Endogenous Collagenases Regulate Osteoclast Fusion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Cells
2.2. Reagents and Antibodies
2.3. Osteoclast Differentiation and Measurement of Cell Size
2.4. Co-Culture of BMMs with Calvarial Cells
2.5. In Vitro Osteoclast Resorption Assay
2.6. The siRNA Knock-Down
2.7. Quantitative Real-Time PCR
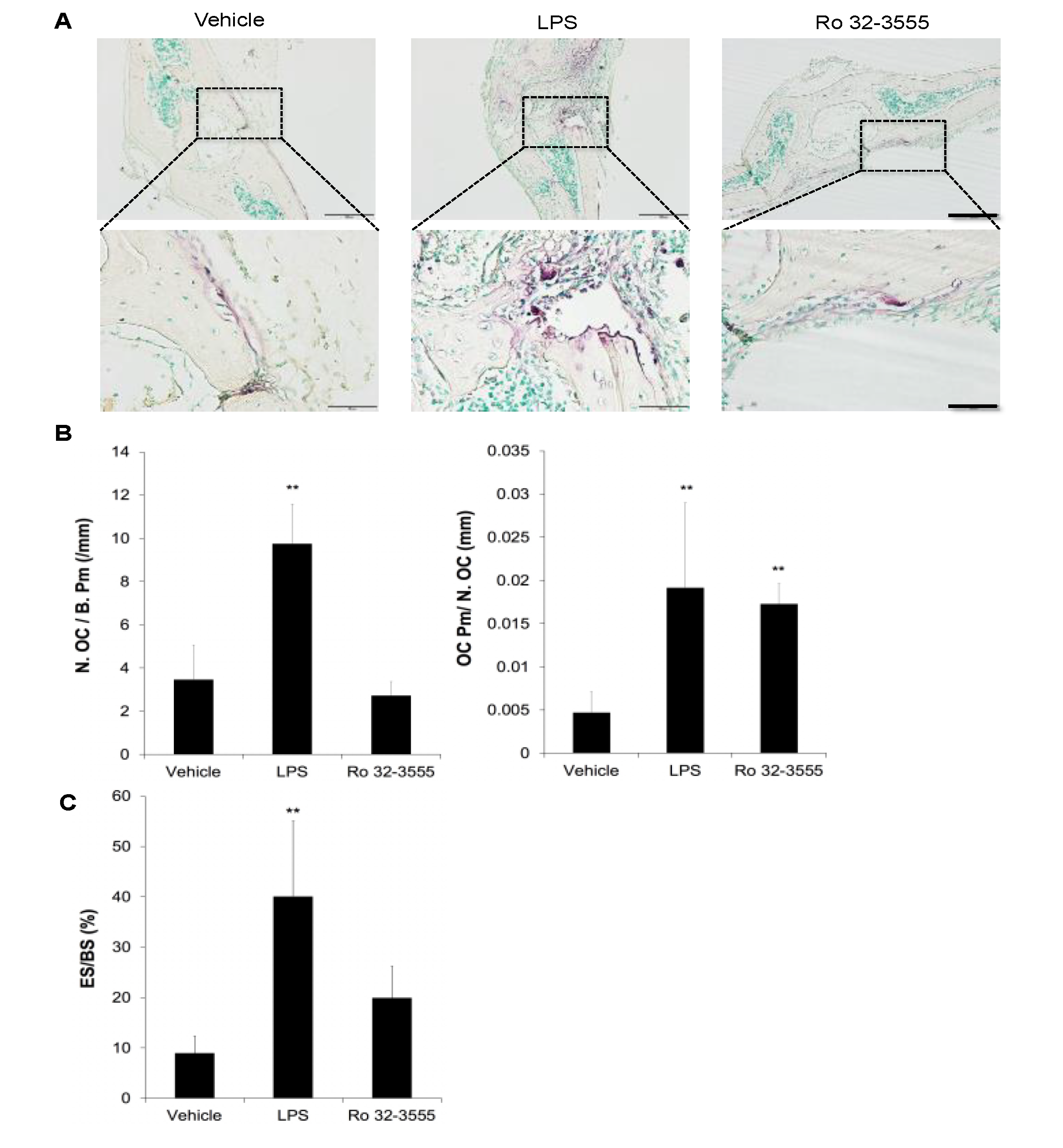
2.8. Measurement of Osteoclasts in Calvarial Bones
2.9. Statistics
3. Results
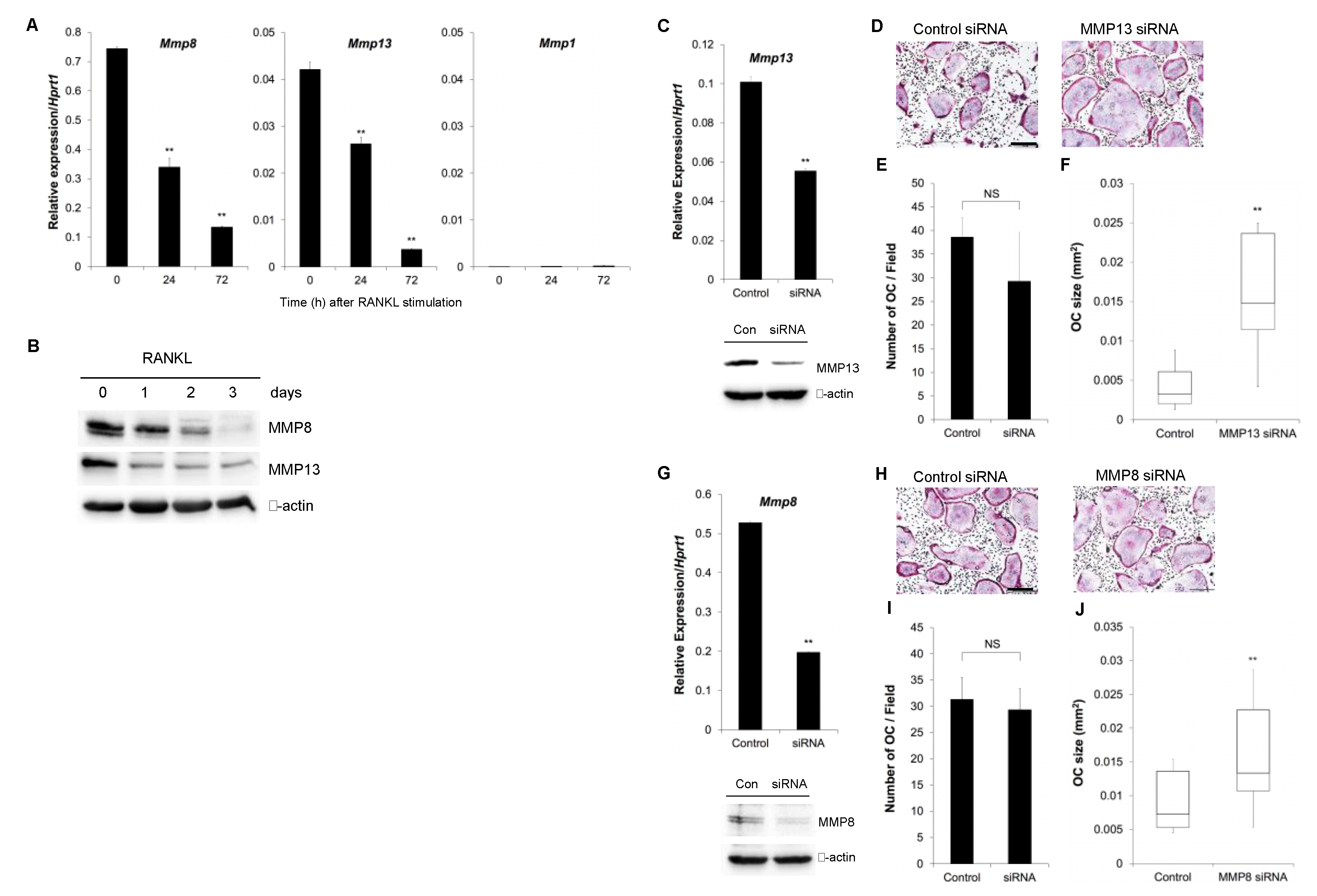
3.1. The Role of Endogenous Collagenases in Osteoclastogenesis
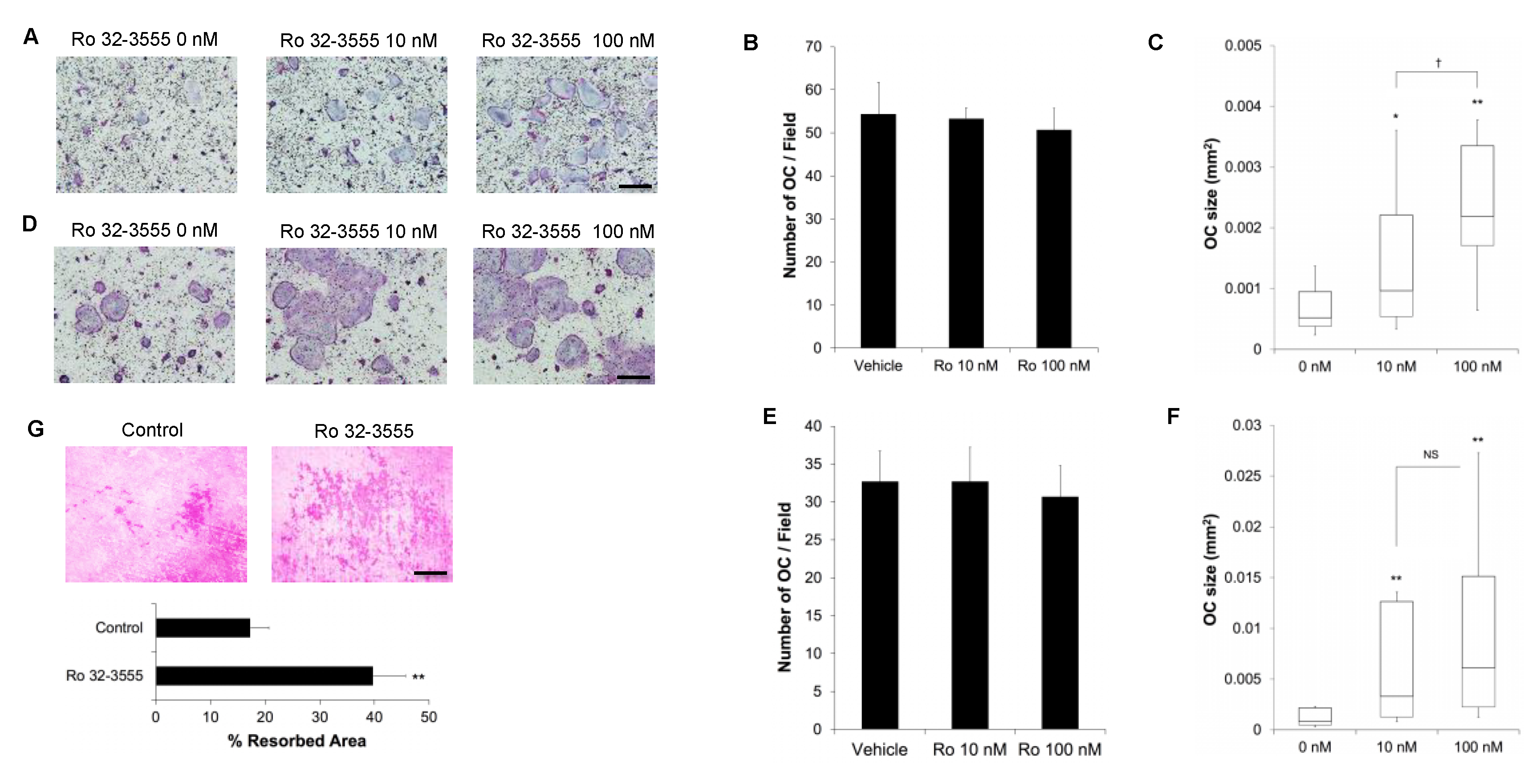
3.2. The Effect of Collagenase Inhibition on Osteoclast Size
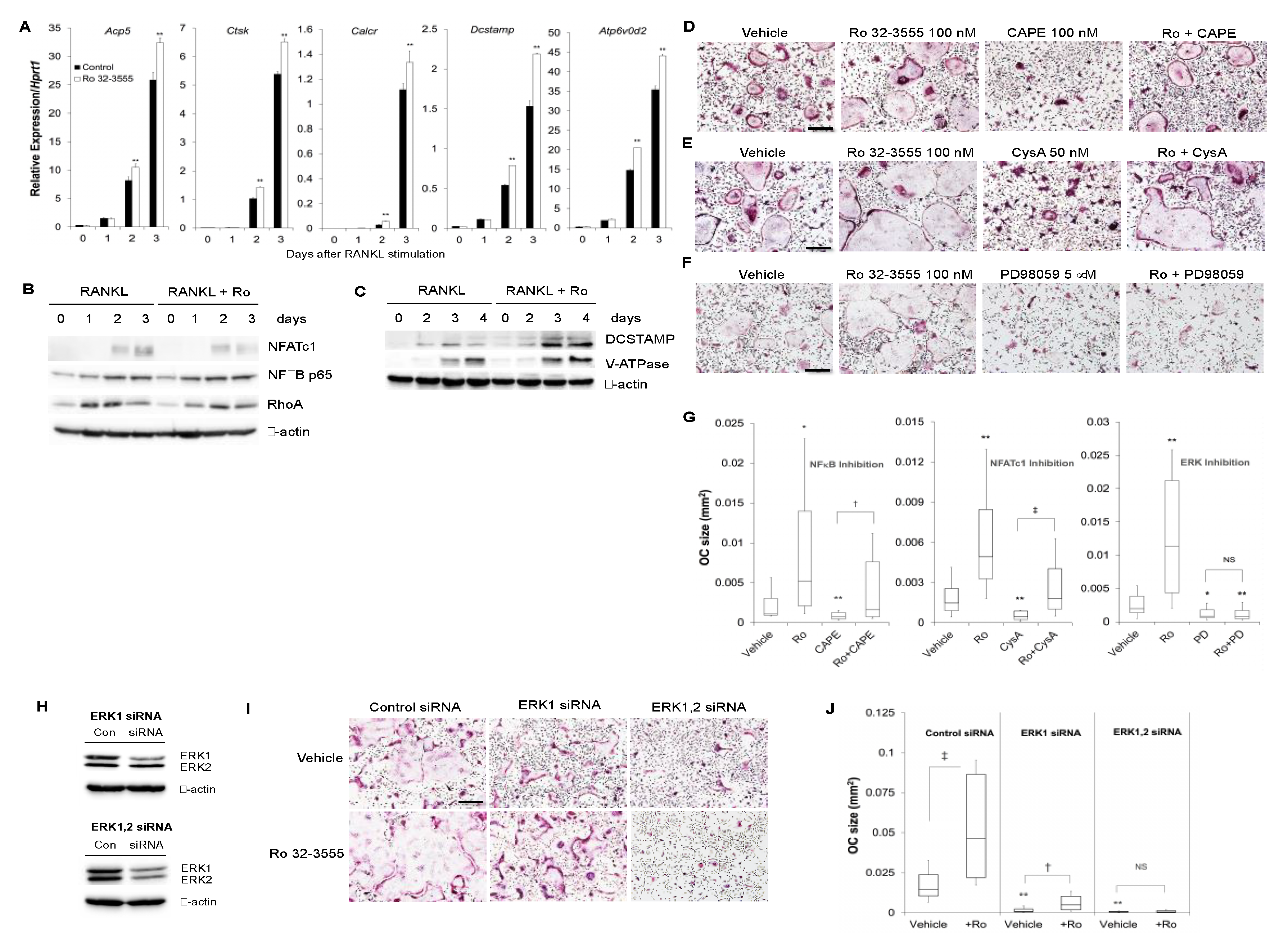
3.3. The Signaling Pathways by which the Inhibition of Collagenase Activities Regulate Osteoclast Size
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Rodan, G.A.; Martin, T.J. Therapeutic approaches to bone diseases. Science 2000, 289, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Shigeyama, Y.; Pap, T.; Kunzler, P.; Simmen, B.R.; Gay, R.E.; Gay, S. Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheumatol. 2000, 43, 2523–2530. [Google Scholar] [CrossRef]
- Bartold, P.M.; Cantley, M.D.; Haynes, D.R. Mechanisms and control of pathologic bone loss in periodontitis. Periodontol 2000 2010, 53, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.H.; Kim, H.H. Signal transduction by receptor activator of nuclear factor kB in osteoclasts. Biochem. Biophys. Res. Commun. 2003, 305, 211–214. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef]
- Xing, L.; Xiu, Y.; Boyce, B.F. Osteoclast fusion and regulation by RANKL-dependent and independent factors. World J. Orthop. 2012, 3, 212–222. [Google Scholar] [CrossRef]
