Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113–122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice

 ,
, 
 , ,
, ,  ,
,  , and
, and 
Abstract
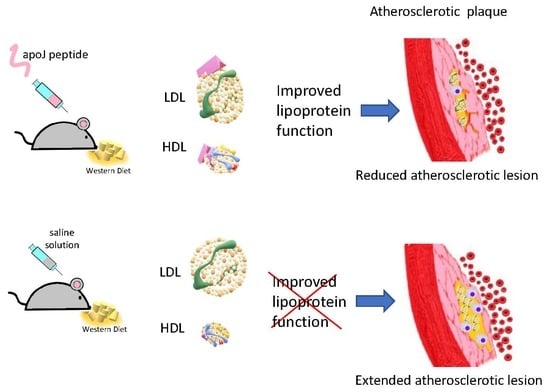
1. Introduction
2. Materials and Methods
2.1. Animal Study Design
2.2. Peptides
2.3. Lipid Analysis
2.4. Lipoprotein Isolation and Characterization
2.5. Susceptibility to Oxidation of Lipoproteins
2.6. Susceptibility to LDL Aggregation
2.7. Electronegativity of LDL
2.8. Cholesterol Efflux Capacity of HDL
2.9. Evaluation of Atherosclerotic Lesions
2.10. Quantitative RT-PCR Analyses
2.11. Statistical Methods
3. Results
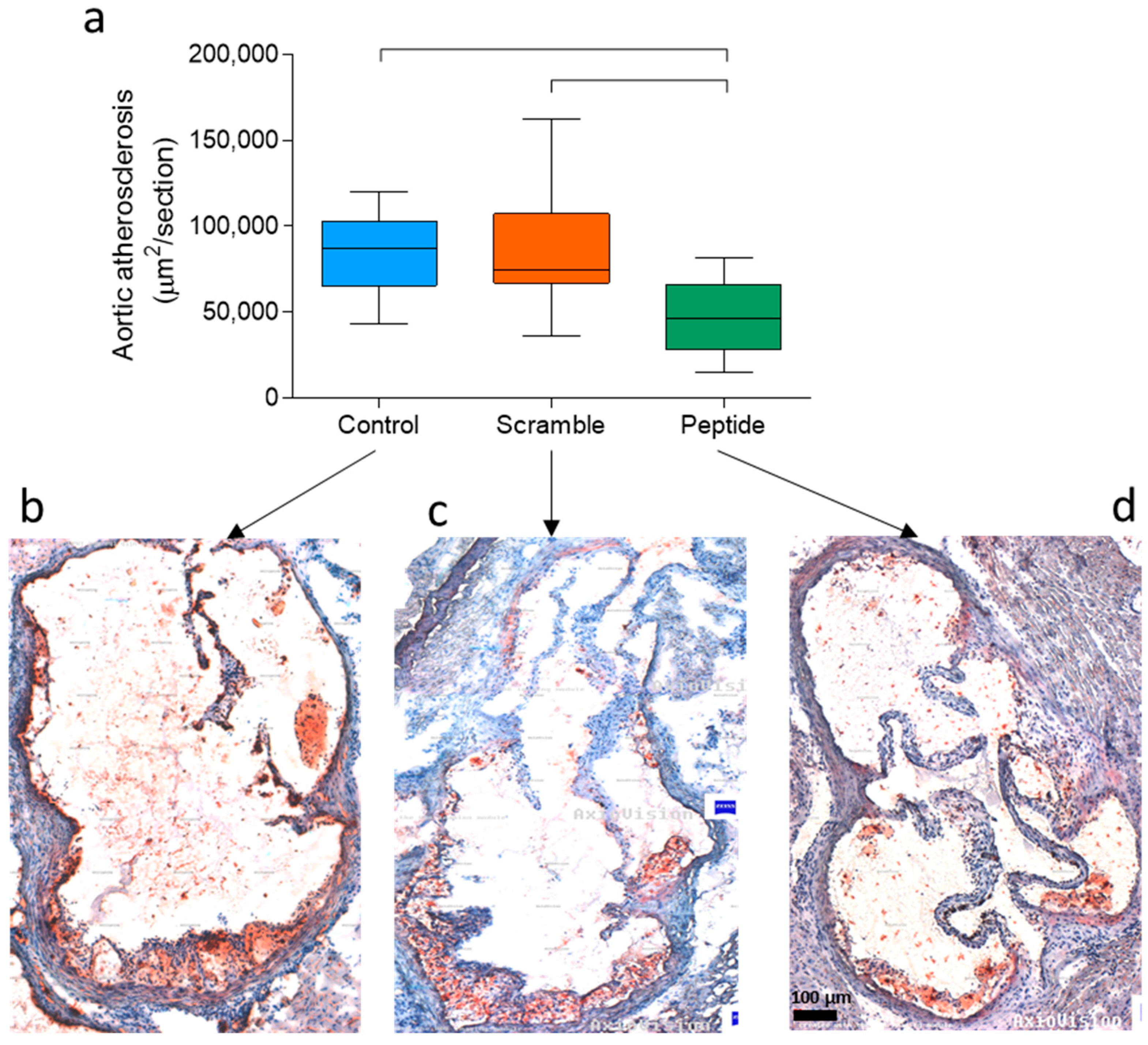
3.1. Subcutaneous Administration of d-[113–122]apoJ Reduces Atherosclerosis
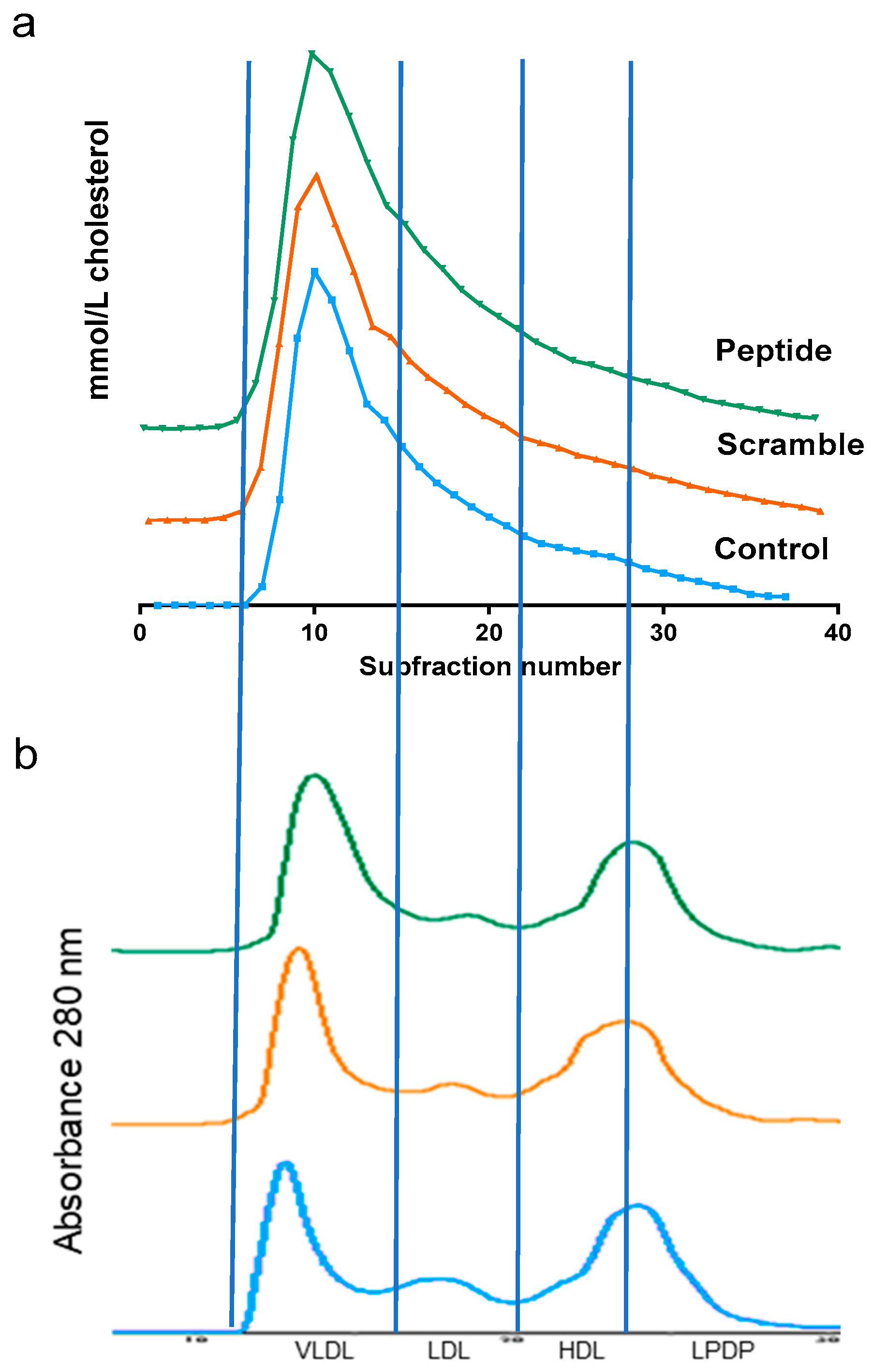
3.2. d-[113–122]apoJ Did Not Alter the Lipid Profile
3.3. d-[113–122]apoJ Favorably Influences the Oxidative Properties of Lipoproteins
3.4. d-[113–122]apoJ Decreases the Susceptibility of LDL to Aggregation
3.5. Electronegativity of LDL Decreased upon d-[113–122]apoJ Treatment
3.6. d-[113–122]apoJ Improves the Cholesterol Efflux Capacity of HDL
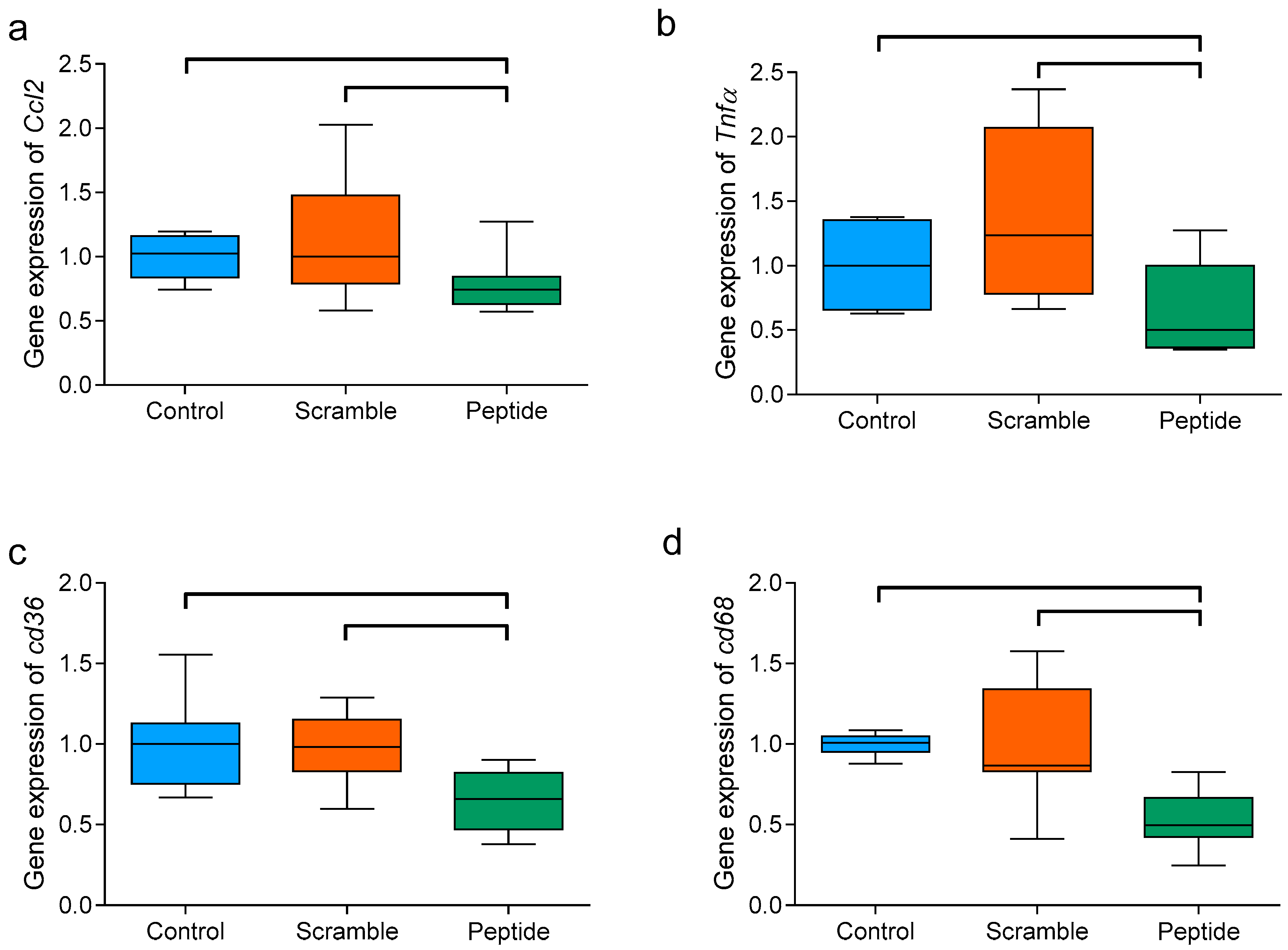
3.7. d-[113–122]apoJ Reduces the Hepatic Expression of Inflammation-Related Genes
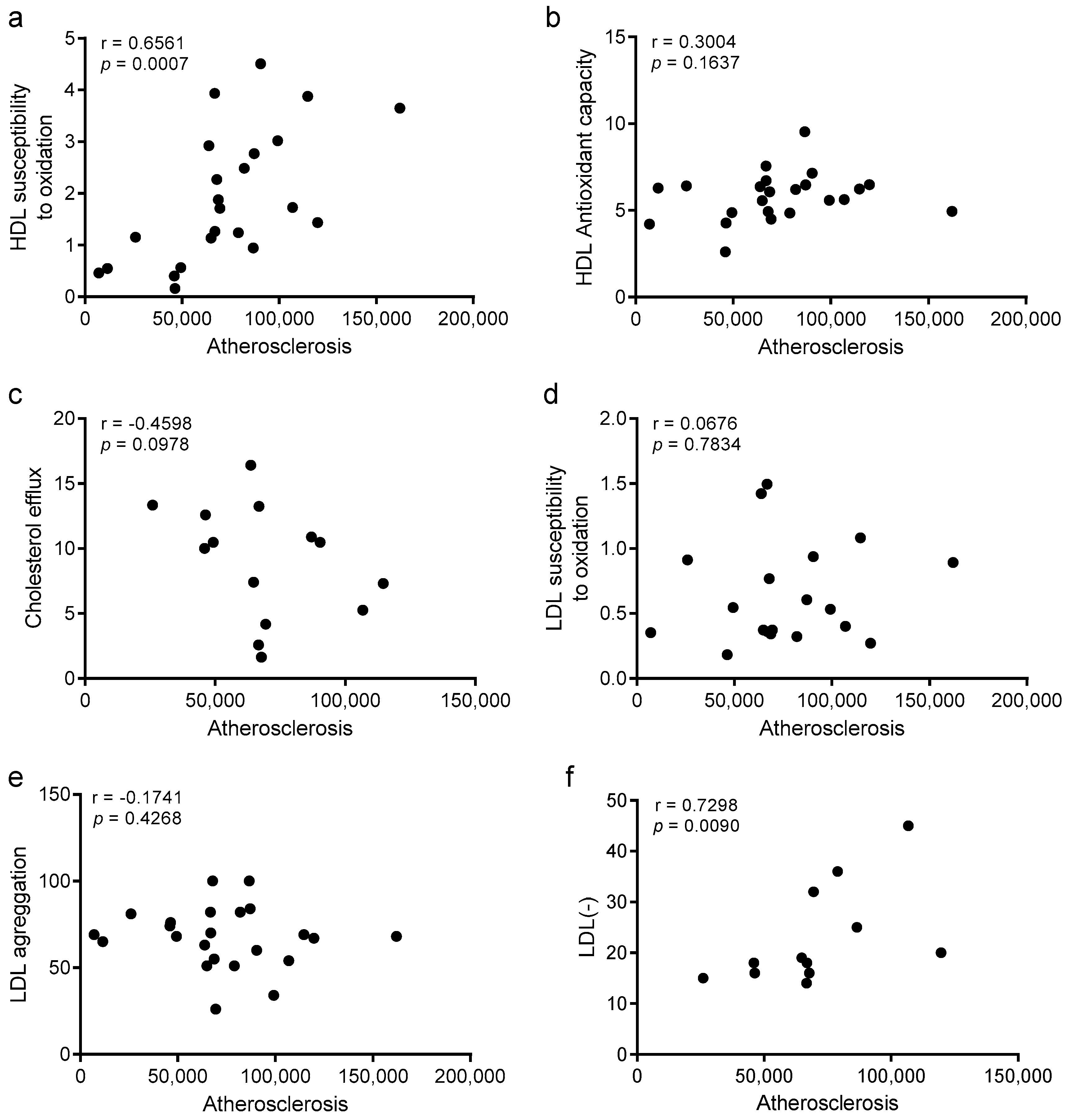
3.8. Improvements in HDL Oxidation and LDL Electronegativity Are Associated with Atherosclerosis Burden
