High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemical Treatments
2.2. Western Blotting Analysis
2.3. Immunofluorescence and Confocal Microscopy
2.4. Transfection of DNA or siRNA
2.5. Flow Cytometry for Determination of Autophagic Flux
2.6. Quantitative Polymerase Chain Reaction (qPCR)
2.7. Measurement of Lysosomal Aacidity and Lysosomal Enzyme Activity
2.8. Statistical Analysis
3. Results
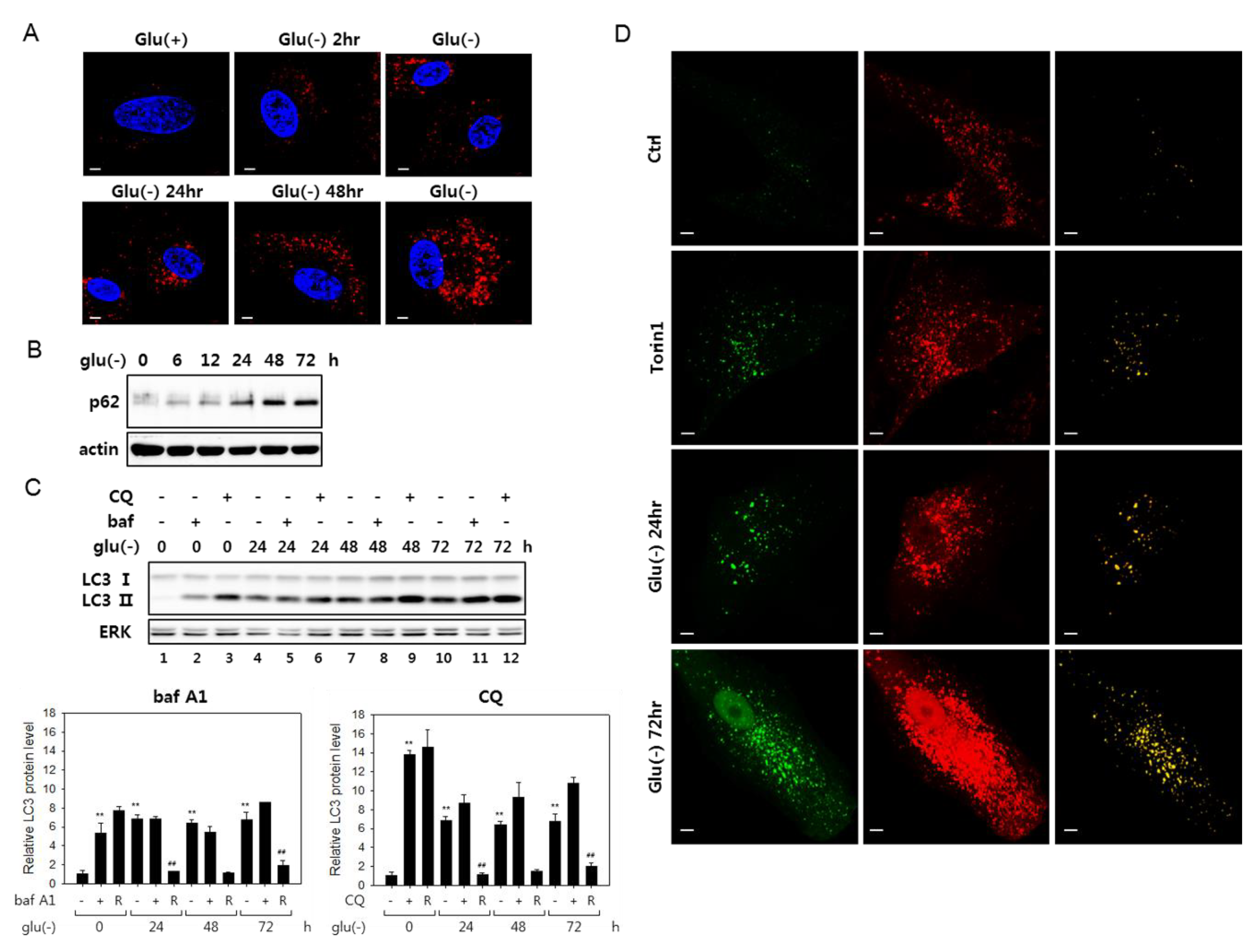
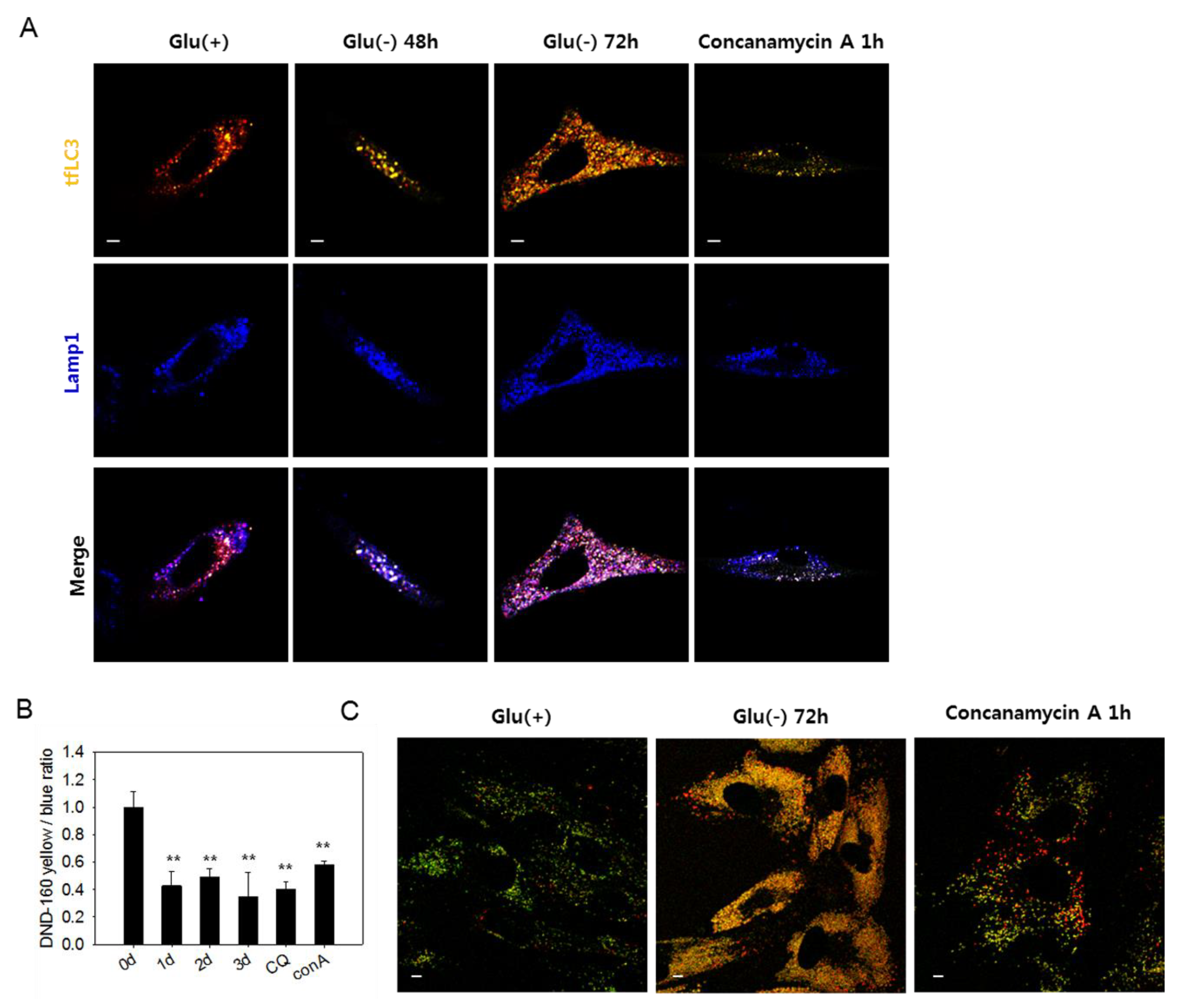
3.1. Autophagic Flux Impairment in Human Fibroblasts Under Glucose Deprivation
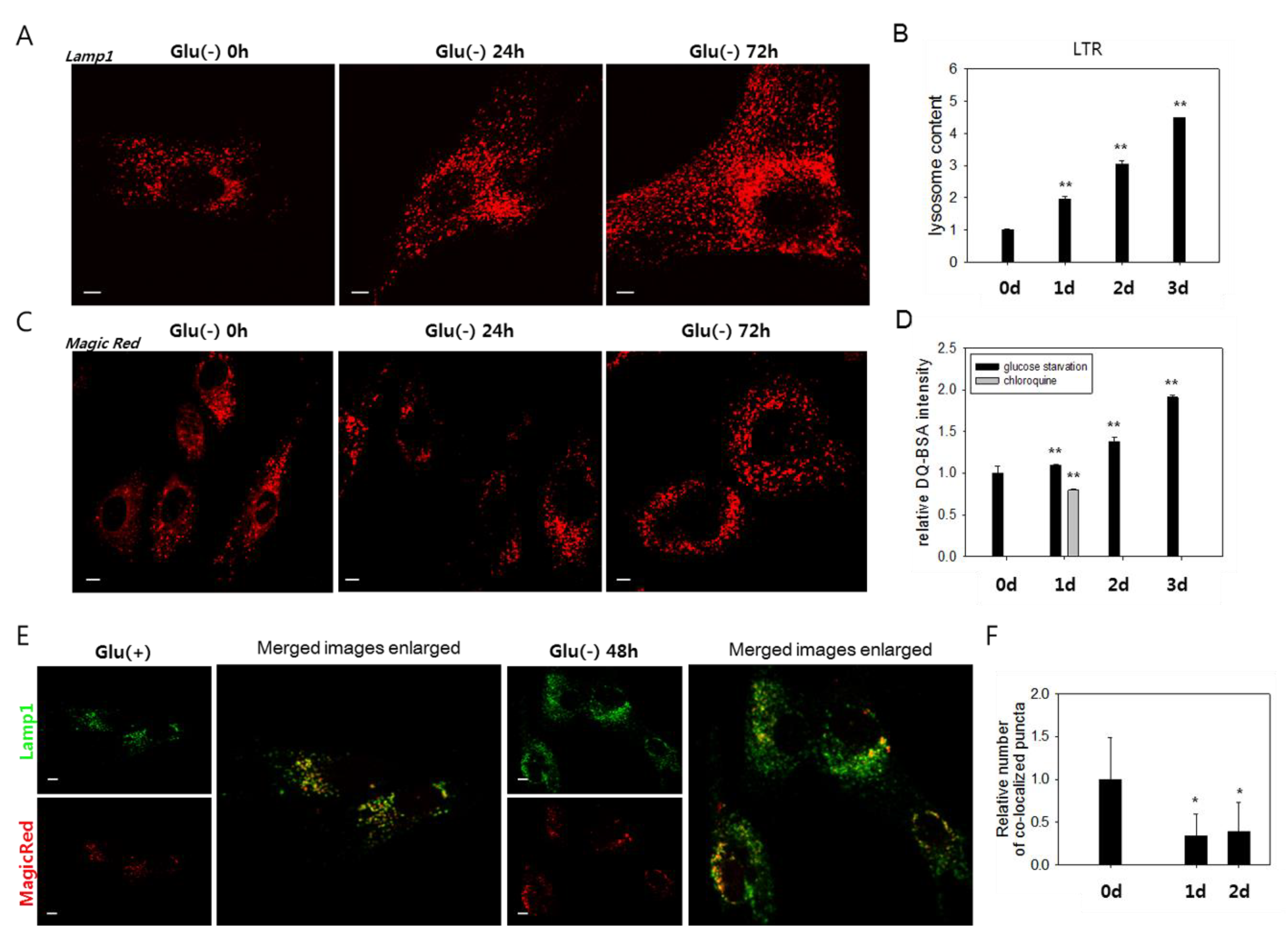
3.2. Lysosomes Accumulate but Many Are Nonfunctional in Glucose-Deprived Cells
3.3. Lysosomes in Glucose-deprived Cells Exhibit Poor Acidity
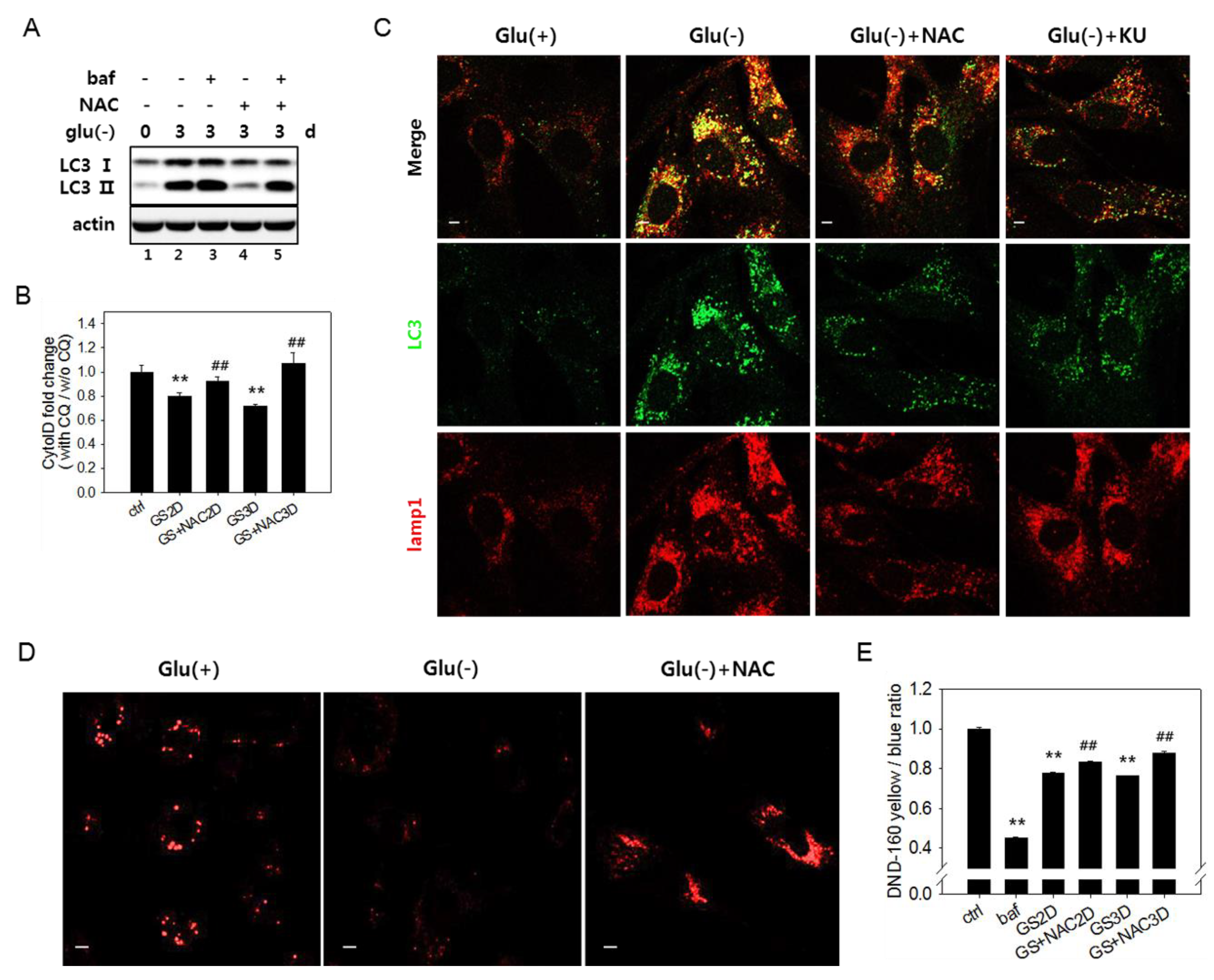
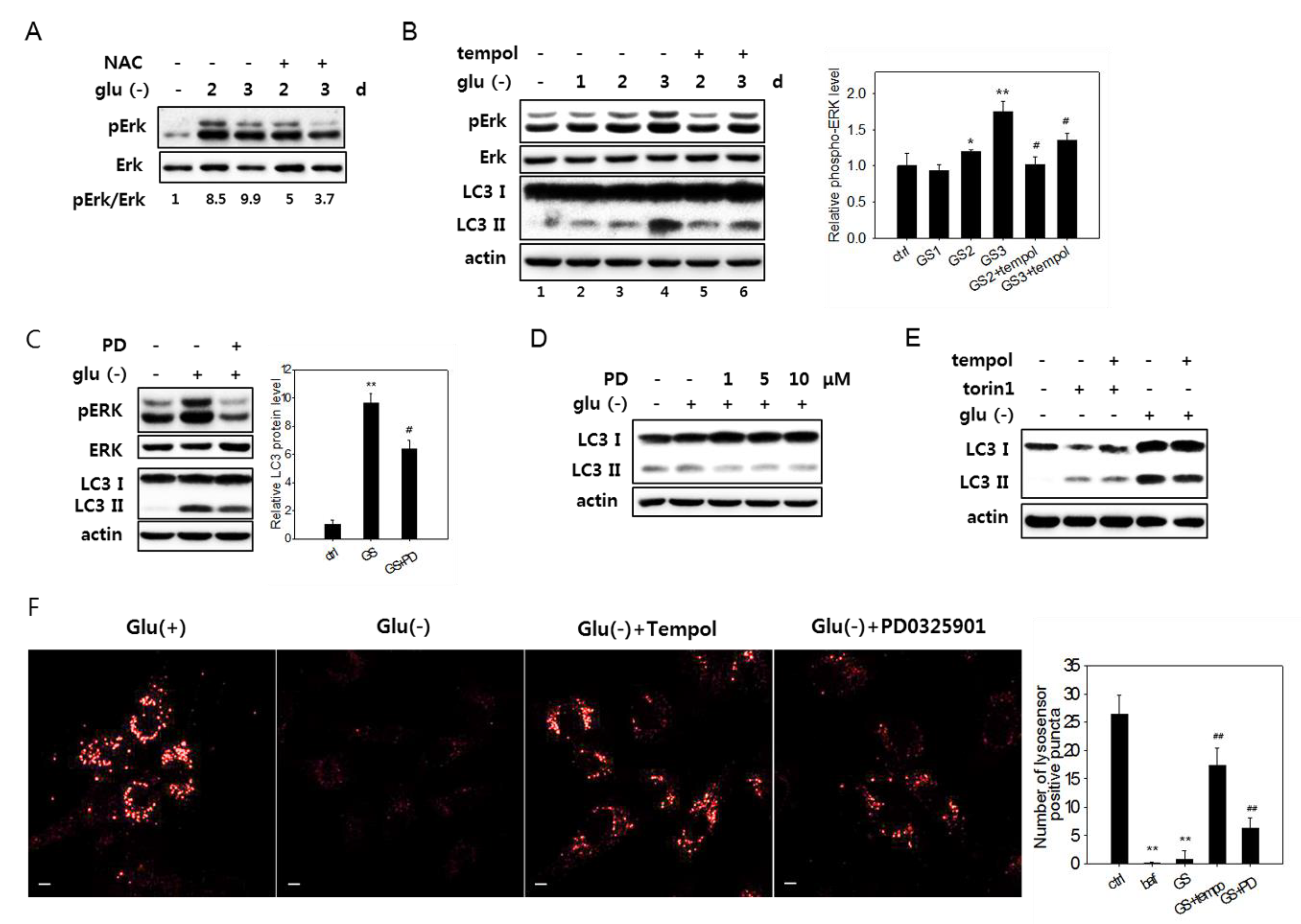
3.4. Increased Reactive Oxygen Species Level Is also Involved in the Failure of Lysosome Acidification and Flux Blockade
3.5. Autophagic Flux Impairment Is Partially Relieved by Inhibition of ATM
3.6. Erk Activation Is Involved in the Impairment of Lysosome Acidity
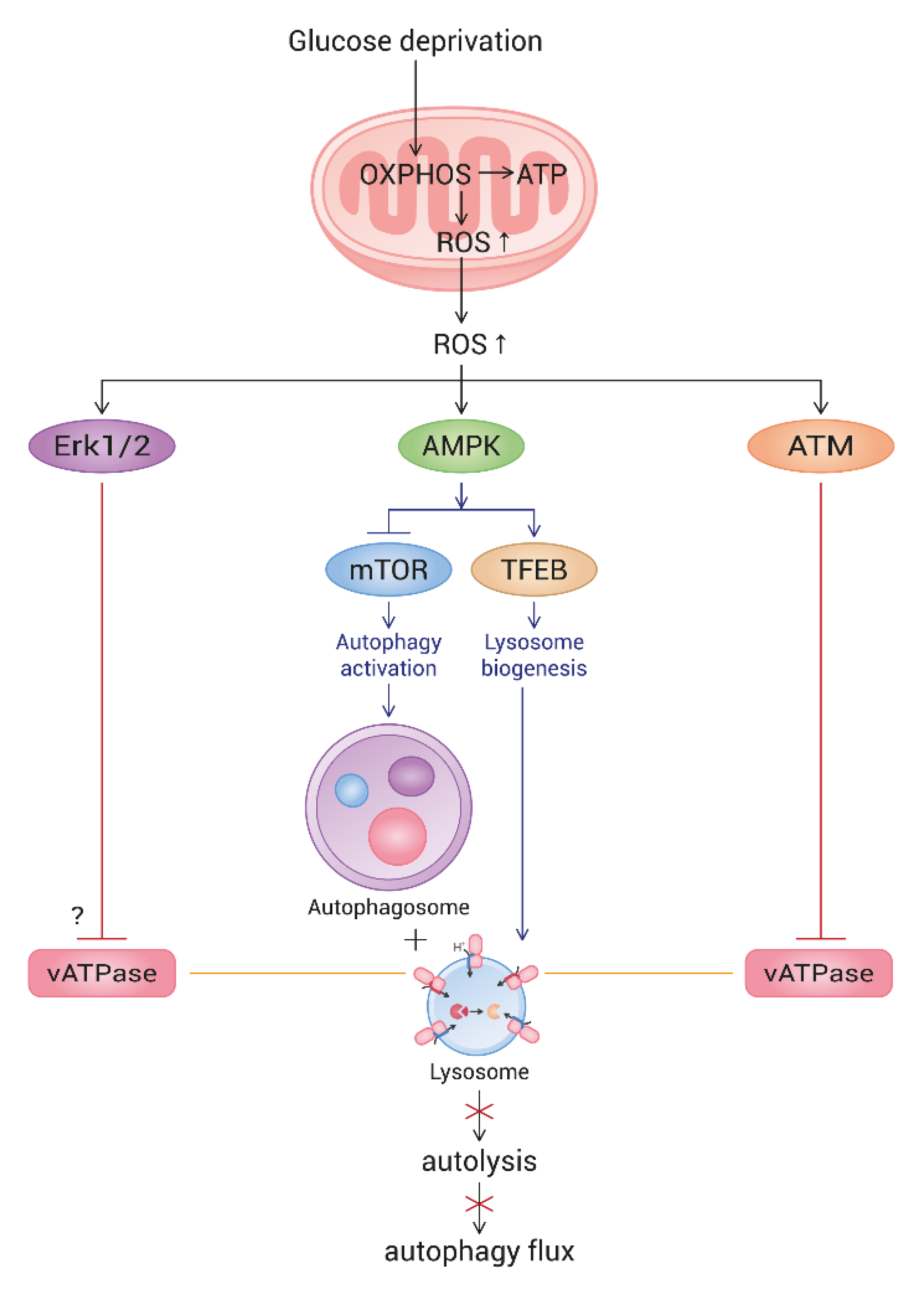
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- White, E. The role for autophagy in cancer. J. Clin. Invest. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of ampk in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. Ulk1.Atg13.Fip200 complex mediates mtor signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ulk1 (hatg1) by amp-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. Ampk and mtor regulate autophagy through direct phosphorylation of ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct vps34 complexes by ampk in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. Ulk1 induces autophagy by phosphorylating