- Pereira, M.; Petretto, E.; Gordon, S.; Bassett, J.H.D.; Williams, G.R.; Behmoaras, J. Common signalling pathways in macrophage and osteoclast multinucleation. J. Cell Sci. 2018, 131, jcs216267. [Google Scholar] [CrossRef]
- Puente, X.S.; Sanchez, L.M.; Overall, C.M.; Lopez-Otin, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed]
- Stahle-Backdahl, M.; Sandstedt, B.; Bruce, K.; Lindahl, A.; Jimenez, M.G.; Vega, J.A.; Lopez-Otin, C. Collagenase-3 (MMP-13) is expressed during human fetal ossification and re-expressed in postnatal bone remodeling and in rheumatoid arthritis. Lab. Invest. 1997, 76, 717–728. [Google Scholar] [PubMed]
- Sasano, Y.; Zhu, J.X.; Tsubota, M.; Takahashi, I.; Onodera, K.; Mizoguchi, I.; Kagayama, M. Gene expression of MMP8 and MMP13 during embryonic development of bone and cartilage in the rat mandible and hind limb. J. Histochem. Cytochem. 2002, 50, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.L.; del Carmen Ovejero, M.; Kirkegaard, T.; Lenhard, T.; Foged, N.T.; Delaisse, J.M. A scrutiny of matrix metalloproteinases in osteoclasts: Evidence for heterogeneity and for the presence of MMPs synthesized by other cells. Bone 2004, 35, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, J.; Ni, J.; Shang, S.; Liu, L.; Xiang, J.; Li, C. Temporal expression of metalloproteinase-8 and -13 and their relationships with extracellular matrix metalloproteinase inducer in the development of ligature-induced periodontitis in rats. J. Periodontal Res. 2013, 48, 411–419. [Google Scholar] [CrossRef]
