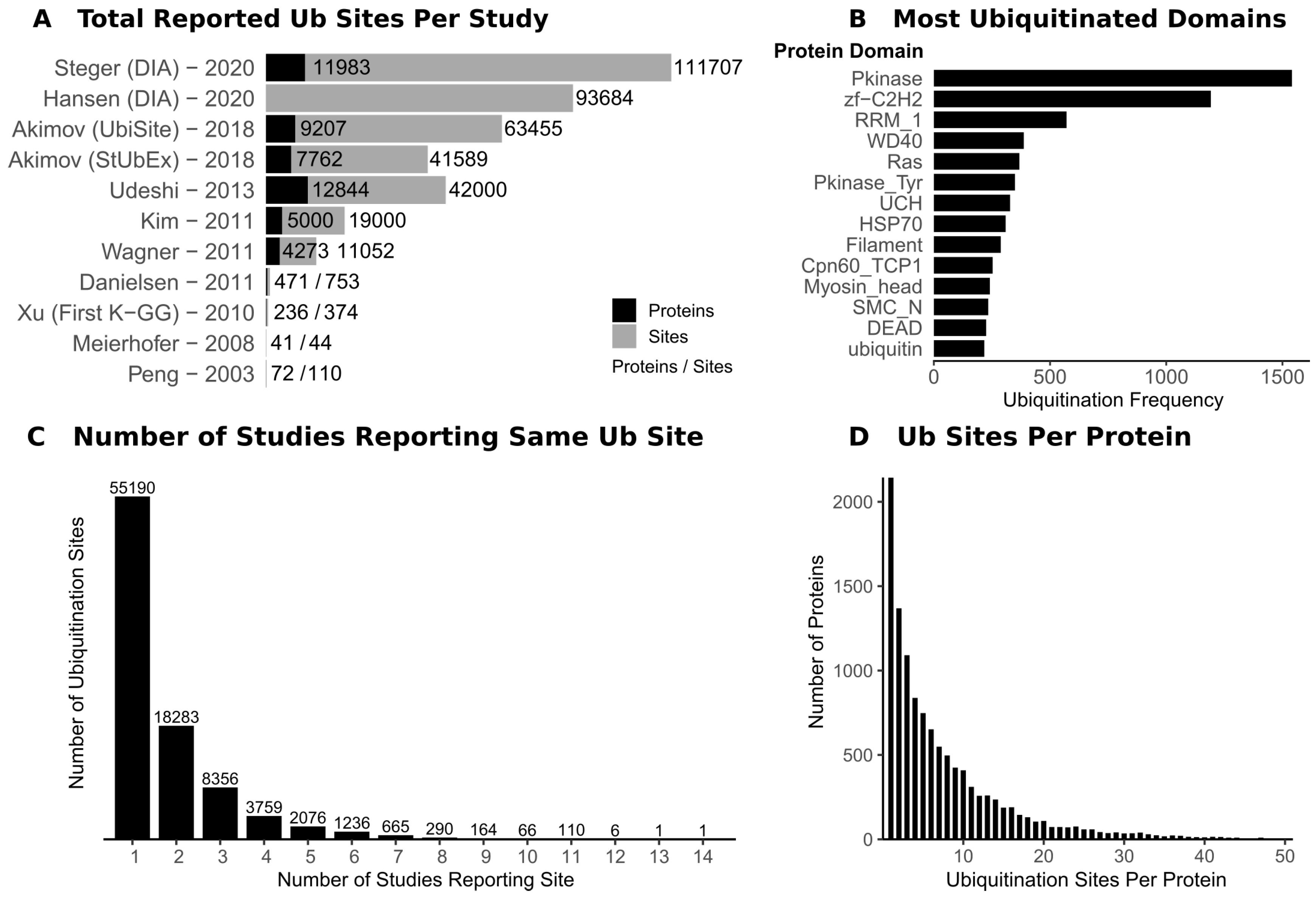
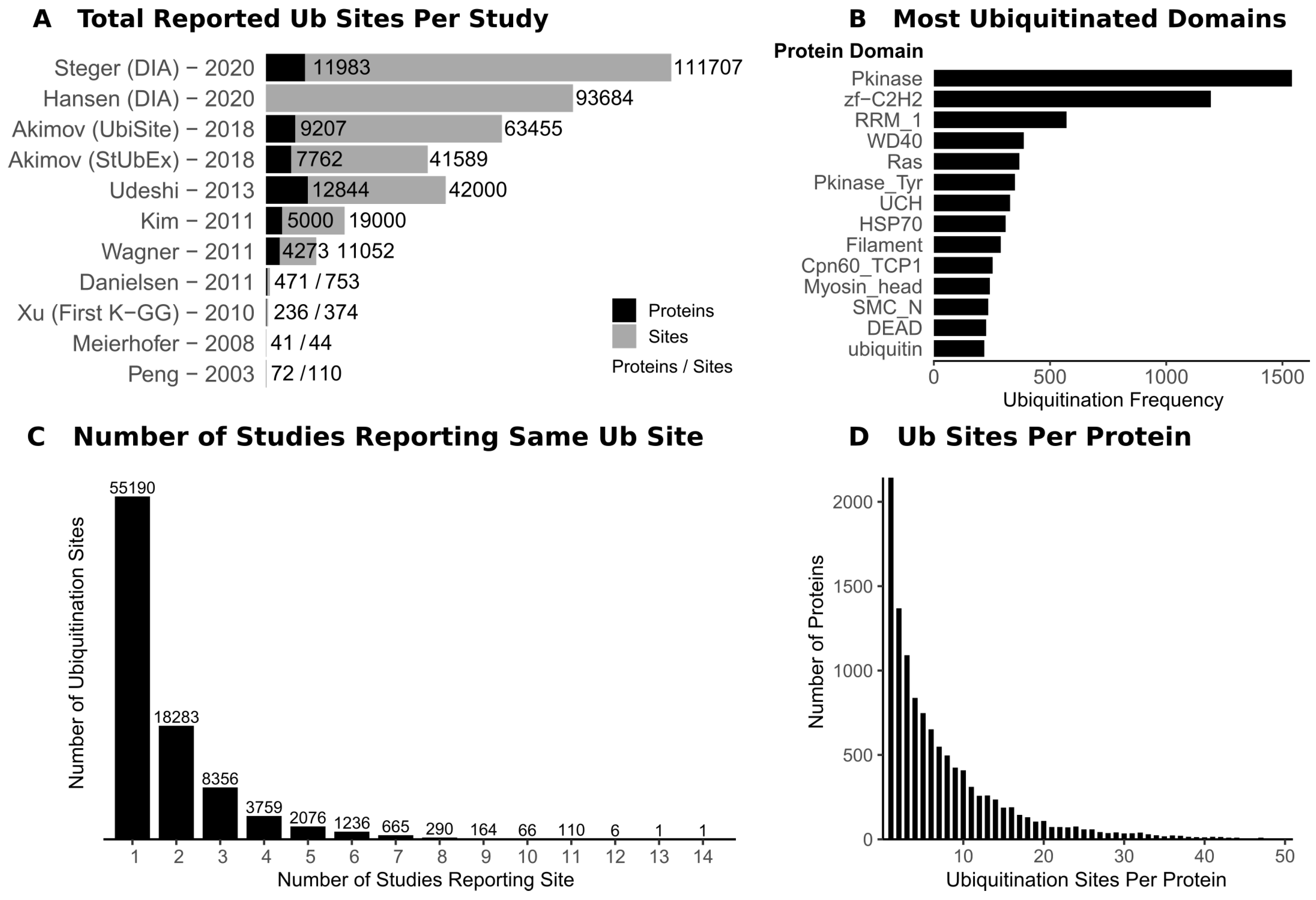
2. Mapping Ubiquitination Sites on Protein Substrates
3. Lessons from Ubiquitin Site Profiling
4. Limitations of Ubiquitin Site Profiling
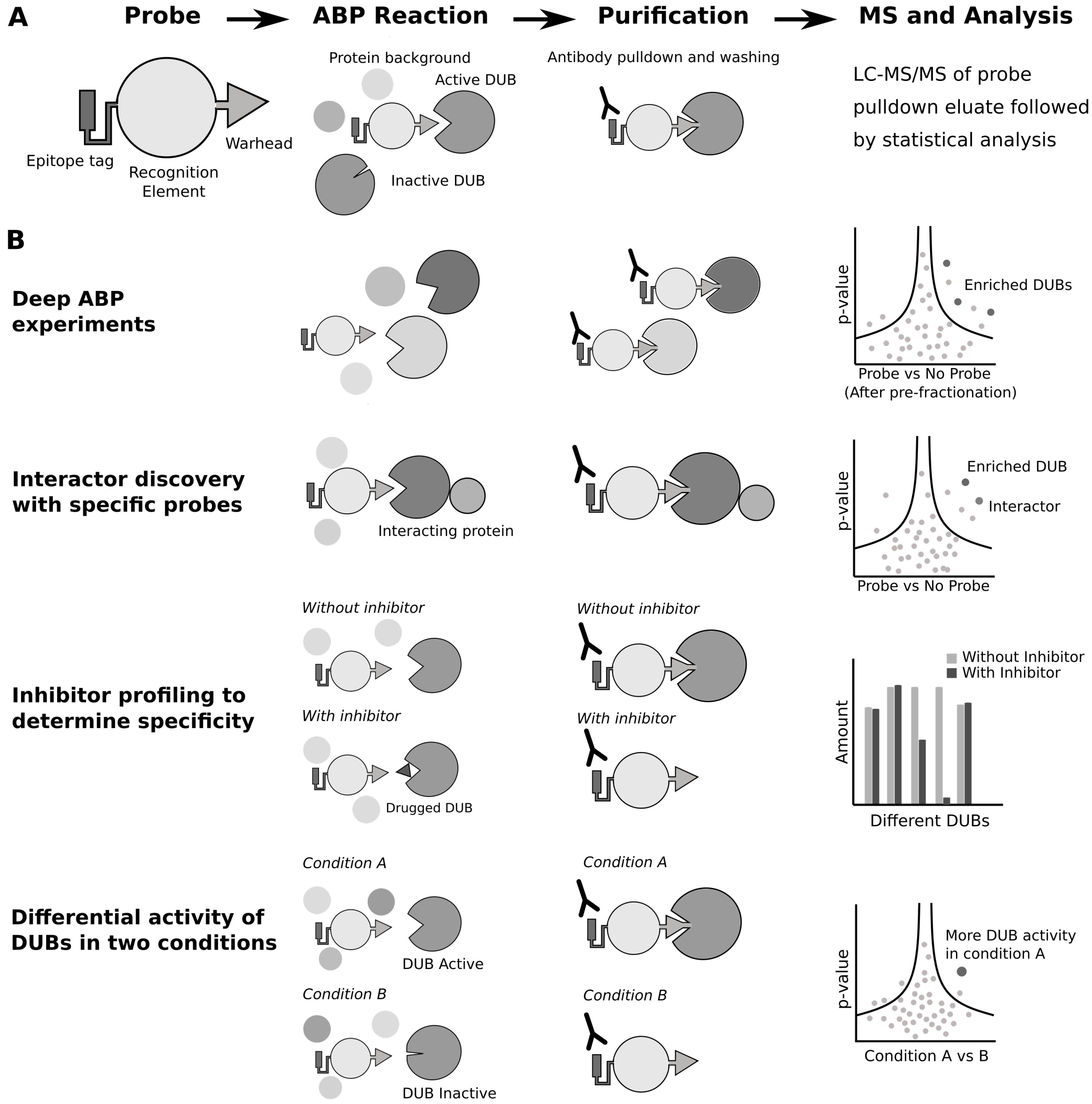
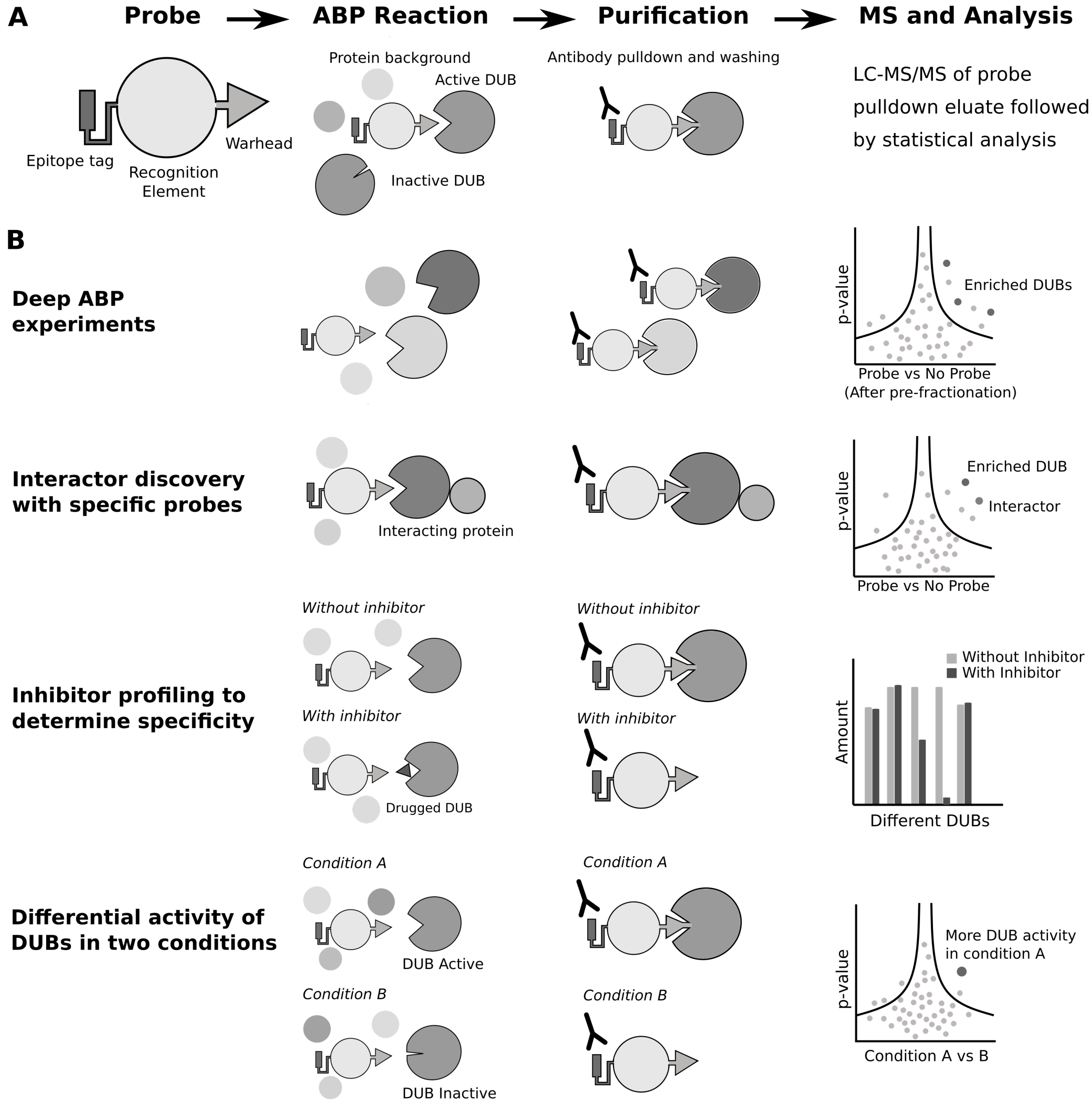
5. Proteomics with Activity-Based Probe Profiling
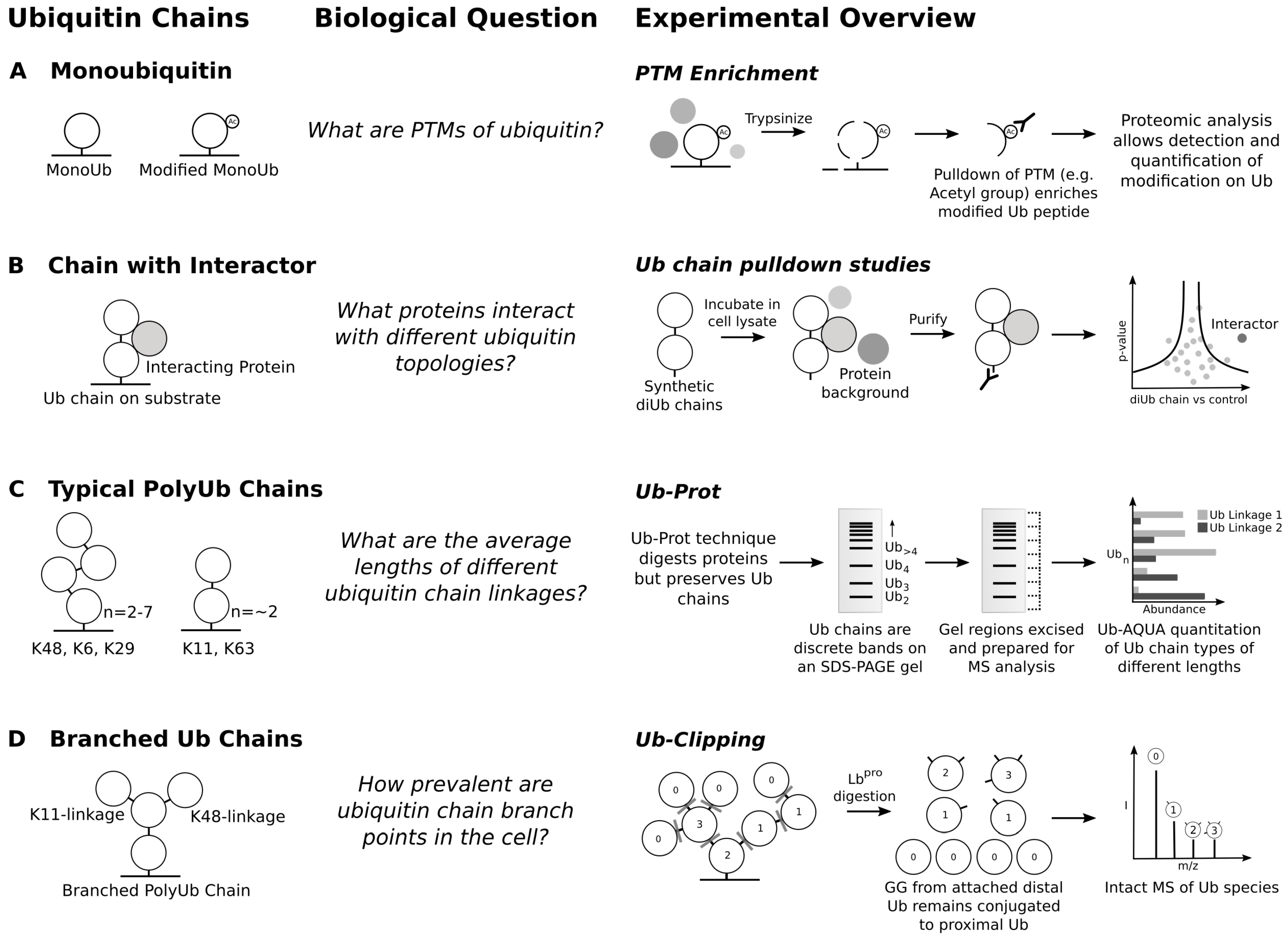
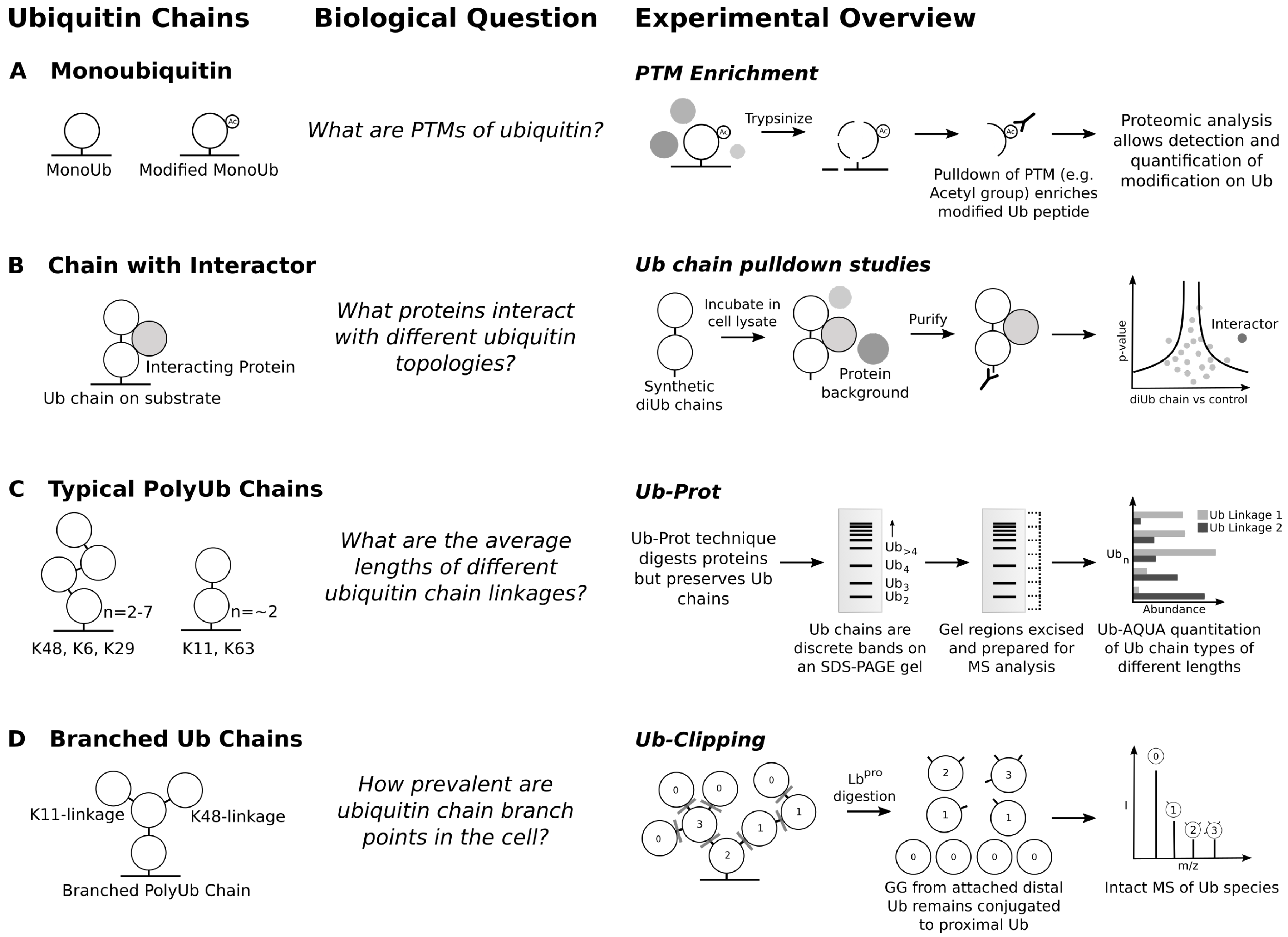
6. Ubiquitin Chain Topology and the Ubiquitin Code Hypothesis
7. Translational Ubiquitomics
8. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, S.; Haendler, B. The Ubiquitin System in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Kliza, K.; Husnjak, K. Resolving the Complexity of Ubiquitin Networks. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, E.; Palaniyappan, N.; Tooth, D.; Layfield, R. Methods for the purification of ubiquitinated proteins. Proteomics 2007, 7, 1016–1022. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Beaudette, P.; Popp, O.; Dittmar, G. Proteomic techniques to probe the ubiquitin landscape. Proteomics 2016, 16, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, J.M.R.; Sylvestersen, K.B.; Bekker-Jensen, S.; Szklarczyk, D.; Poulsen, J.W.; Horn, H.; Jensen, L.J.; Mailand, N.; Nielsen, M.L. Mass Spectrometric Analysis of Lysine Ubiquitylation Reveals Promiscuity at Site Level. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lectez, B.; Migotti, R.; Lee, S.Y.; Ramirez, J.; Beraza, N.; Mansfield, B.; Sutherland, J.D.; Martinez-Chantar, M.L.; Dittmar, G.; Mayor, U. Ubiquitin profiling in liver using a transgenic mouse with biotinylated ubiquitin. J. Proteome Res. 2014, 13, 3016–3026. [Google Scholar] [CrossRef]
- Meierhofer, D.; Wang, X.; Huang, L.; Kaiser, P. Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J. Proteome Res. 2008, 7, 4566–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, V.; Olsen, L.C.; Hansen, S.V.; Barrio-Hernandez, I.; Puglia, M.; Jensen, S.S.; Solov’Yov, I.A.; Kratchmarova, I.; Blagoev, B. StUbEx PLUS—A Modified Stable Tagged Ubiquitin Exchange System for Peptide Level Purification and In-Depth Mapping of Ubiquitination Sites. J. Proteome Res. 2018, 17, 296–304. [Google Scholar] [CrossRef]
- Stes, E.; Laga, M.; Walton, A.; Samyn, N.; Timmerman, E.; De Smet, I.; Goormachtig, S.; Gevaert, K. A COFRADIC protocol to study protein ubiquitination. J. Proteome Res. 2014, 13, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 2010, 28, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (K-ϵ-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Schölz, C.; Kelstrup, C.D.; Young, C.; Nielsen, M.L.; Olsen, J.V.; Brakebusch, C.; Choudhary, C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol. Cell. Proteom. 2012, 11, 1578–1585. [Google Scholar] [CrossRef] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and n-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Hansen, F.M.; Tanzer, M.C.; Brüning, F.; Bludau, I.; Schulman, B.A.; Robles, M.S.; Karayel, O.; Mann, M. Data-independent acquisition method for ubiquitinome analysis reveals regulation of circadian biology. bioRxiv 2020. [Google Scholar] [CrossRef]
- Steger, M.; Ihmor, P.; Backman, M.; Müller, S.; Daub, H. Deep ubiquitination site profiling by single-shot data-independent acquisition mass spectrometry. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.E.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef]