3.9. The Atherosclerosis Burden Correlates with Hepatic Inflammation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Davignon, J.; Hermans, M.P.; Al-Rubeaan, K.; Amarenco, P.; Assmann, G.; Barter, P.; Betteridge, J.; Bruckert, E.; Cuevas, A.; et al. Residual macrovascular risk in 2013: What have we learned? Cardiovasc. Diabetol. 2014, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Witztum, J.L.; Steinberg, D. The oxidative modification hypothesis of atherosclerosis: Does it hold for humans? Trends Cardiovasc. Med. 2001, 11, 93–102. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Ruuth, M.; Nguyen, S.D.; Vihervaara, T.; Hilvo, M.; Laajala, T.D.; Kondadi, P.K.; Gistera, A.; Lahteenmaki, H.; Kittila, T.; Huusko, J.; et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur. Heart J. 2018, 39, 2562–2573. [Google Scholar] [CrossRef]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis From the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef]
- Gallone, G.; Baldetti, L.; Pagnesi, M.; Latib, A.; Colombo, A.; Libby, P.; Giannini, F. Medical Therapy for Long-Term Prevention of Atherothrombosis Following an Acute Coronary Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2886–2903. [Google Scholar] [CrossRef]
- Recio, C.; Maione, F.; Iqbal, A.J.; Mascolo, N.; De Feo, V. The Potential Therapeutic Application of Peptides and Peptidomimetics in Cardiovascular Disease. Front. Pharmacol. 2016, 7, 526. [Google Scholar] [CrossRef]
- Uehara, Y.; Chiesa, G.; Saku, K. High-Density Lipoprotein-Targeted Therapy and Apolipoprotein A-I Mimetic Peptides. Circ. J. 2015, 79, 2523–2528. [Google Scholar] [CrossRef]
- White, C.R.; Garber, D.W.; Anantharamaiah, G.M. Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: A review. J. Lipid Res. 2014, 55, 2007–2021. [Google Scholar] [CrossRef]
