Ticagrelor Prevents Endothelial Cell Apoptosis through the Adenosine Signalling Pathway in the Early Stages of Hypoxia

 , , ,
, , ,
Abstract

1. Introduction
2. Results
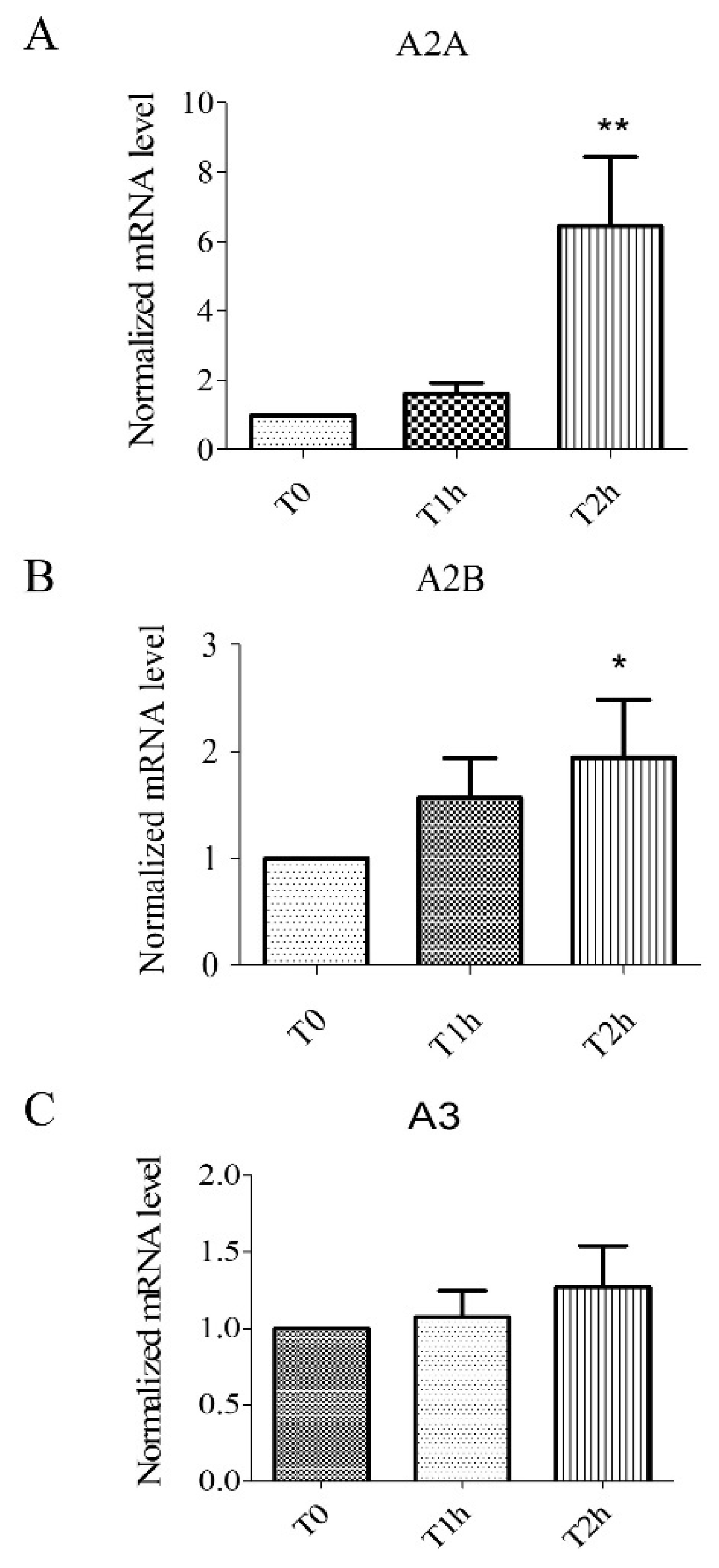
2.1. Hypoxia Stress-Induced Overexpression of A2A, A2B Receptors but Not A3 Receptors
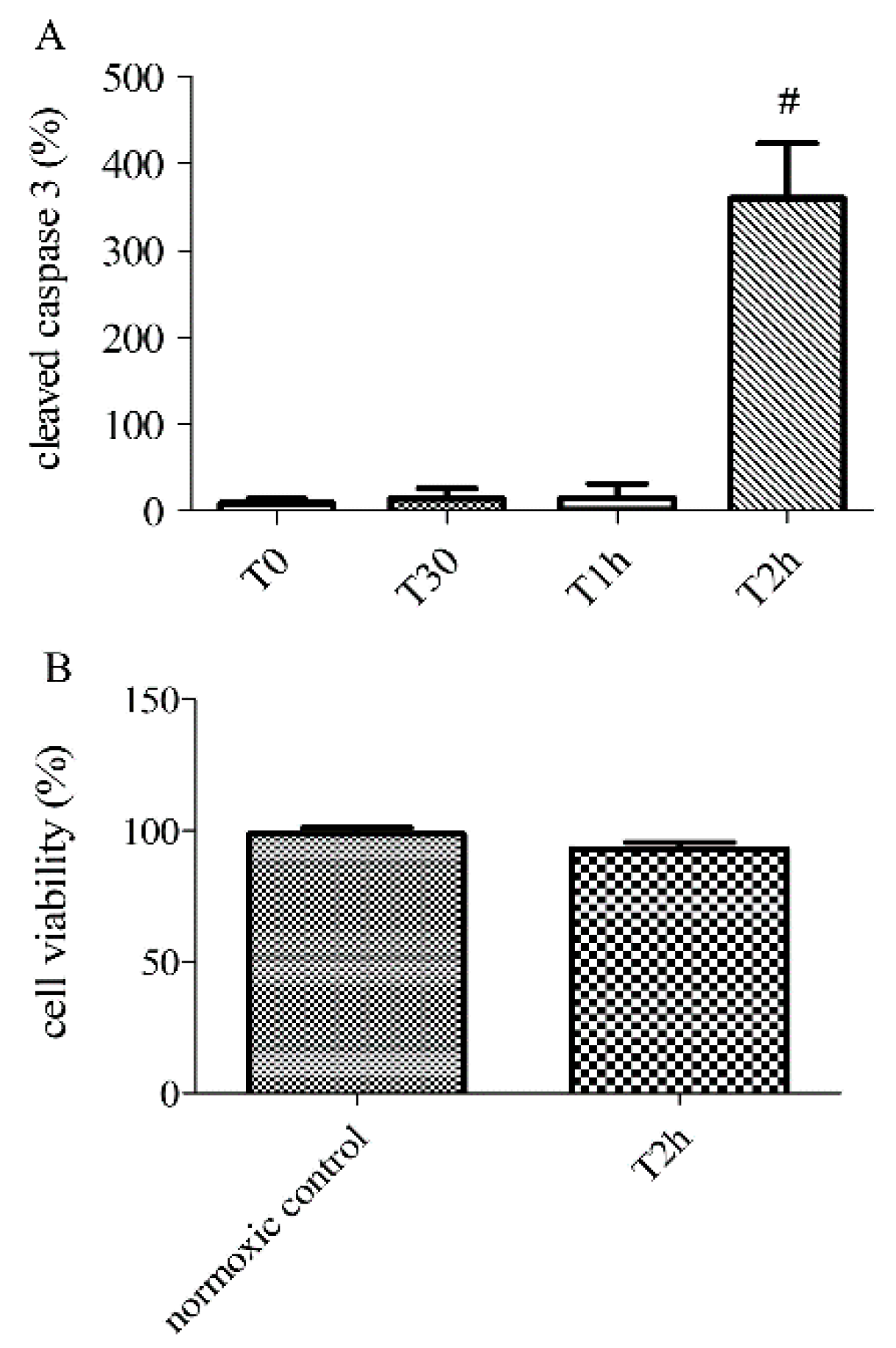
2.2. Effect of Hypoxia-Reoxygenation Stress on Apoptosis Using Different Markers
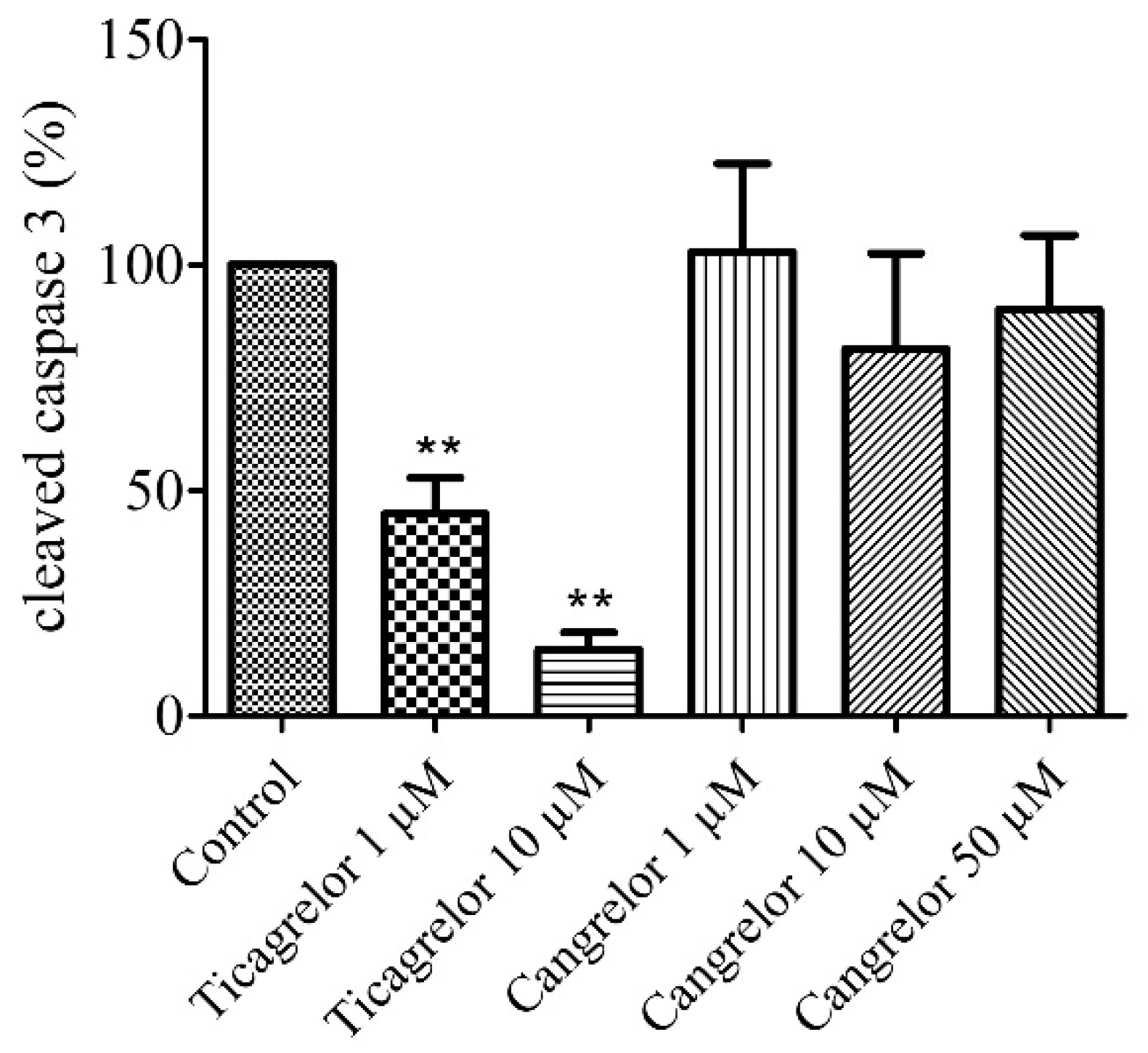
2.3. Pretreatment with Ticagrelor and Its Effect on Apoptosis
2.4. Pretreatment with Cangrelor and Its Effect on Apoptosis
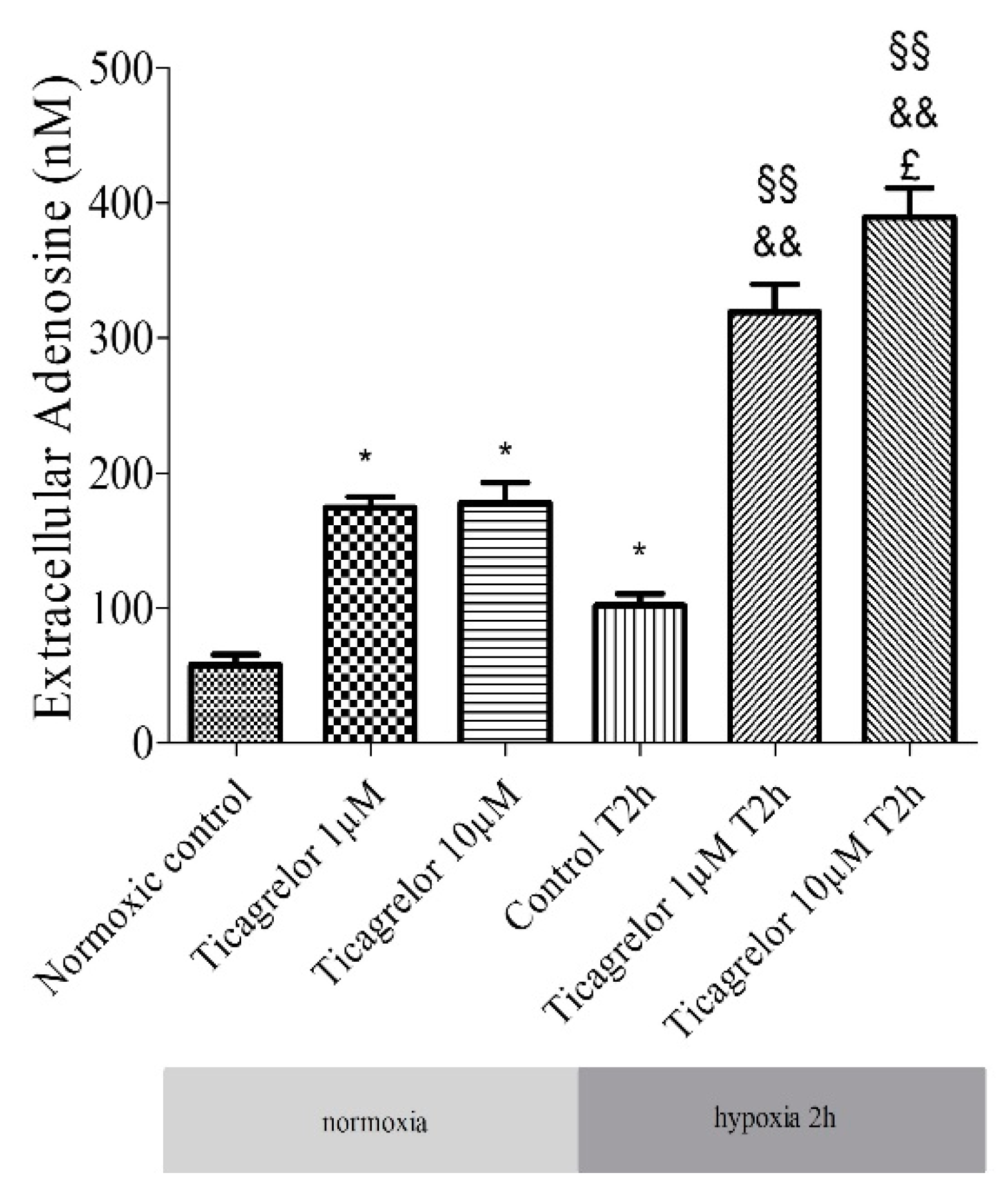
2.5. Pretreatment with Ticagrelor and Its Effects on Adenosine Extracellular Concentration
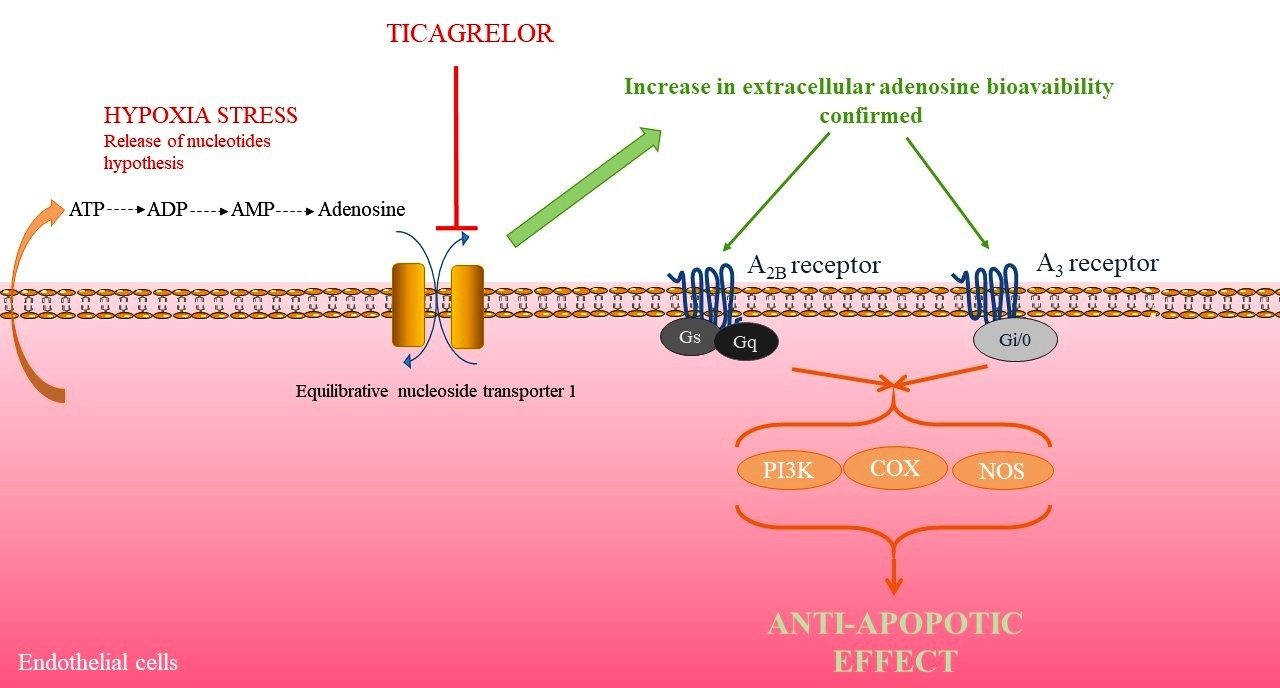
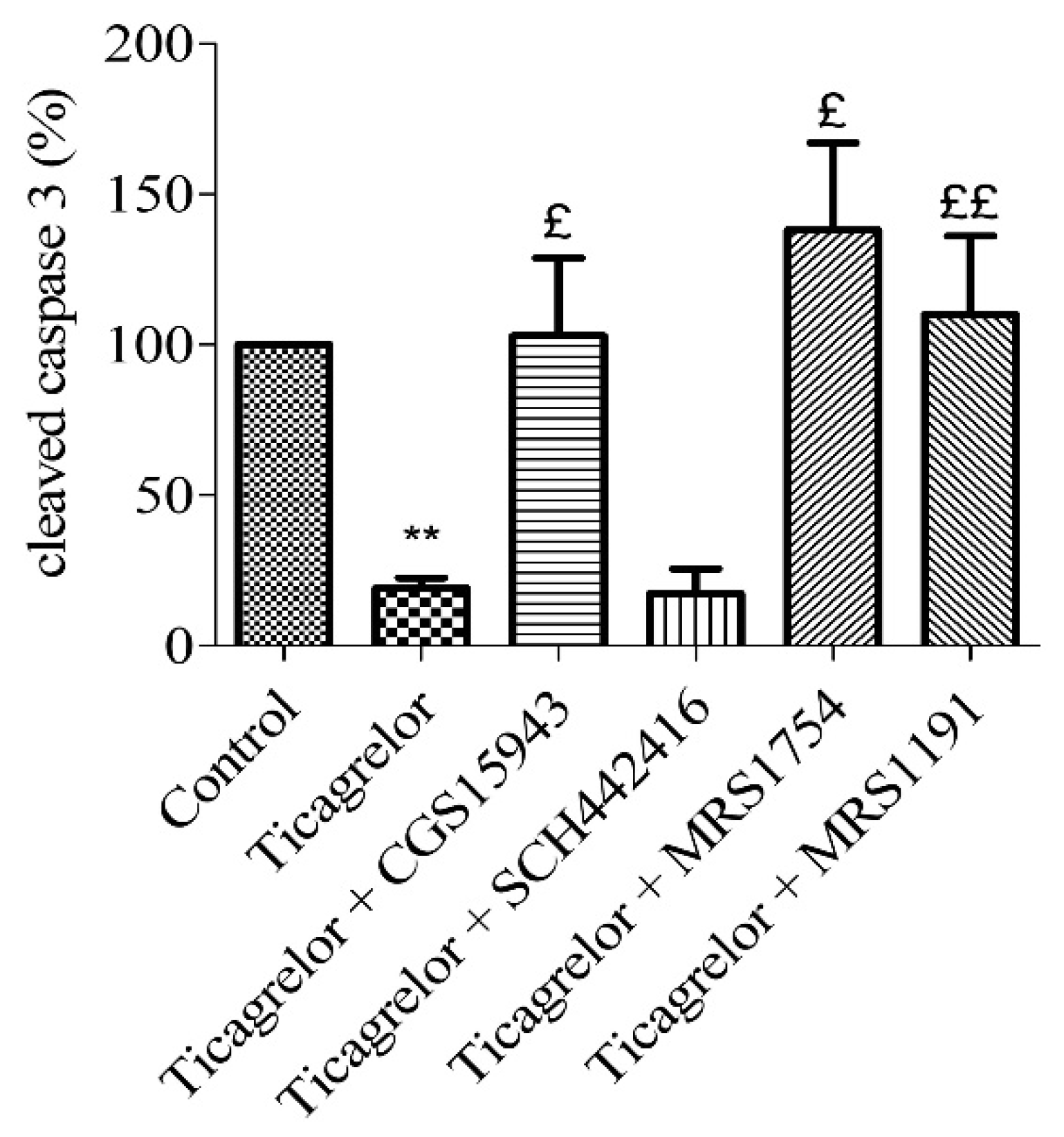
2.6. Involvement of Adenosine Receptors in Ticagrelor-Induced-Anti-Apoptotic Effects against Damaging Effects of Hypoxia in Endothelial Cells
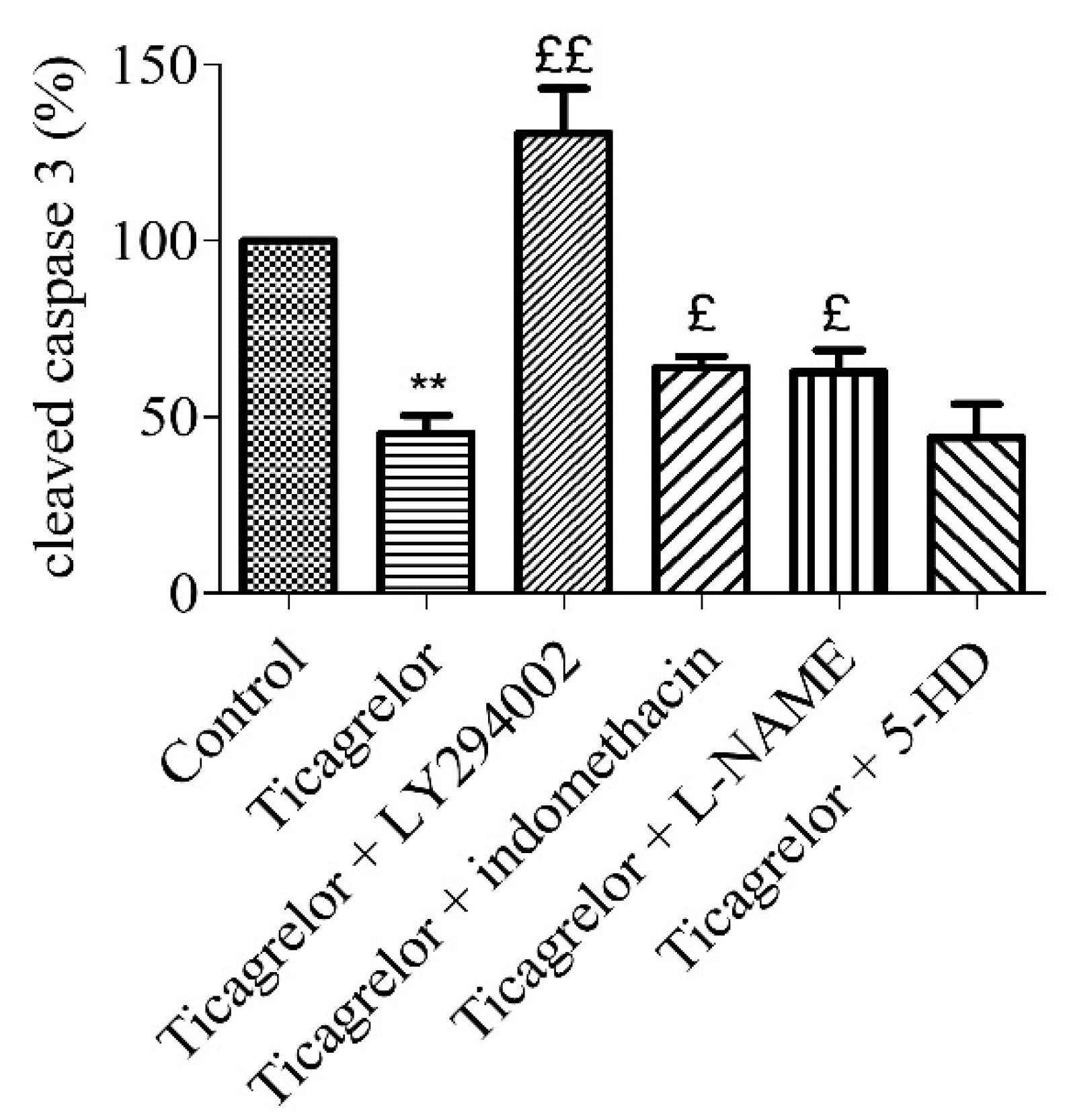
2.7. Anti-Apoptotic Effect of Ticagrelor Involves pi3k, Nos, and Cox Pathways
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Experimental Protocol
4.3. RT-PCR
4.4. Quantification of Adenosine in Extracellular Medium Liquid Chromatography Coupled with a High-Resolution Mass Spectrometer
4.5. Immunoblotting
4.6. PrestoBlue Assays
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and Its Alterations in Cardiovascular Diseases: Life Style Intervention. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M. Injuries to the vascular endothelium: Vascular wall and endothelial dysfunction. Rev. Neurol. Dis. 2008, 5 (Suppl. S1), S4–S11. [Google Scholar]
- Mason, J.C. Cytoprotective pathways in the vascular endothelium. Do they represent a viable therapeutic target? Vascul. Pharmacol. 2016, 86, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.K.; Symons, J.D.; Boudina, S.; Jaishy, B.; Shiu, Y.-T. Role of Endothelial Cells in Myocardial Ischemia-Reperfusion Injury. Vasc. Dis. Prev. 2010, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Scarabelli, T.; Stephanou, A.; Rayment, N.; Pasini, E.; Comini, L.; Curello, S.; Ferrari, R.; Knight, R.; Latchman, D. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation 2001, 104, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Parolari, A.; Rubini, P.; Cannata, A.; Bonati, L.; Alamanni, F.; Tremoli, E.; Biglioli, P. Endothelial damage during myocardial preservation and storage. Ann. Thorac. Surg. 2002, 73, 682–690. [Google Scholar] [CrossRef]
- Djerada, Z.; Feliu, C.; Richard, V.; Millart, H. Current knowledge on the role of P2Y receptors in cardioprotection against ischemia-reperfusion. Pharmacol. Res. 2016, 118, 5–18. [Google Scholar] [CrossRef]