beclin-1 and activating vps34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small molecule inhibition of the autophagy kinase ulk1 and identification of ulk1 substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; Deberardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef]
- Lee, Y.J.; Galoforo, S.S.; Berns, C.M.; Chen, J.C.; Davis, B.H.; Sim, J.E.; Corry, P.M.; Spitz, D.R. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J. Biol. Chem. 1998, 273, 5294–5299. [Google Scholar] [CrossRef]
- Ahmad, I.M.; Aykin-Burns, N.; Sim, J.E.; Walsh, S.A.; Higashikubo, R.; Buettner, G.R.; Venkataraman, S.; Mackey, M.A.; Flanagan, S.W.; Oberley, L.W.; et al. Mitochondrial o2*- and h2o2 mediate glucose deprivation-induced stress in human cancer cells. J. Biol. Chem. 2005, 280, 4254–4263. [Google Scholar] [CrossRef] [PubMed]
- Jelluma, N.; Yang, X.; Stokoe, D.; Evan, G.I.; Dansen, T.B.; Haas-Kogan, D.A. Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol. Cancer Res. 2006, 4, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, X.D.; Liu, W.; Zhang, T.Y.; Zuo, J. Glucose deprivation induces mitochondrial dysfunction and oxidative stress in pc12 cell line. J. Cell. Mol. Med. 2003, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals 2009, 2, 150–167. [Google Scholar] [CrossRef]
- Blackburn, R.V.; Spitz, D.R.; Liu, X.; Galoforo, S.S.; Sim, J.E.; Ridnour, L.A.; Chen, J.C.; Davis, B.H.; Corry, P.M.; Lee, Y.J. Metabolic oxidative stress activates signal transduction and gene expression during glucose deprivation in human tumor cells. Free Radic. Biol. Med. 1999, 26, 419–430. [Google Scholar] [CrossRef]
- Gold, A.J.; Yaffe, S.R. Effects of prolonged starvation on cardiac energy metabolism in the rat. J. Nutr. 1978, 108, 410–416. [Google Scholar] [CrossRef]