- Inada, M.; Wang, Y.; Byrne, M.H.; Rahman, M.U.; Miyaura, C.; Lopez-Otin, C.; Krane, S.M. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Natl. Acad. Sci. USA 2004, 101, 17192–17197. [Google Scholar] [CrossRef]
- Stickens, D.; Behonick, D.J.; Ortega, N.; Heyer, B.; Hartenstein, B.; Yu, Y.; Fosang, A.J.; Schorpp-Kistner, M.; Angel, P.; Werb, Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 2004, 131, 5883–5895. [Google Scholar] [CrossRef]
- Tang, S.Y.; Herber, R.P.; Ho, S.P.; Alliston, T. Matrix metalloproteinase-13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. J. Bone Miner. Res. 2012, 27, 1936–1950. [Google Scholar] [CrossRef]
- Kuula, H.; Salo, T.; Pirila, E.; Tuomainen, A.M.; Jauhiainen, M.; Uitto, V.J.; Tjaderhane, L.; Pussinen, P.J.; Sorsa, T. Local and systemic responses in matrix metalloproteinase 8-deficient mice during Porphyromonas gingivalis-induced periodontitis. Infect. Immun. 2009, 77, 850–859. [Google Scholar] [CrossRef]
- Garcia, S.; Forteza, J.; Lopez-Otin, C.; Gomez-Reino, J.J.; Gonzalez, A.; Conde, C. Matrix metalloproteinase-8 deficiency increases joint inflammation and bone erosion in the K/BxN serum-transfer arthritis model. Arthritis Res. Ther. 2010, 12, R224. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Park, J.W.; Lee, J.M.; Suh, J.Y.; Lee, J.K.; Chang, B.S.; Um, H.S.; Kim, J.Y.; Lee, Y. IL-17 inhibits osteoblast differentiation and bone regeneration in rat. Arch. Oral Biol. 2014, 59, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-DDCT) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Huang, H.; Kim, H.J.; Park, C.K.; Kim, H.H. The phosphatidylinositol 3-kinase-mediated production of interferon-beta is critical for the lipopolysaccharide inhibition