- Wolska, A.; Lo, L.; Sviridov, D.O.; Pourmousa, M.; Pryor, M.; Ghosh, S.S.; Kakkar, R.; Davidson, M.; Wilson, S.; Pastor, R.W.; et al. A dual apolipoprotein C-II mimetic-apolipoprotein C-III antagonist peptide lowers plasma triglycerides. Sci. Transl. Med. 2020, 12, eaaw7905. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Wagner, A.C.; Hama, S.; Hough, G.; Bachini, E.; Garber, D.W.; Mishra, V.K.; et al. An oral apoJ peptide renders HDL antiinflammatory in mice and monkeys and dramatically reduces atherosclerosis in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1932–1937. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Apoprotein E and Reverse Cholesterol Transport. Int. J. Mol. Sci. 2018, 19, 3479. [Google Scholar] [CrossRef]
- Tabet, F.; Remaley, A.T.; Segaliny, A.I.; Millet, J.; Yan, L.; Nakhla, S.; Barter, P.J.; Rye, K.A.; Lambert, G. The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Bielicki, J.K.; Zhang, H.; Cortez, Y.; Zheng, Y.; Narayanaswami, V.; Patel, A.; Johansson, J.; Azhar, S. A new HDL mimetic peptide that stimulates cellular cholesterol efflux with high efficiency greatly reduces atherosclerosis in mice. J. Lipid Res. 2010, 51, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, H.; Liu, M.; Li, F.; Liu, L.; Du, F.; Fan, D.; Yu, H. A human apolipoprotein E mimetic peptide reduces atherosclerosis in aged apolipoprotein E null mice. Am. J. Transl. Res. 2016, 8, 3482–3492. [Google Scholar]
- Karalis, I.; Jukema, J.W. HDL Mimetics Infusion and Regression of Atherosclerosis: Is It Still Considered a Valid Therapeutic Option? Curr. Cardiol. Rep. 2018, 20, 66. [Google Scholar] [CrossRef]