- Song, S.B.; Hwang, E.S. A rise in atp, ros, and mitochondrial content upon glucose withdrawal correlates with a dysregulated mitochondria turnover mediated by the activation of the protein deacetylase sirt1. Cells 2018, 8, 11. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ros signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of mitochondrial reactive oxygen species in the activation of cellular signals, molecules, and function. Handb. Exp. Pharmacol. 2017, 240, 439–456. [Google Scholar]
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to ampk activation. Cell. Signal. 2013, 25, 50–65. [Google Scholar] [CrossRef]
- Ramirez-Peinado, S.; Leon-Annicchiarico, C.L.; Galindo-Moreno, J.; Iurlaro, R.; Caro-Maldonado, A.; Prehn, J.H.; Ryan, K.M.; Munoz-Pinedo, C. Glucose-starved cells do not engage in prosurvival autophagy. J. Biol. Chem. 2013, 288, 30387–30398. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Ntsapi, C.; Swart, C.; Lumkwana, D.; Loos, B. Autophagic flux failure in neurodegeneration: Identifying the defect and compensating flux offset. In Autophagy in Current Trends in Cellular Physiology and Pathology; Gorbunov, N.V., Schneider, M., Eds.; IntechOpen: London, UK, 2016. [Google Scholar]
- Cherra, S.J., 3rd; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008, 3, 309–323. [Google Scholar] [PubMed]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of foxo by sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin a1 disrupts autophagic flux by inhibiting both v-atpase-dependent acidification and ca-p60a/serca-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification-the emerging role of v-atpase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef]