- Rose, C.M.; Isasa, M.; Ordureau, A.; Prado, M.A.; Beausoleil, S.A.; Jedrychowski, M.P.; Finley, D.J.; Harper, J.W.; Gygi, S.P. Highly Multiplexed Quantitative Mass Spectrometry Analysis of Ubiquitylomes. Cell Syst. 2016, 3, 395–403.e4. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Mani, D.C.; Satpathy, S.; Fereshetian, S.; Gasser, J.A.; Svinkina, T.; Olive, M.E.; Ebert, B.L.; Mertins, P.; Carr, S.A. Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Elia, A.E.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Ordureau, A.; Paulo, J.A.; Zhang, J.; An, H.; Swatek, K.N.; Cannon, J.R.; Wan, Q.; Komander, D.; Harper, J.W. Global Landscape and Dynamics of Parkin and USP30-Dependent Ubiquitylomes in iNeurons during Mitophagic Signaling. Mol. Cell 2020, 77, 1124–1142.e10. [Google Scholar] [CrossRef]
- Theurillat, J.P.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Chapman, J.D.; Goodlett, D.R.; Masselon, C.D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 2014, 33, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 min. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Gorman, A.W.; Vogel, C.; Silva, G.M. Site-Specific K63 Ubiquitinomics Provides Insights into Translation Regulation under Stress. J. Proteome Res. 2019, 18, 309–318. [Google Scholar] [CrossRef]
- Minguez, P.; Parca, L.; Diella, F.; Mende, D.R.; Kumar, R.; Helmer-Citterich, M.; Gavin, A.C.; Van Noort, V.; Bork, P. Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 2012, 8, 599. [Google Scholar] [CrossRef]
- Fouad, S.; Wells, O.S.; Hill, M.A.; D’Angiolella, V. Cullin Ring Ubiquitin Ligases (CRLs) in Cancer: Responses to Ionizing Radiation (IR) Treatment. Front. Physiol. 2019, 10, 1144. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genom. 2017, 44, 243–250. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications – writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Saeki, Y.; Sakamoto, K.; Ohtake, K.; Nishikawa, H.; Tsuchiya, H.; Ohta, T.; Tanaka, K.; Kanno, J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 2015, 16, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhou, T.; He, B.; Yu, H.; Guo, X.; Song, X.; Sha, J. mUbiSiDa: A comprehensive database for protein ubiquitination sites in mammals. PLoS ONE 2014, 9, e85744. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Liu, Z.; Wang, Y.; Cheng, H.; Yang, Q.; Guo, A.; Ren, J.; Xue, Y. UUCD: A family-based database of ubiquitin and ubiquitin-like conjugation. Nucleic Acids Res. 2013, 41, D445–D451. [Google Scholar] [CrossRef] [Green Version]
- Kennelly, P.J.; Krebs, E.G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 1991, 266, 15555–15558. [Google Scholar]
- Gavel, Y.; Heijne, G.V. Sequence differences between glycosylated and non-glycosylated asn-x-thr/ser acceptor sites: Implications for protein engineering. Protein Eng. Des. Sel. 1990, 3, 433–442. [Google Scholar] [CrossRef]
- Jadhav, T.; Wooten, M.W. Defining an embedded code for protein Ubiquitination. J. Proteom. Bioinform. 2009, 2, 316–333. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhou, Y.; Zhang, Z.; Song, J. Towards more accurate prediction of ubiquitination sites: A comprehensive review of current methods, tools and features. Brief. Bioinform. 2014, 16, 640–657. [Google Scholar] [CrossRef] [Green Version]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins Struct. Funct. Bioinform. 