- Chen, Z.M.; Jiang, L.; Chen, Y.; Xie, J.X.; Pan, H.; Peto, R.; Collins, R.; S Liu, L. COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45 852 patients with acute myocardial infarction: Randomised placebo-controlled trial. Lancet 2005, 366, 1607–1621. [Google Scholar] [CrossRef]
- Montalescot, G.; Wiviott, S.D.; Braunwald, E.; Murphy, S.A.; Gibson, C.M.; McCabe, C.H.; Antman, E.M. TRITON-TIMI 38 investigators Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): Double-blind, randomised controlled trial. Lancet Lond. Engl. 2009, 373, 723–731. [Google Scholar] [CrossRef]
- Yang, X.-M.; Liu, Y.; Cui, L.; Yang, X.; Liu, Y.; Tandon, N.; Kambayashi, J.; Downey, J.M.; Cohen, M.V. Platelet P2Y12 blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 251–262. [Google Scholar] [CrossRef]
- Yang, X.-M.; Liu, Y.; Cui, L.; Yang, X.; Liu, Y.; Tandon, N.; Kambayashi, J.; Downey, J.M.; Cohen, M.V. Two classes of anti-platelet drugs reduce anatomical infarct size in monkey hearts. Cardiovasc. Drugs Ther. 2013, 27, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Cui, L.; Alhammouri, A.; Downey, J.M.; Cohen, M.V. Triple therapy greatly increases myocardial salvage during ischemia/reperfusion in the in situ rat heart. Cardiovasc. Drugs Ther. 2013, 27, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Sivaraman, V.; Kunuthur, S.P.; Cohen, M.V.; Downey, J.M.; Yellon, D.M. Cardioprotective Properties of the Platelet P2Y12 Receptor Inhibitor, Cangrelor: Protective in Diabetics and Reliant Upon the Presence of Blood. Cardiovasc. Drugs Ther. Spons. Int. Soc. Cardiovasc. Pharmacother. 2015, 29, 415–418. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, X.; Huang, Y.; Khalil, M.; Wiktor, D.; van Giezen, J.J.J.; Penn, M.S. Adjunctive treatment with ticagrelor, but not clopidogrel, added to tPA enables sustained coronary artery recanalisation with recovery of myocardium perfusion in a canine coronary thrombosis model. Thromb. Haemost. 2010, 104, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, G.; Gutiérrez, M.; Casani, L.; Lambert, C.; Mendieta, G.; Ben-Aicha, S.; Capdevila, A.; Pons-Lladó, G.; Carreras, F.; Carlsson, L.; et al. P2Y12 antagonists and cardiac repair post-myocardial infarction: Global and regional heart function analysis and molecular assessments in pigs. Cardiovasc. Res. 2018, 114, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Vilahur, G.; Gutiérrez, M.; Casani, L.; Varela, L.; Capdevila, A.; Pons-Lladó, G.; Carreras, F.; Carlsson, L.; Hidalgo, A.; Badimon, L. Protective Effects of Ticagrelor on Myocardial Injury After Infarction. Circulation 2016, 134, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Birnbaum, G.D.; Perez-Polo, J.R.; Nanhwan, M.K.; Nylander, S.; Birnbaum, Y. Ticagrelor protects the heart against reperfusion injury and improves remodeling after myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1805–1814. [Google Scholar] [CrossRef]