- Khanna, K.K.; Lavin, M.F.; Jackson, S.P.; Mulhern, T.D. Atm, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001, 8, 1052–1065. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, J.H.; Paull, T.T.; Gehrke, S.; D’Alessandro, A.; Dou, Q.; Gladyshev, V.N.; Schroeder, E.A.; Steyl, S.K.; Christian, B.E.; et al. Mitochondrial redox sensing by the kinase atm maintains cellular antioxidant capacity. Sci. Signal. 2018, 11, eaaq0702. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies atm as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of map kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar]
- Bhagatte, Y.; Lodwick, D.; Storey, N. Mitochondrial ros production and subsequent erk phosphorylation are necessary for temperature preconditioning of isolated ventricular myocytes. Cell Death Dis. 2012, 3, e345. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, K.Y.; Park, S.G.; Yu, S.N.; Kim, Y.W.; Nam, H.W.; An, H.H.; Kim, Y.W.; Ahn, S.C. Mitochondrial ros activates erk/autophagy pathway as a protected mechanism against deoxypodophyllotoxin-induced apoptosis. Oncotarget 2017, 8, 111581–111596. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Chao, A.; Tsai, C.L.; Chuang, W.C.; Huang, W.P.; Chen, G.C.; Lin, C.Y.; Wang, T.H.; Wang, H.S.; Lai, C.H. Bortezomib enhances cancer cell death by blocking the autophagic flux through stimulating erk phosphorylation. Cell Death Dis. 2014, 5, e1510. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An atp-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mtorc1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Huss, M.; Ingenhorst, G.; Konig, S.; Gassel, M.; Drose, S.; Zeeck, A.; Altendorf, K.; Wieczorek, H. Concanamycin a, the specific inhibitor of v-atpases, binds to the v(o) subunit c. J. Biol. Chem. 2002, 277, 40544–40548. [Google Scholar] [CrossRef] [PubMed]