2010, 78, 365–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Qiu, J.D.; Shi, S.P.; Suo, S.B.; Huang, S.Y.; Liang, R.P. Incorporating key position and amino acid residue features to identify general and species-specific Ubiquitin conjugation sites. Bioinformatics 2013, 29, 1614–1622. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Yang, Y.; Wang, X.; Wang, H.; Xu, Y. DeepUbi: A deep learning framework for prediction of ubiquitination sites in proteins. BMC Bioinform. 2019, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The predator becomes the prey: Regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Iconomou, M.; Saunders, D.N. Systematic approaches to identify E3 ligase Substrates. Biochem. J. 2016, 473, 4083–4101. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Verdine, G.L.; Walensky, L.D. The challenge of drugging undruggable targets in cancer: Lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 2007, 13, 7264–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, D.; Anderton, B.; Goga, A. Taking on Challenging Targets: Making MYC Druggable. Am. Soc. Clin. Oncol. Educ. B 2014, 34, e497–e502. [Google Scholar] [CrossRef] [Green Version]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, T.; Li, Z.; Sun, K.; Fu, Y.; Cheng, T.; Guo, J.; Yu, B.; Shi, X.; Liu, H. Discovery of [1,2,3]triazolo[4,5-d] pyrimidine derivatives as highly potent, selective, and cellularly active USP28 inhibitors. Acta Pharm. Sin. B 2020, 10, 1476–1491. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.; Zhang, L.; Yang, Z.; Chen, X.; Luo, J.; Zhou, Z.; Mei, X.; Yu, X.; Shao, Z.; et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–77. [Google Scholar] [CrossRef]
- Pinto-Fernandez, A.; Kessler, B.M. DUBbing cancer: Deubiquitylating enzymes involved in epigenetics, DNA damage and the cell cycle as therapeutic targets. Front. Genet. 2016, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.W.; Nagel, J.; Hoving, S.; Gerrits, B.; Bauer, A.; Thomas, J.R.; Kirschner, M.W.; Schirle, M.; Luchansky, S.J. Quantitative Lys-ϵ-Gly-Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin-mediated proteolysis of DNA damage-inducible transcript 4 (DDIT4) by the E3 Ligase HUWE1. J. Biol. Chem. 2014, 289, 28942–28955. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Saeki, Y.; Murakami, A.; Kawawaki, J.; Tsuchiya, H.; Yoshihara, H.; Shindo, M.; Tanaka, K. A comprehensive method for detecting ubiquitinated substrates using TR-TUBE. Proc. Natl. Acad. Sci. USA 2015, 112, 4630–4635. [Google Scholar] [CrossRef] [Green Version]
- Potu, H.; Peterson, L.F.; Kandarpa, M.; Pal, A.; Sun, H.; Durham, A.; Harms, P.W.; Hollenhorst, P.C.; Eskiocak, U.; Talpaz, M.; et al. Usp9x regulates Ets-1 ubiquitination and stability to control NRAS expression and tumorigenicity in melanoma. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, J.J.; Vasilevskaya, I.A.; Neupane, N.P.; Shafi, A.A.; McNair, C.; Dylgjeri, E.; Mandigo, A.C.; Schiewer, M.J.; Schrecengost, R.S.; Gallagher, P.; et al. USP22 functions as an oncogenic driver in prostate cancer by regulating cell proliferation and DNA repair. Cancer Res. 2020, 80, 430–443. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Phu, L.; Rose, C.M.; Tea, J.S.; Wall, C.E.; Verschueren, E.; Cheung, T.K.; Kirkpatrick, D.S.; Bingol, B. Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol. Cell 2020, 77, 1107–1123.e10. [Google Scholar] [CrossRef] [PubMed]