- Nguyen, S.D.; Javanainen, M.; Rissanen, S.; Zhao, H.; Huusko, J.; Kivela, A.M.; Yla-Herttuala, S.; Navab, M.; Fogelman, A.M.; Vattulainen, I.; et al. Apolipoprotein A-I mimetic peptide 4F blocks sphingomyelinase-induced LDL aggregation. J. Lipid Res. 2015, 56, 1206–1221. [Google Scholar] [CrossRef]
- Mishra, V.K.; Palgunachari, M.N.; Hudson, J.S.; Shin, R.; Keenum, T.D.; Krishna, N.R.; Anantharamaiah, G.M. Structure and lipid interactions of an anti-inflammatory and anti-atherogenic 10-residue class G(*) apolipoprotein J peptide using solution NMR. Biochim. Biophys. Acta 2011, 1808, 498–507. [Google Scholar] [CrossRef]
- Martinez-Bujidos, M.; Rull, A.; Gonzalez-Cura, B.; Perez-Cuellar, M.; Montoliu-Gaya, L.; Villegas, S.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2015, 29, 1688–1700. [Google Scholar] [CrossRef]
- Rivas-Urbina, A.; Rull, A.; Montoliu-Gaya, L.; Perez-Cuellar, M.; Ordonez-Llanos, J.; Villegas, S.; Sanchez-Quesada, J.L. Low-density lipoprotein aggregation is inhibited by apolipoprotein J-derived mimetic peptide d-[113-122]apoJ. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158541. [Google Scholar] [CrossRef]
- Oppi, S.; Luscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research-Which Is My Line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef]
- Mansukhani, N.A.; Wang, Z.; Shively, V.P.; Kelly, M.E.; Vercammen, J.M.; Kibbe, M.R. Sex Differences in the LDL Receptor Knockout Mouse Model of Atherosclerosis. Artery Res. 2017, 20, 8–11. [Google Scholar] [CrossRef]