- Cotgreave, I.A.; Moldeus, P. Lung protection by thiol-containing antioxidants. Bull. Eur. Physiopathol. Respir. 1987, 23, 275–277. [Google Scholar]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007, 42, 1632–1650. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged lc3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef]
- Nwadike, C.; Williamson, L.E.; Gallagher, L.E.; Guan, J.L.; Chan, E.Y.W. Ampk inhibits ulk1-dependent autophagosome formation and lysosomal acidification via distinct mechanisms. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/sqstm1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy in neurodegenerative disease: Friend, foe or turncoat? Trends Neurosci. 2006, 29, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Ham, B.; Mogridge, J.; Saftig, P.; Lin, S.; Kim, S.O. Cathepsin b-mediated autophagy flux facilitates the anthrax toxin receptor 2-mediated delivery of anthrax lethal factor into the cytoplasm. J. Biol. Chem. 2010, 285, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding uvrag targets the class c vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Theaker, J.; Pelegrin, P.; Clifton, A.D.; Braddock, M.; Surprenant, A. P2x(7) receptor-mediated release of cathepsins from macrophages is a cytokine-independent mechanism potentially involved in joint diseases. J. Immunol. 2010, 185, 2611–2619. [Google Scholar] [CrossRef]
- Katayama, H.; Yamamoto, A.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. Gfp-like proteins stably accumulate in lysosomes. Cell Struct. Funct. 2008, 33, 1–12. [Google Scholar] [CrossRef]
- Rao, V.T.; Khan, D.; Jones, R.G.; Nakamura, D.S.; Kennedy, T.E.; Cui, Q.L.; Rone, M.B.; Healy, L.M.; Watson, R.; Ghosh, S.; et al. Potential benefit of the charge-stabilized nanostructure saline rns60 for myelin maintenance and repair. Sci. Rep. 2016, 6, 30020. [Google Scholar] [CrossRef]