- Roubille, F.; Lairez, O.; Mewton, N.; Rioufol, G.; Ranc, S.; Sanchez, I.; Cung, T.T.; Elbaz, M.; Piot, C.; Ovize, M. Cardioprotection by clopidogrel in acute ST-elevated myocardial infarction patients: A retrospective analysis. Basic Res. Cardiol. 2012, 107, 275. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.-M.; White, J.; Yellon, D.M.; Bell, R.M.; Downey, J.M. Cangrelor-Mediated Cardioprotection Requires Platelets and Sphingosine Phosphorylation. Cardiovasc. Drugs Ther. 2016, 30, 229–232. [Google Scholar] [CrossRef]
- Yang, X.-M.; Gadde, S.; Audia, J.P.; Alvarez, D.F.; Downey, J.M.; Cohen, M.V. Ticagrelor Does Not Protect Isolated Rat Hearts, Thus Clouding Its Proposed Cardioprotective Role Through ENT 1 in Heart Tissue. J. Cardiovasc. Pharmacol. Ther. 2019, 1074248419829169. [Google Scholar] [CrossRef]
- Korybalska, K.; Rutkowski, R.; Luczak, J.; Czepulis, N.; Karpinski, K.; Witowski, J. The role of purinergic P2Y12 receptor blockers on the angiogenic properties of endothelial cells: An in vitro study. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2018, 69. [Google Scholar] [CrossRef]
- Ganbaatar, B.; Fukuda, D.; Salim, H.M.; Nishimoto, S.; Tanaka, K.; Higashikuni, Y.; Hirata, Y.; Yagi, S.; Soeki, T.; Sata, M. Ticagrelor, a P2Y12 antagonist, attenuates vascular dysfunction and inhibits atherogenesis in apolipoprotein-E-deficient mice. Atherosclerosis 2018, 275, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Huang, Y.; Ji, X.; Sun, J.; Fu, G. Ticagrelor and clopidogrel suppress NF-κB signaling pathway to alleviate LPS-induced dysfunction in vein endothelial cells. BMC Cardiovasc. Disord. 2019, 19, 318. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2 × 7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef] [PubMed]
- YU, J.; HUANG, X.; WU, Q.; WANG, J.; YU, X.; ZHAO, P. Effect of A2A receptor antagonist (SCH 442416) on the mRNA expression of glutamate aspartate transporter and glutamine synthetase in rat retinal Müller cells under hypoxic conditions in vitro. Exp. Ther. Med. 2012, 3, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Salie, R.; Moolman, J.A.; Lochner, A. The mechanism of beta-adrenergic preconditioning: Roles for adenosine and ROS during triggering and mediation. Basic Res. Cardiol. 2012, 107, 281. [Google Scholar] [CrossRef]
- Maddock, H.L.; Broadley, K.J.; Bril, A.; Khandoudi, N. Role of endothelium in ischaemia-induced myocardial dysfunction of isolated working hearts: Cardioprotection by activation of adenosine A(2A) receptors. J. Auton. Pharmacol. 2001, 21, 263–271. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Irenius, E.; Kull, B.; Schulte, G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells11Abbreviations: cAMP, cyclic adenosine 3′,5′-monophosphate; CHO, Chinese hamster ovary; NBMPR, nitrobenzylthioinosine; and NECA, 5′-N-ethyl carboxamido adenosine. Biochem. Pharmacol. 2001, 61, 443–448. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Dawicki, D.D.; Chatterjee, D.; Wyche, J.; Rounds, S. Extracellular ATP and adenosine cause apoptosis of pulmonary artery endothelial cells. Am. J. Physiol. 1997, 273, L485–L494. [Google Scholar] [CrossRef]