- Melchionna, M.; Styan, K.E.; Marchesan, S. The Unexpected Advantages of Using D-Amino Acids for Peptide Self- Assembly into Nanostructured Hydrogels for Medicine. Curr. Top. Med. Chem. 2016, 16, 2009–2018. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Havel, R.J.; Eder, H.A.; Bragdon, J.H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Investig. 1955, 34, 1345–1353. [Google Scholar] [CrossRef]
- de Juan-Franco, E.; Perez, A.; Ribas, V.; Sanchez-Hernandez, J.A.; Blanco-Vaca, F.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Standardization of a method to evaluate the antioxidant capacity of high-density lipoproteins. Int. J. Biomed Sci. 2009, 5, 402–410. [Google Scholar]
- Ribas, V.; Sanchez-Quesada, J.L.; Anton, R.; Camacho, M.; Julve, J.; Escola-Gil, J.C.; Vila, L.; Ordonez-Llanos, J.; Blanco-Vaca, F. Human apolipoprotein A-II enrichment displaces paraoxonase from HDL and impairs its antioxidant properties: A new mechanism linking HDL protein composition and antiatherogenic potential. Circ. Res. 2004, 95, 789–797. [Google Scholar] [CrossRef]
- Benitez, S.; Sanchez-Quesada, J.L.; Lucero, L.; Arcelus, R.; Ribas, V.; Jorba, O.; Castellvi, A.; Alonso, E.; Blanco-Vaca, F.; Ordonez-Llanos, J. Changes in low-density lipoprotein electronegativity and oxidizability after aerobic exercise are related to the increase in associated non-esterified fatty acids. Atherosclerosis 2002, 160, 223–232. [Google Scholar] [CrossRef]