of osteoclastogenesis. Life Sci. 2008, 83, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Lee, Y.; Kim, H.H. CXCL2 mediates lipopolysaccharide-induced osteoclastogenesis in RANKL-primed precursors. Cytokine 2011, 55, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kB. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef]
- Flanagan, W.M.; Corthesy, B.; Bram, R.J.; Crabtree, G.R. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature 1991, 352, 803–807. [Google Scholar] [CrossRef]
- Alessi, D.R.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A.R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [CrossRef]
- Walker, D.G.; Lapiere, C.M.; Gross, J. A collagenolytic factor in rat bone promoted by parathyroid extract. Biochem. Biophys. Res. Commun. 1964, 15, 397–402. [Google Scholar] [CrossRef]
- Johansson, N.; Saarialho-Kere, U.; Airola, K.; Herva, R.; Nissinen, L.; Westermarck, J.; Vuorio, E.; Heino, J.; Kahari, V.M. Collagenase-3 (MMP-13) is expressed by hypertrophic chondrocytes, periosteal cells, and osteoblasts during human fetal bone development. Dev. Dyn. 1997, 208, 387–397. [Google Scholar] [CrossRef]
- Rifas, L.; Arackal, S. T cells regulate the expression of matrix metalloproteinase in human osteoblasts via a dual mitogen-activated protein kinase mechanism. Arthritis Rheumatol. 2003, 48, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Shin, J.M.; Bae, Y.S.; Cho, H.J.; Park, K.K.; Choe, J.Y.; Han, S.M.; Moon, S.K.; Kim, W.J.; Choi, Y.H.; et al. Melittin has a chondroprotective effect by inhibiting MMP-1 and MMP-8 expressions via blocking NF-kB and AP-1 signaling pathway in chondrocytes. Int. Immunopharmacol. 