- Xiang, X.Y.; Yang, X.C.; Su, J.; Kang, J.S.; Wu, Y.; Xue, Y.N.; Dong, Y.T.; Sun, L.K. Inhibition of autophagic flux by ros promotes apoptosis during dtt-induced er/oxidative stress in hela cells. Oncol. Rep. 2016, 35, 3471–3479. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, Y.; Peng, T.; Li, L.; Zhu, W.; Liu, F.; Liu, S.; An, X.; Luo, R.; Cheng, J.; et al. Mitochondrial ros-induced lysosomal dysfunction impairs autophagic flux and contributes to m1 macrophage polarization in a diabetic condition. Clin. Sci. 2019, 133, 1759–1777. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Sharma, P.K.; Tiwari, R.; Rayavarapu, R.G.; Shankar, J.; Chauhan, L.K.S.; Pandey, A.K. Impaired lysosomal activity mediated autophagic flux disruption by graphite carbon nanofibers induce apoptosis in human lung epithelial cells through oxidative stress and energetic impairment. Part. Fibre Toxicol. 2017, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Autophagy: Eat thyself, sustain thyself. Nat. Methods 2015, 12, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liang, Y.; Murphy, S.F.; Huang, A.; Shen, H.; Kelly, D.F.; Sobrado, P.; Sheng, Z. A rapid and high content assay that measures cyto-id-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 2015, 11, 560–572. [Google Scholar] [CrossRef]
- Nishi, T.; Forgac, M. The vacuolar (h+)-atpases--nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef]