- Yu, Y.; Si, Y.; Song, G.; Luo, T.; Wang, J.; Qin, S. Ethanolic extract of propolis promotes reverse cholesterol transport and the expression of ATP-binding cassette transporter A1 and G1 in mice. Lipids 2011, 46, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Leman, L.J.; Maryanoff, B.E.; Ghadiri, M.R. Molecules that mimic apolipoprotein A-I: Potential agents for treating atherosclerosis. J. Med. Chem. 2014, 57, 2169–2196. [Google Scholar] [CrossRef] [PubMed]
- Stoekenbroek, R.M.; Stroes, E.S.; Hovingh, G.K. ApoA-I mimetics. Handb. Exp. Pharmacol. 2015, 224, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Urbina, A.; Rull, A.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Electronegative LDL: An Active Player in Atherogenesis or a By- Product of Atherosclerosis? Curr. Med. Chem. 2019, 26, 1665–1679. [Google Scholar] [CrossRef] [PubMed]
- Diffenderfer, M.R.; Schaefer, E.J. The composition and metabolism of large and small LDL. Curr. Opin. Lipidol. 2014, 25, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Bubb, K. The mystery of evacetrapib-why are CETP inhibitors failing? Expert Rev. Cardiovasc. Ther. 2020, 18, 127–130. [Google Scholar] [CrossRef]
- Tall, A.R. Plasma high density lipoproteins: Therapeutic targeting and links to atherogenic inflammation. Atherosclerosis 2018, 276, 39–43. [Google Scholar] [CrossRef]
- Tribble, D.L. Lipoprotein oxidation in dyslipidemia: Insights into general mechanisms affecting lipoprotein oxidative behavior. Curr. Opin. Lipidol. 1995, 6, 196–208. [Google Scholar] [CrossRef]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.A.; Chapman, M.J.; Kontush, A. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: Relevance to inflammation and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordonez Llanos, J.; Benitez, S. Electronegative LDL: A circulating modified LDL with a role in inflammation. Mediat. Inflamm. 