- Nanhwan, M.K.; Ling, S.; Kodakandla, M.; Nylander, S.; Ye, Y.; Birnbaum, Y. Chronic treatment with ticagrelor limits myocardial infarct size: An adenosine and cyclooxygenase-2-dependent effect. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2078–2085. [Google Scholar] [CrossRef]
- Wang, D.; Yang, X.-H.; Zhang, J.-D.; Li, R.-B.; Jia, M.; Cui, X.-R. Compared efficacy of clopidogrel and ticagrelor in treating acute coronary syndrome: A meta-analysis. BMC Cardiovasc. Disord. 2018, 18, 217. [Google Scholar] [CrossRef] [PubMed]
- Wittfeldt, A.; Emanuelsson, H.; Brandrup-Wognsen, G.; van Giezen, J.J.J.; Jonasson, J.; Nylander, S.; Gan, L.-M. Ticagrelor enhances adenosine-induced coronary vasodilatory responses in humans. J. Am. Coll. Cardiol. 2013, 61, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Van Giezen, J.J.J.; Sidaway, J.; Glaves, P.; Kirk, I.; Björkman, J.-A. Ticagrelor inhibits adenosine uptake in vitro and enhances adenosine-mediated hyperemia responses in a canine model. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Torngren, K.; Ohman, J.; Salmi, H.; Larsson, J.; Erlinge, D. Ticagrelor improves peripheral arterial function in patients with a previous acute coronary syndrome. Cardiology 2013, 124, 252–258. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Li, D.; Zhang, Y.; Sun, D.; Liu, G.; Pan, Y.; Shi, J.; Li, Y.; Yin, S.; Li, Y. Effects of different doses of ticagrelor on platelet aggregation and endothelial function in diabetic patients with stable coronary artery disease. Platelets 2018, 1–10. [Google Scholar] [CrossRef]
- Cattaneo, M.; Schulz, R.; Nylander, S. Adenosine-mediated effects of ticagrelor: Evidence and potential clinical relevance. J. Am. Coll. Cardiol. 2014, 63, 2503–2509. [Google Scholar] [CrossRef]
- Armstrong, D.; Summers, C.; Ewart, L.; Nylander, S.; Sidaway, J.E.; van Giezen, J.J.J. Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 209–219. [Google Scholar] [CrossRef]
- Nylander, S.; Femia, E.A.; Scavone, M.; Berntsson, P.; Asztély, A.-K.; Nelander, K.; Löfgren, L.; Nilsson, R.G.; Cattaneo, M. Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y12 antagonism. J. Thromb. Haemost. JTH 2013, 11, 1867–1876. [Google Scholar] [CrossRef]
- Bonello, L.; Laine, M.; Kipson, N.; Mancini, J.; Helal, O.; Fromonot, J.; Gariboldi, V.; Condo, J.; Thuny, F.; Frere, C.; et al. Ticagrelor increases adenosine plasma concentration in patients with an acute coronary syndrome. J. Am. Coll. Cardiol. 2014, 63, 872–877. [Google Scholar] [CrossRef]