- Soliman, M.; Seo, J.Y.; Kim, D.S.; Kim, J.Y.; Park, J.G.; Alfajaro, M.M.; Baek, Y.B.; Cho, E.H.; Kwon, J.; Choi, J.S.; et al. Activation of pi3k, akt, and erk during early rotavirus infection leads to v-atpase-dependent endosomal acidification required for uncoating. PLoS Pathog. 2018, 14, e1006820. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. Tfeb links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Kumar, S.; Hari, K.; Jolly, M.K.; Rangarajan, A. Feedback loops involving ampk, erk and tfeb in matrix detachment leads to nongenetic heterogeneity. BioRxiv 2019, 736546. [Google Scholar] [CrossRef]
- Heo, J.I.; Oh, S.J.; Kho, Y.J.; Kim, J.H.; Kang, H.J.; Park, S.H.; Kim, H.S.; Shin, J.Y.; Kim, M.J.; Kim, M.; et al. Atm mediates interdependent activation of p53 and erk through formation of a ternary complex with p-p53 and p-erk in response to DNA damage. Mol. Biol. Rep. 2012, 39, 8007–8014. [Google Scholar] [CrossRef]
- Dubbelhuis, P.F.; Meijer, A.J. Hepatic amino acid-dependent signaling is under the control of amp-dependent protein kinase. FEBS Lett. 2002, 521, 39–42. [Google Scholar] [CrossRef]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. Amp-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mtor) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef] [PubMed]
- Krause, U.; Bertrand, L.; Hue, L. Control of p70 ribosomal protein s6 kinase and acetyl-coa carboxylase by amp-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur. J. Biochem. 2002, 269, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Tokunaga, C.; Dalal, S.; Richardson, C.; Yoshino, K.; Hara, K.; Kemp, B.E.; Witters, L.A.; Mimura, O.; Yonezawa, K. A possible linkage between amp-activated protein kinase (ampk) and mammalian target of rapamycin (mtor) signalling pathway. Genes Cells 2003, 8, 65–79. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between autophagy and neurodegenerative diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Steele, J.W.; Fan, E.; Kelahmetoglu, Y.; Tian, Y.; Bustos, V. Modulation of autophagy as a therapeutic target for alzheimer’s disease. Postdoc. J. 2013, 1, 21–34. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Manufacturer | Cat # | dose used | Expected effect/property |