2013, 2013, 181324. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chen, F.Y.; Lee, A.S.; Ting, K.H.; Chang, C.M.; Hsu, J.F.; Lee, W.S.; Sheu, J.R.; Chen, C.H.; Shen, M.Y. Sesamol reduces the atherogenicity of electronegative L5 LDL in vivo and in vitro. J. Nat. Prod. 2015, 78, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.S.; Yang, T.C.; Chang, P.Y.; Chang, S.F.; Ho, S.L.; Chen, H.L.; Lu, S.C. Electronegative LDL is linked to high-fat, high-cholesterol diet-induced nonalcoholic steatohepatitis in hamsters. J. Nutr. Biochem. 2016, 30, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef]
- Koonen, D.P.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.L.; Ong, H.; Vance, D.E.; Dyck, J.R. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls | Scramble | d-[113–122]apoJ | |
|---|---|---|---|
| Total cholesterol (mM) | 52.5 ± 8.2 | 50.7 ± 7.7 | 51.9 ± 7.6 |
| VLDL-c (mM) | 39.3 ± 8.2 | 36.5 ± 7.5 | 34.9 ± 8.0 |
| LDL-c (mM) | 9.6 ± 2.0 | 10.4 ± 3.8 | 13.6 ± 4.0 |
| HDL-c (mM) | 3.5 ± 1.5 | 3.3 ± 0.9 | 3.5 ± 1.8 |
| Triglycerides (mM) | 9.6 ± 2.2 | 7.8 ± 3.1 | 7.9 ± 3.5 |
| Phospholipids (mM) | 10.8 ± 0.5 | 10.6 ± 1.5 | 10.2 ± 1.8 |
| AST (U/L) | 107.4 ± 71.4 | 110.3 ± 70.9 | 86.8 ± 47.6 |
| ALT (U/L) | 29.3 ± 20.2 | 38.4 ± 25.5 | 22.1 ±11.0 |
| Liver weight (g) | 1.48 ± 0.23 | 1.34 ± 0.32 | 1.33 ± 0.22 |
| Liver cholesterol (µmol/g liver) | 9.50 ± 4.67 | 11.57 ± 4.25 | 10.84 ± 3.63 |
| Liver triglycerides (µmol/g liver) | 12.42 ± 3.98 | 15.43 ± 3.05 | 11.51 ± 3.89 |