- Alexopoulos, D.; Moulias, A.; Koutsogiannis, N.; Xanthopoulou, I.; Kakkavas, A.; Mavronasiou, E.; Davlouros, P.; Hahalis, G. Differential effect of ticagrelor versus prasugrel on coronary blood flow velocity in patients with non-ST-elevation acute coronary syndrome undergoing percutaneous coronary intervention: An exploratory study. Circ. Cardiovasc. Interv. 2013, 6, 277–283. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ortega-Paz, L.; Brugaletta, S.; Ariotti, S.; Akkerhuis, K.M.; Karagiannis, A.; Windecker, S.; Valgimigli, M. Adenosine and Ticagrelor Plasma Levels in Patients With and Without Ticagrelor-Related Dyspnea. Circulation 2018, 138, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Rabani, V.; Montange, D.; Meneveau, N.; Davani, S. Impact of ticagrelor on P2Y1 and P2Y12 localization and on cholesterol levels in platelet plasma membrane. Platelets 2018, 29, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, F.; Rabani, V.; Pais-De-Barros, J.-P.; Davani, S. Reorganization of platelet membrane sphingomyelins by adenosine diphosphate and ticagrelor. Chem. Phys. Lipids 2018, 216, 25–29. [Google Scholar] [CrossRef]
- Dobesh, P.P.; Oestreich, J.H. Ticagrelor: Pharmacokinetics, Pharmacodynamics, Clinical Efficacy, and Safety. Pharmacotherapy 2014, 34, 1077–1090. [Google Scholar] [CrossRef]
- Sandinge, A.-S.; Janefeldt, A.; Pehrsson, S.; Nylander, S. Quantification of unbound concentration of ticagrelor in plasma as a proof of mechanism biomarker of the reversal agent, MEDI2452. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Löfgren, L.; Pehrsson, S.; Hägglund, G.; Tjellström, H.; Nylander, S. Accurate measurement of endogenous adenosine in human blood. PLoS ONE 2018, 13, e0205707. [Google Scholar] [CrossRef]
- Feliu, C.; Peyret, H.; Poitevin, G.; Cazaubon, Y.; Oszust, F.; Nguyen, P.; Millart, H.; Djerada, Z. Complementary Role of P2 and Adenosine Receptors in ATP Induced-Anti-Apoptotic Effects Against Hypoxic Injury of HUVECs. Int. J. Mol. Sci. 2019, 20, 1446. [Google Scholar] [CrossRef]
- Rounds, S.; Yee, W.L.; Dawicki, D.D.; Harrington, E.; Parks, N.; Cutaia, M.V. Mechanism of extracellular ATP- and adenosine-induced apoptosis of cultured pulmonary artery endothelial cells. Am. J. Physiol. 1998, 275, L379–L388. [Google Scholar] [CrossRef]
- Harrison, G.J.; Willis, R.J.; Headrick, J.P. Extracellular adenosine levels and cellular energy metabolism in ischemically preconditioned rat heart. Cardiovasc. Res. 1998, 40, 74–87. [Google Scholar] [CrossRef]