- Rusilowicz-Jones, E.V.; Jardine, J.; Kallinos, A.; Pinto-Fernandez, A.; Guenther, F.; Giurrandino, M.; Barone, F.G.; McCarron, K.; Burke, C.J.; Murad, A.; et al. USP30 sets a trigger threshold for PINK1-PARKIN amplification of mitochondrial ubiquitylation. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Sapmaz, A.; Berlin, I.; Bos, E.; Wijdeven, R.H.; Janssen, H.; Konietzny, R.; Akkermans, J.J.; Erson-Bensan, A.E.; Koning, R.I.; Kessler, B.M.; et al. USP32 regulates late endosomal transport and recycling through deubiquitylation of Rab7. Nat. Commun. 2019, 10, 1454. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.A.; Satpathy, S.; Beli, P.; Choudhary, C. SPATA 2 links CYLD to the TNF-α receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 2016, 35, 1868–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unsworth, A.J.; Bombik, I.; Pinto-Fernandez, A.; McGouran, J.F.; Konietzny, R.; Zahedi, R.P.; Watson, S.P.; Kessler, B.M.; Pears, C.J. Human Platelet Protein Ubiquitylation and Changes following GPVI Activation. Thromb. Haemost. 2019, 119, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.; Mace, Y.; Drouet, F.; Bony, E.; Boidot, R.; Draoui, N.; Lobysheva, I.; Corbet, C.; Polet, F.; Martherus, R.; et al. A new ER-specific photosensitizer unravels 1O2-driven protein oxidation and inhibition of deubiquitinases as a generic mechanism for cancer PDT. Oncogene 2016, 35, 3976–3985. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Mani, D.R.; Eisenhaure, T.; Mertins, P.; Jaffe, J.D.; Clauser, K.R.; Hacohen, N.; Carr, S.A. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol. Cell. Proteom. 2012, 11, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Aravamudhan, S.; Nolte, H.; Türk, C.; Hölper, S.; Mü Ller, S.; Gü Nther, S.; Blaauw, B.; Braun, T.; Krüger, M. Dynamic changes in the mouse skeletal muscle proteome during denervation-induced atrophy. Dis. Model. Mech. 2017, 10, 881–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.; Charles, P.D.; He, L.; Mowlds, P.; Kessler, B.M.; Fischer, R. Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis. J. Proteome Res. 2017, 16, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, L.; Bezstarosti, K.; Sap, K.A.; Dekkers, D.H.; Rijkers, E.; Mientjes, E.; Elgersma, Y.; Demmers, J.A. Improvement of ubiquitylation site detection by Orbitrap mass spectrometry. J. Proteom. 2018, 172, 49–56. [Google Scholar] [CrossRef]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227.e8. [Google Scholar] [CrossRef] [Green Version]
- Sobott, F.; Watt, S.J.; Smith, J.; Edelmann, M.J.; Kramer, H.B.; Kessler, B.M. Comparison of CID Versus ETD Based MS/MS Fragmentation for the Analysis of Protein Ubiquitination. J. Am. Soc. Mass Spectrom. 2009, 20, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Porras-Yakushi, T.R.; Sweredoski, M.J.; Hess, S. ETD Outperforms CID and HCD in the Analysis of the Ubiquitylated Proteome. J. Am. Soc. Mass Spectrom. 2015, 26, 1580–1587. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Geladaki, A.; Kočevar Britovšek, N.; Breckels, L.M.; Smith, T.S.; Vennard, O.L.; Mulvey, C.M.; Crook, O.M.; Gatto, L.; Lilley, K.S. Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gillotin, S.; Davies, J.D.; Philpott, A. Subcellular localisation modulates ubiquitylation and degradation of Ascl1. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelsall, I.R.; Zhang, J.; Knebel, A.; Arthur, S.J.; Cohen, P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl. Acad. Sci. USA 2019, 116, 13293–13298. [Google Scholar] [CrossRef] [Green Version]