| Liver phospholipids (µmol/g liver) | 4.62 ±1.97 | 4.30 ± 1.75 | 4.64 ±1.43 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas-Urbina, A.; Rull, A.; Aldana-Ramos, J.; Santos, D.; Puig, N.; Farre-Cabrerizo, N.; Benitez, S.; Perez, A.; de Gonzalo-Calvo, D.; Escola-Gil, J.C.; et al. Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113–122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice. Biomolecules 2020, 10, 829. https://doi.org/10.3390/biom10060829
Rivas-Urbina A, Rull A, Aldana-Ramos J, Santos D, Puig N, Farre-Cabrerizo N, Benitez S, Perez A, de Gonzalo-Calvo D, Escola-Gil JC, et al. Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113–122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice. Biomolecules. 2020; 10(6):829. https://doi.org/10.3390/biom10060829
Chicago/Turabian StyleRivas-Urbina, Andrea, Anna Rull, Joile Aldana-Ramos, David Santos, Nuria Puig, Nuria Farre-Cabrerizo, Sonia Benitez, Antonio Perez, David de Gonzalo-Calvo, Joan Carles Escola-Gil, and et al. 2020. "Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113–122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice" Biomolecules 10, no. 6: 829. https://doi.org/10.3390/biom10060829
APA StyleRivas-Urbina, A., Rull, A., Aldana-Ramos, J., Santos, D., Puig, N., Farre-Cabrerizo, N., Benitez, S., Perez, A., de Gonzalo-Calvo, D., Escola-Gil, J. C., Julve, J., Ordoñez-Llanos, J., & Sanchez-Quesada, J. L. (2020). Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113–122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice. Biomolecules, 10(6), 829. https://doi.org/10.3390/biom10060829