- Djerada, Z.; Peyret, H.; Dukic, S.; Millart, H. Extracellular NAADP affords cardioprotection against ischemia and reperfusion injury and involves the P2Y11-like receptor. Biochem. Biophys. Res. Commun. 2013, 434, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Ralevic, V. Purinergic signaling and blood vessels in health and disease. Pharmacol. Rev. 2014, 66, 102–192. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Headrick, J.P.; Peart, J. A3 adenosine receptor-mediated protection of the ischemic heart. Vascul. Pharmacol. 2005, 42, 271–279. [Google Scholar] [CrossRef]
- Rothermel Beverly, A.; Hill Joseph, A. Adenosine A3 Receptor and Cardioprotection. Circulation 2008, 118, 1691–1693. [Google Scholar] [CrossRef]
- Black, R.G.; Guo, Y.; Ge, Z.-D.; Murphree, S.S.; Prabhu, S.D.; Jones, W.K.; Bolli, R.; Auchampach, J.A. Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ. Res. 2002, 91, 165–172. [Google Scholar] [CrossRef]
- Maddock, H.L.; Mocanu, M.M.; Yellon, D.M. Adenosine A(3) receptor activation protects the myocardium from reperfusion/reoxygenation injury. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1307–H1313. [Google Scholar] [CrossRef]
- Stambaugh, K.; Jacobson, K.A.; Jiang, J.L.; Liang, B.T. A novel cardioprotective function of adenosine A1 and A3 receptors during prolonged simulated ischemia. Am. J. Physiol. 1997, 273, H501–H505. [Google Scholar] [CrossRef][Green Version]
- Chanyshev, B.; Shainberg, A.; Isak, A.; Litinsky, A.; Chepurko, Y.; Tosh, D.K.; Phan, K.; Gao, Z.-G.; Hochhauser, E.; Jacobson, K.A. Anti-ischemic effects of multivalent dendrimeric A3 adenosine receptor agonists in cultured cardiomyocytes and in the isolated rat heart. Pharmacol. Res. 2012, 65, 338–346. [Google Scholar] [CrossRef][Green Version]
- Wan, T.C.; Tampo, A.; Kwok, W.-M.; Auchampach, J.A. Ability of CP-532,903 to protect mouse hearts from ischemia/reperfusion injury is dependent on expression of A3 adenosine receptors in cardiomyoyctes. Biochem. Pharmacol. 2019, 163, 21–31. [Google Scholar] [CrossRef]
- Palmer, T.M.; Gettys, T.W.; Stiles, G.L. Differential interaction with and regulation of multiple G-proteins by the rat A3 adenosine receptor. J. Biol. Chem. 1995, 270, 16895–16902. [Google Scholar] [CrossRef]
- Peart, J.N.; Headrick, J.P. Adenosinergic cardioprotection: Multiple receptors, multiple pathways. Pharmacol. Ther. 2007, 114, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Methner, C.; Schmidt, K.; Cohen, M.V.; Downey, J.M.; Krieg, T. Both A2a and A2b adenosine receptors at reperfusion are necessary to reduce infarct size in mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1262–H1264. [Google Scholar] [CrossRef]