- Pao, K.C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; Van Aalten, D.M.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 2018, 556, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Liu, X.; Xia, T.; Tekcham, D.S.; Wang, W.; Chen, H.; Li, T.; Lu, C.; Ning, Z.; Liu, X.; et al. A Multidimensional Characterization of E3 Ubiquitin Ligase and Substrate Interaction Network. iScience 2019, 16, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Needham, E.J.; Parker, B.L.; Burykin, T.; James, D.E.; Humphrey, S.J. Illuminating the dark phosphoproteome. Sci. Signal. 2019, 12, eaau8645. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Xu, P.; Qin, J. Ubiquitinated proteome: Ready for global? Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.L.; Vermeulen, M.; Bonaldi, T.; Cox, J.; Moroder, L.; Mann, M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 2008, 5, 459–460. [Google Scholar] [CrossRef]
- Sanman, L.E.; Bogyo, M. Activity-Based Profiling of Proteases. Annu. Rev. Biochem. 2014, 83, 249–273. [Google Scholar] [CrossRef] [Green Version]
- Borodovsky, A.; Kessler, B.M.; Casagrande, R.; Overkleeft, H.S.; Wilkinson, K.D.; Ploegh, H.L. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001, 20, 5187–5196. [Google Scholar] [CrossRef] [Green Version]
- Hewings, D.S.; Flygare, J.A.; Bogyo, M.; Wertz, I.E. Activity-based probes for the ubiquitin conjugation- deconjugation machinery: New chemistries, new tools, and new insights. FEBS J. 2017, 284, 1555–1576. [Google Scholar] [CrossRef] [Green Version]
- Byrne, R.; Mund, T.; Licchesi, J.D. Activity-Based Probes for HECT E3 Ubiquitin Ligases. ChemBioChem 2017, 18, 1415–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, S.; Fletcher, A.J.; Branigan, E.; Hay, R.T.; Virdee, S. Photocrosslinking Activity-Based Probes for Ubiquitin RING E3 Ligases. Cell Chem. Biol. 2020, 27, 74–82.e6. [Google Scholar] [CrossRef] [Green Version]
- Ekkebus, R.; Van Kasteren, S.I.; Kulathu, Y.; Scholten, A.; Berlin, I.; Geurink, P.P.; De Jong, A.; Goerdayal, S.; Neefjes, J.; Heck, A.J.; et al. On terminal alkynes that can react with active-site cysteine nucleophiles in proteases. J. Am. Chem. Soc. 2013, 135, 2867–2870. [Google Scholar] [CrossRef] [PubMed]
- Hameed, D.S.; Sapmaz, A.; Burggraaff, L.; Amore, A.; Slingerland, C.J.; van Westen, G.J.; Ovaa, H. Development of Ubiquitin-Based Probe for Metalloprotease Deubiquitinases. Angew. Chem. Int. Ed. 2019, 58, 14477–14482. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Fernández, A.; Davis, S.; Schofield, A.B.; Scott, H.C.; Zhang, P.; Salah, E.; Mathea, S.; Charles, P.D.; Damianou, A.; Bond, G.; et al. Comprehensive Landscape of Active Deubiquitinating Enzymes Profiled by Advanced Chemoproteomics. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Mulder, M.P.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.P.; Chang, J.G.; Merkx, R.; Bialas, J.; Groettrup, M.; Vertegaal, A.C.; et al. A cascading activity-based probe sequentially targets E1-E2-E3 ubiquitin enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef]
- Hewings, D.S.; Heideker, J.; Ma, T.P.; Ahyoung, A.P.; El Oualid, F.; Amore, A.; Costakes, G.T.; Kirchhofer, D.; Brasher, B.; Pillow, T.; et al. Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Kwasna, D.; Abdul Rehman, S.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell 2018, 70, 150–164.e6. [Google Scholar] [CrossRef] [Green Version]