2015, 25, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Saiganesh, S.; Saathvika, R.; Udhaya, V.; Arumugam, B.; Vishal, M.; Selvamurugan, N. Matrix metalloproteinase-13: A special focus on its regulation by signaling cascades and microRNAs in bone. Int. J. Biol. Macromol. 2018, 109, 338–349. [Google Scholar]
- Lee, S.H.; Rho, J.; Jeong, D.; Sul, J.Y.; Kim, T.; Kim, N.; Kang, J.S.; Miyamoto, T.; Suda, T.; Lee, S.K.; et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef]
- Yagi, M.; Miyamoto, T.; Sawatani, Y.; Iwamoto, K.; Hosogane, N.; Fujita, N.; Morita, K.; Ninomiya, K.; Suzuki, T.; Miyamoto, K.; et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005, 202, 345–351. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.H.; Ha Kim, J.; Choi, Y.; Kim, N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol. Endocrinol. 2008, 22, 176–185. [Google Scholar] [CrossRef]
- He, Y.; Staser, K.; Rhodes, S.D.; Liu, Y.; Wu, X.; Park, S.J.; Yuan, J.; Yang, X.; Li, X.; Jiang, L.; et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS ONE 2011, 6, e24780. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, H.S.; Yeon, J.T.; Choi, S.W.; Chun, C.H.; Kwak, H.B.; Oh, J. GM-CSF regulates fusion of mononuclear osteoclasts into bone-resorbing osteoclasts by activating the Ras/ERK pathway. J. Immunol. 2009, 183, 3390–3399. [Google Scholar] [CrossRef]
- Oh, J.H.; Lee, J.Y.; Park, J.H.; No, J.H.; Lee, N.K. Obatoclax regulates the proliferation and fusion of osteoclast precursors through the inhibition of ERK activation by RANKL. Mol. Cells 2015, 38, 279–284. [Google Scholar] [CrossRef]