- Orru, M.; Bakešová, J.; Brugarolas, M.; Quiroz, C.; Beaumont, V.; Goldberg, S.R.; Lluís, C.; Cortés, A.; Franco, R.; Casadó, V.; et al. Striatal Pre- and Postsynaptic Profile of Adenosine A2A Receptor Antagonists. PLoS ONE 2011, 6, e16088. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Schulz, R. Effects of P2Y12 receptor antagonists beyond platelet inhibition – comparison of ticagrelor with thienopyridines. Br. J. Pharmacol. 2016, 7, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Driscoll, E.M.; Gonzales, M.L.; Park, A.M.; Lucchesi, B.R. Prevention of Arterial Thrombosis by Intravenously Administered Platelet P2T Receptor Antagonist AR-C69931MX in a Canine Model. J. Pharmacol. Exp. Ther. 2000, 295, 492–499. [Google Scholar]
- Srinivasan, S.; Mir, F.; Huang, J.-S.; Khasawneh, F.T.; Lam, S.C.-T.; Breton, G.C.L. The P2Y12 Antagonists, 2-Methylthioadenosine 5′-Monophosphate Triethylammonium Salt and Cangrelor (ARC69931MX), Can Inhibit Human Platelet Aggregation through a Gi-independent Increase in cAMP Levels. J. Biol. Chem. 2009, 284, 16108–16117. [Google Scholar] [CrossRef]
- Urban, D.; Härtel, F.V.; Gadiraju, K.; Gündüz, D.; Aslam, M.; Piper, H.M.; Noll, T. Extracellular ATP attenuates ischemia-induced caspase-3 cleavage in human endothelial cells. Biochem. Biophys. Res. Commun. 2012, 425, 230–236. [Google Scholar] [CrossRef]
- Wee, S.; Peart, J.N.; Headrick, J.P. P2 purinoceptor-mediated cardioprotection in ischemic-reperfused mouse heart. J. Pharmacol. Exp. Ther. 2007, 323, 861–867. [Google Scholar] [CrossRef]
- Millart, H.; Alouane, L.; Oszust, F.; Chevallier, S.; Robinet, A. Involvement of P2Y receptors in pyridoxal-5′-phosphate-induced cardiac preconditioning. Fundam. Clin. Pharmacol. 2009, 23, 279–292. [Google Scholar] [CrossRef]
- Alm, R.; Edvinsson, L.; Malmsjö, M. Organ culture: A new model for vascular endothelium dysfunction. BMC Cardiovasc. Disord. 2002, 2, 8. [Google Scholar] [CrossRef]
- Cohen, S.; Megherbi, M.; Jordheim, L.P.; Lefebvre, I.; Perigaud, C.; Dumontet, C.; Guitton, J. Simultaneous analysis of eight nucleoside triphosphates in cell lines by liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2009, 877, 3831–3840. [Google Scholar] [CrossRef] [PubMed]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Seifar, R.M.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 2009, 81, 7379–7389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Walker, A.D.; Lin, Z.; Han, X.; Blatnik, M.; Steenwyk, R.C.; Groeber, E.A. Strategies for quantitation of endogenous adenine nucleotides in human plasma using novel ion-pair hydrophilic interaction chromatography coupled with tandem mass spectrometry. J. Chromatogr. A 2014, 1325, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Feliu, C.; Millart, H.; Guillemin, H.; Vautier, D.; Binet, L.; Fouley, A.; Djerada, Z. Validation of a fast UPLC-MS/MS method for quantitative analysis of opioids, cocaine, amphetamines (and their derivatives) in human whole blood. Bioanalysis 2015, 7, 2685–2700. [Google Scholar] [CrossRef]