- Artavanis-Tsakonas, K.; Misaghi, S.; Comeaux, C.A.; Catic, A.; Spooner, E.; Duraisingh, M.T.; Ploegh, H.L. Identification by functional proteomics of a deubiquitinating/deNeddylating enzyme in Plasmodium falciparum. Mol. Microbiol. 2006, 61, 1187–1195. [Google Scholar] [CrossRef]
- Damianou, A.; Burge, R.J.; Catta-Preta, C.M.; Geoghegan, V.; Nievas, Y.R.; Newling, K.; Brown, E.; Burchmore, R.; Rodenko, B.; Mottram, J.C. Essential roles for deubiquitination in Leishmania life cycle progression. PLoS Pathog. 2020, 16, e1008455. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and Cellular Roles of Ubiquitin-Specific Deubiquitinating Enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummari, E.; Alugubelly, N.; Hsu, C.Y.; Dong, B.; Nanduri, B.; Edelmann, M.J. Activity-based proteomic profiling of deubiquitinating enzymes in salmonella-infected macrophages leads to identification of putative function of UCH-L5 in inflammasome regulation. PLoS ONE 2015, 10, e0135531. [Google Scholar] [CrossRef]
- Zhang, Z.; Fang, X.; Wu, X.; Ling, L.; Chu, F.; Li, J.; Wang, S.; Zang, J.; Zhang, B.; Ye, S.; et al. Acetylation- Dependent Deubiquitinase OTUD3 Controls MAVS Activation in Innate Antiviral Immunity. Mol. Cell 2020, 79, 304–319.e7. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; González-Prieto, R.; Zhang, M.; Geurink, P.P.; Kooij, R.; Iyengar, P.V.; van Dinther, M.; Bos, E.; Zhang, X.; Le Dévédec, S.E.; et al. Deubiquitinase activity profiling identifies UCHL1 as a candidate oncoprotein that promotes TGFβ-induced breast cancer metastasis. Clin. Cancer Res. 2020, 26, 1460–1473. [Google Scholar] [CrossRef] [Green Version]
- McGouran, J.F.; Gaertner, S.R.; Altun, M.; Kramer, H.B.; Kessler, B.M. Deubiquitinating enzyme specificity for ubiquitin chain topology profiled by di-ubiquitin activity probes. Chem. Biol. 2013, 20, 1447–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stangl, A.; Elliott, P.R.; Pinto-Fernandez, A.; Bonham, S.; Harrison, L.; Schaub, A.; Kutzner, K.; Keusekotten, K.; Pfluger, P.T.; El Oualid, F.; et al. Regulation of the endosomal SNX27-retromer by OTULIN. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.M.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014, 5, 4763. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Pérez Berrocal, D.A.; Witting, K.F.; Ovaa, H.; Mulder, M.P. Hybrid Chains: A Collaboration of Ubiquitin and Ubiquitin-Like Modifiers Introducing Cross-Functionality to the Ubiquitin Code. Front. Chem. 2020, 7, 931. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Smits, A.H.; van Tilburg, G.B.; Jansen, P.W.; Makowski, M.M.; Ovaa, H.; Vermeulen, M. An Interaction Landscape of Ubiquitin Signaling. Mol. Cell 2017, 65, 941–955.e8. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Lutz, J.; Höllmüller, E.; Scheffner, M.; Marx, A.; Stengel, F. Identification of Proteins Interacting with Ubiquitin Chains. Angew. Chem. Int. Ed. 2017, 56, 15764–15768. [Google Scholar] [CrossRef]
- Radley, E.H.; Long, J.; Gough, K.C.; Layfield, R. The ‘dark matter’ of ubiquitin-mediated processes: Opportunities and challenges in the identification of ubiquitin-binding domains. Biochem. Soc. Trans. 2019, 47, 1949–1962. [Google Scholar] [CrossRef]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Cui, J.; Yao, Q.; Li, S.; Ding, X.; Lu, Q.; Mao, H.; Liu, L.; Zheng, N.; Chen, S.; Shao, F. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science 2010, 329, 1215–1218. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattern, M.; Sutherland, J.; Kadimisetty, K.; Barrio, R.; Rodriguez, M.S. Using Ubiquitin Binders to Decipher the Ubiquitin Code. Trends Biochem. Sci. 2019, 44, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN Restricts Met1-Linked Ubiquitination to Control Innate Immune Signaling. Mol. Cell 2013, 50, 818–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, G.M.; Finley, D.; Vogel, C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat. Struct. Mol. Biol. 2015, 22, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Phu, L.; Izrael-Tomasevic, A.; Matsumoto, M.L.; Bustos, D.; Dynek, J.N.; Fedorova, A.V.; Bakalarski, C.E.; Arnott, D.; Deshayes, K.; Dixit, V.M.; et al. Improved quantitative mass spectrometry methods for characterizing complex ubiquitin signals. Mol. Cell. Proteom. 2011, 10, M110.003756. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, H.; Burana, D.; Ohtake, F.; Arai, N.; Kaiho, A.; Komada, M.; Tanaka, K.; Saeki, Y. Ub-ProT reveals global length and composition of protein ubiquitylation in cells. Nat. Commun. 2018, 9, 524. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef] [Green Version]
- Boname, J.M.; Thomas, M.; Stagg, H.R.; Xu, P.; Peng, J.; Lehner, P.J. Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 2010, 11, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Yamanaka, Y.; Ishikawa, A.; Aoki-Kawasumi, M.; Mito-Yoshida, M.; Ohmura-Hoshino, M.; Matsuki, Y.; Kajikawa, M.; Hirano, H.; Ishido, S. Contribution of lysine 11-linked ubiquitination to MIR2-mediated major histocompatibility complex class I internalization. J. Biol. Chem. 2010, 285, 35311–35319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.J.; Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hospenthal, M.K.; Mevissen, T.E.; Komander, D. Deubiquitinase-based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest). Nat. Protoc. 2015, 10, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Peng, J. Characterization of polyubiquitin chain structure by middle-down mass spectrometry. Anal. Chem. 2008, 80, 3438–3444. [Google Scholar] [CrossRef] [Green Version]
- Valkevich, E.M.; Sanchez, N.A.; Ge, Y.; Strieter, E.R. Middle-Down mass spectrometry enables characterization of branched ubiquitin chains. Biochemistry 2014, 53, 4979–4989. [Google Scholar] [CrossRef]
- Crowe, S.O.; Rana, A.S.; Deol, K.K.; Ge, Y.; Strieter, E.R. Ubiquitin Chain Enrichment Middle-Down Mass Spectrometry Enables Characterization of Branched Ubiquitin Chains in Cellulo. Anal. Chem. 2017, 89, 4428–4434. [Google Scholar] [CrossRef] [Green Version]
- Swatek, K.N.; Usher, J.L.; Kueck, A.F.; Gladkova, C.; Mevissen, T.E.T.; Pruneda, J.N.; Skern, T.; Komander, D. Insights into ubiquitin chain architecture using Ub-clipping. Nature 2019, 572, 533–537. [Google Scholar] [CrossRef]
- Mathis, B.; Lai, Y.; Qu, C.; Janicki, J.; Cui, T. CYLD-Mediated Signaling and Diseases. Curr. Drug Targets 2014, 16, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, M.E.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TOR CH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- Cowey, C.L.; Rathmell, W.K. VHL gene mutations in renal cell carcinoma: Role as a biomarker of disease outcome and drug efficacy. Curr. Oncol. Rep. 2009, 11, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25, S32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thery, F.; Wu, N.C.; Luhmann, E.K.; Dussurget, O.; Foecke, M.; Bredow, C.; Jiménez-Fernández, D.; Leandro, K.; Beling, A.; et al. The in vivo ISGylome links ISG15 to metabolic pathways and autophagy upon Listeria monocytogenes infection. Nat. Commun. 2019, 10, 5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Aim | Sample Requirement | Enrichment Method | MS Factors and Analysis | Example |