- Zhang, C.; Dou, C.E.; Xu, J.; Dong, S. DC-STAMP, the key fusion-mediating molecule in osteoclastogenesis. J. Cell Physiol. 2014, 229, 1330–1335. [Google Scholar] [CrossRef]
- Piper, K.; Boyde, A.; Jones, S.J. The relationship between the number of nuclei of an osteoclast and its resorptive capability in vitro. Anat. Embryol. 1992, 186, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Lees, R.L.; Sabharwal, V.K.; Heersche, J.N. Resorptive state and cell size influence intracellular pH regulation in rabbit osteoclasts cultured on collagen-hydroxyapatite films. Bone 2001, 28, 187–194. [Google Scholar] [CrossRef]
- Boissy, P.; Saltel, F.; Bouniol, C.; Jurdic, P.; Machuca-Gayet, I. Transcriptional activity of nuclei in multinucleated osteoclasts and its modulation by calcitonin. Endocrinology 2002, 143, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Li, S.; Feng, R.; Ma, H.; Sabeh, F.; Roodman, G.D.; Wang, J.; Robinson, S.; Guo, X.E.; Lund, T.; et al. Multiple myeloma-derived MMP-13 mediates osteoclast fusogenesis and osteolytic disease. J. Clin. Invest. 2016, 126, 1759–1772. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.J.; Lee, Y. Endogenous Collagenases Regulate Osteoclast Fusion. Biomolecules 2020, 10, 705. https://doi.org/10.3390/biom10050705
Kim HJ, Lee Y. Endogenous Collagenases Regulate Osteoclast Fusion. Biomolecules. 2020; 10(5):705. https://doi.org/10.3390/biom10050705
Chicago/Turabian StyleKim, Hyo Jeong, and Youngkyun Lee. 2020. "Endogenous Collagenases Regulate Osteoclast Fusion" Biomolecules 10, no. 5: 705. https://doi.org/10.3390/biom10050705
APA StyleKim, H. J., & Lee, Y. (2020). Endogenous Collagenases Regulate Osteoclast Fusion. Biomolecules, 10(5), 705. https://doi.org/10.3390/biom10050705