- Djerada, Z.; Feliu, C.; Tournois, C.; Vautier, D.; Binet, L.; Robinet, A.; Marty, H.; Gozalo, C.; Lamiable, D.; Millart, H. Validation of a fast method for quantitative analysis of elvitegravir, raltegravir, maraviroc, etravirine, tenofovir, boceprevir and 10 other antiretroviral agents in human plasma samples with a new UPLC-MS/MS technology. J. Pharm. Biomed. Anal. 2013, 86, 100–111. [Google Scholar] [CrossRef]
- Djerada, Z.; Feliu, C.; Cazaubon, Y.; Smati, F.; Gomis, P.; Guerrot, D.; Charbit, B.; Fernandes, O.; Malinovsky, J.-M. Population Pharmacokinetic-Pharmacodynamic Modeling of Ropivacaine in Spinal Anesthesia. Clin. Pharmacokinet. 2018, 57, 1135–1147. [Google Scholar] [CrossRef]
- Djerada, Z.; Fournet-Fayard, A.; Gozalo, C.; Lelarge, C.; Lamiable, D.; Millart, H.; Malinovsky, J.-M. Population pharmacokinetics of nefopam in elderly, with or without renal impairment, and its link to treatment response. Br. J. Clin. Pharmacol. 2014, 77, 1027–1038. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds Name | Target | Concentration | Reference |
|---|---|---|---|
| CGS15943 | Adenosine receptors antagonist | 1 µM | Avanzato et al. [24] |
| SCH442416 | selective receptor antagonist A2A | 10 µM | Yu et al. [25] |
| MRS1754 | selective receptor antagonist A2B | 0.1 µM | Salie et al. [26] |
| MRS1191 | selective receptor antagonist A3 | 10 µM | Salie et al. [26] |
| LY294002 | PI3K inhibitor | 10 µM | Urban et al. [27] |
| 5-HD | mitoKATP inhibitor | 100 µM | Millart et al. [28] |
| L-NAME | NOS inhibitor | 10 µM | Millart et al. [28] |
| indomethacin | COX inhibitor | 5 µM | Alm et al. [29] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feliu, C.; Peyret, H.; Brassart-Pasco, S.; Oszust, F.; Poitevin, G.; Nguyen, P.; Millart, H.; Djerada, Z. Ticagrelor Prevents Endothelial Cell Apoptosis through the Adenosine Signalling Pathway in the Early Stages of Hypoxia. Biomolecules 2020, 10, 740. https://doi.org/10.3390/biom10050740
Feliu C, Peyret H, Brassart-Pasco S, Oszust F, Poitevin G, Nguyen P, Millart H, Djerada Z. Ticagrelor Prevents Endothelial Cell Apoptosis through the Adenosine Signalling Pathway in the Early Stages of Hypoxia. Biomolecules. 2020; 10(5):740. https://doi.org/10.3390/biom10050740
Chicago/Turabian StyleFeliu, Catherine, Hélène Peyret, Sylvie Brassart-Pasco, Floriane Oszust, Gaël Poitevin, Philippe Nguyen, Hervé Millart, and Zoubir Djerada. 2020. "Ticagrelor Prevents Endothelial Cell Apoptosis through the Adenosine Signalling Pathway in the Early Stages of Hypoxia" Biomolecules 10, no. 5: 740. https://doi.org/10.3390/biom10050740
APA StyleFeliu, C., Peyret, H., Brassart-Pasco, S., Oszust, F., Poitevin, G., Nguyen, P., Millart, H., & Djerada, Z. (2020). Ticagrelor Prevents Endothelial Cell Apoptosis through the Adenosine Signalling Pathway in the Early Stages of Hypoxia. Biomolecules, 10(5), 740. https://doi.org/10.3390/biom10050740