|---|---|---|---|---|
| chloroquine | Sigma-Aldrich, St. Louis, MO, USA | C6628 | 50 μM | Inhibition of autophagosome-lysosome fusion [35] |
| bafilomycin A1 | Enzo Life Science, Farmingdale, NY, USA | BML-CM110-0100 | 200 nM | Inhibition of autophagosome-lysosome fusion and lysosome acidification [26] |
| torin1 | Biorbyt, Cambridge, UK | orb146133 | 250 nM | Induction of autophagy by inhibiting mTOR [36] |
| concanamycin A | Sigma-Aldrich | C9705 | 200 nM | Inhibition of vATPase [37] |
| KU60019 | Selleckchem, Houston, TX, USA | S1570 | 0.5 μM | Inhibition of ATM [30] |
| n-acetylcysteine | Sigma-Aldrich | A7250 | 5 mM | Antioxidant [38] |
| 4-hydroxy-2,2,6,6-tetramethyl-piperidin-1-oxyl | Sigma-Aldrich | 176141 | 1 mM | A membrane-permeable free radical scavenger [39] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.B.; Hwang, E.S. High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways. Biomolecules 2020, 10, 761. https://doi.org/10.3390/biom10050761
Song SB, Hwang ES. High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways. Biomolecules. 2020; 10(5):761. https://doi.org/10.3390/biom10050761
Chicago/Turabian StyleSong, Seon Beom, and Eun Seong Hwang. 2020. "High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways" Biomolecules 10, no. 5: 761. https://doi.org/10.3390/biom10050761
APA StyleSong, S. B., & Hwang, E. S. (2020). High Levels of ROS Impair Lysosomal Acidity and Autophagy Flux in Glucose-Deprived Fibroblasts by Activating ATM and Erk Pathways. Biomolecules, 10(5), 761. https://doi.org/10.3390/biom10050761