|---|---|---|---|---|
| Deep ubiquitome | Up to 50 mg cell culture | UbiSite | Prefractionation to increase depth | [21] |
| Multiple PTMs | 1–20 mg cell culture | UbiSite or K-GG with additional PTM pulldowns | Multi-Omic data analysis | [31] |
| Multiple conditions | 0.5–20 mg cell culture | K-GG or TUBE | Use of SILAC or TMT— in-solution or on-bead | [19,29] |
| Chain type specific | 1–200 mg cell culture/yeast | TUBE, possible to combine with K-GG | - | [38] |
| Low abundance modifications | <1 mg lysate | K-GG | Use of DIA to increase MS sensitivity | [22,23] |
| Databases | Information | Reference |
|---|---|---|
| PhosphositePlus Database | Most comprehensive database for protein ubiquitination including most recent studies | [24] |
| Protein Lysine Modification Database (PLMD) | Contains information on lysine ubiquitination and on other lysine modifications. Potential for investigating PTM crosstalk | [41] |
| Mammalian Ubiquitination Site Database (mUbiSiDa) | A database of ubiquitination sites assembled in 2013 | [44] |
| Ubiquitin and Ubiquitin-like conjugation Database (UUCD) | A database of actual and predicted ubiquitin and Ubl associated machinery in several species | [45] |
| Ubiquitin Modifying Enzyme | Study Details | Reference |
|---|---|---|
| Cullin Ring Ligases | K-GG, CRL inhibition | [18] |
| SPOP | K-GG, SILAC, mutant and overexpression | [33] |
| Parkin | K-GG, inactive mutant | [28] |
| K-GG, inactive mutant | [32] | |
| LZTR1 | K-GG, knockout | [63] |
| HUWE1 | K-GG, knockdown | [64] |
| Skp2 | TUBE, overexpression | [65] |
| USP7 | K-GG, DIA-MS, inhibitor | [23] |
| USP9X | K-GG, knockdown | [66] |
| USP22 | K-GG, knockdown and overexpression | [67] |
| USP30 | K-GG, knockdown | [68] |
| K-GG, knockout | [32] | |
| K-GG, inhibitor | [69] | |
| K-GG, knockout and inhibitor | [70] | |
| USP32 | TUBE, knockdown | [71] |
| Stimulus | ||
| UV-induced DNA damage | K-GG, SILAC | [27] |
| UV- and radiation-induced DNA damage | K-GG, SILAC | [31] |
| TNF signalling | K-GG, SILAC | [72] |
| Cell cycle synchronisation | K-GG, DIA-MS | [23] |
| Lenalidomide treatment | K-GG, UbiFast/TMT | [29] |
| CRP-XL signalling | K-GG | [73] |
| Photosensitiser treatment | K-GG | [74] |
| Proteasome inhibition | K-GG, SILAC | [18] |
| K-GG, SILAC | [75] | |
| UbiSite | [21] | |
| Muscle atrophy | K-GG, time course examining mouse muscle ubiquitome following atrophy | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vere, G.; Kealy, R.; Kessler, B.M.; Pinto-Fernandez, A. Ubiquitomics: An Overview and Future. Biomolecules 2020, 10, 1453. https://doi.org/10.3390/biom10101453
Vere G, Kealy R, Kessler BM, Pinto-Fernandez A. Ubiquitomics: An Overview and Future. Biomolecules. 2020; 10(10):1453. https://doi.org/10.3390/biom10101453
Chicago/Turabian StyleVere, George, Rachel Kealy, Benedikt M. Kessler, and Adan Pinto-Fernandez. 2020. "Ubiquitomics: An Overview and Future" Biomolecules 10, no. 10: 1453. https://doi.org/10.3390/biom10101453
APA StyleVere, G., Kealy, R., Kessler, B. M., & Pinto-Fernandez, A. (2020). Ubiquitomics: An Overview and Future. Biomolecules, 10(10), 1453. https://doi.org/10.3390/biom10101453