The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors


 ,
,  and
and 
Abstract
1. Introduction
2. Immunotherapy in Solid Tumors
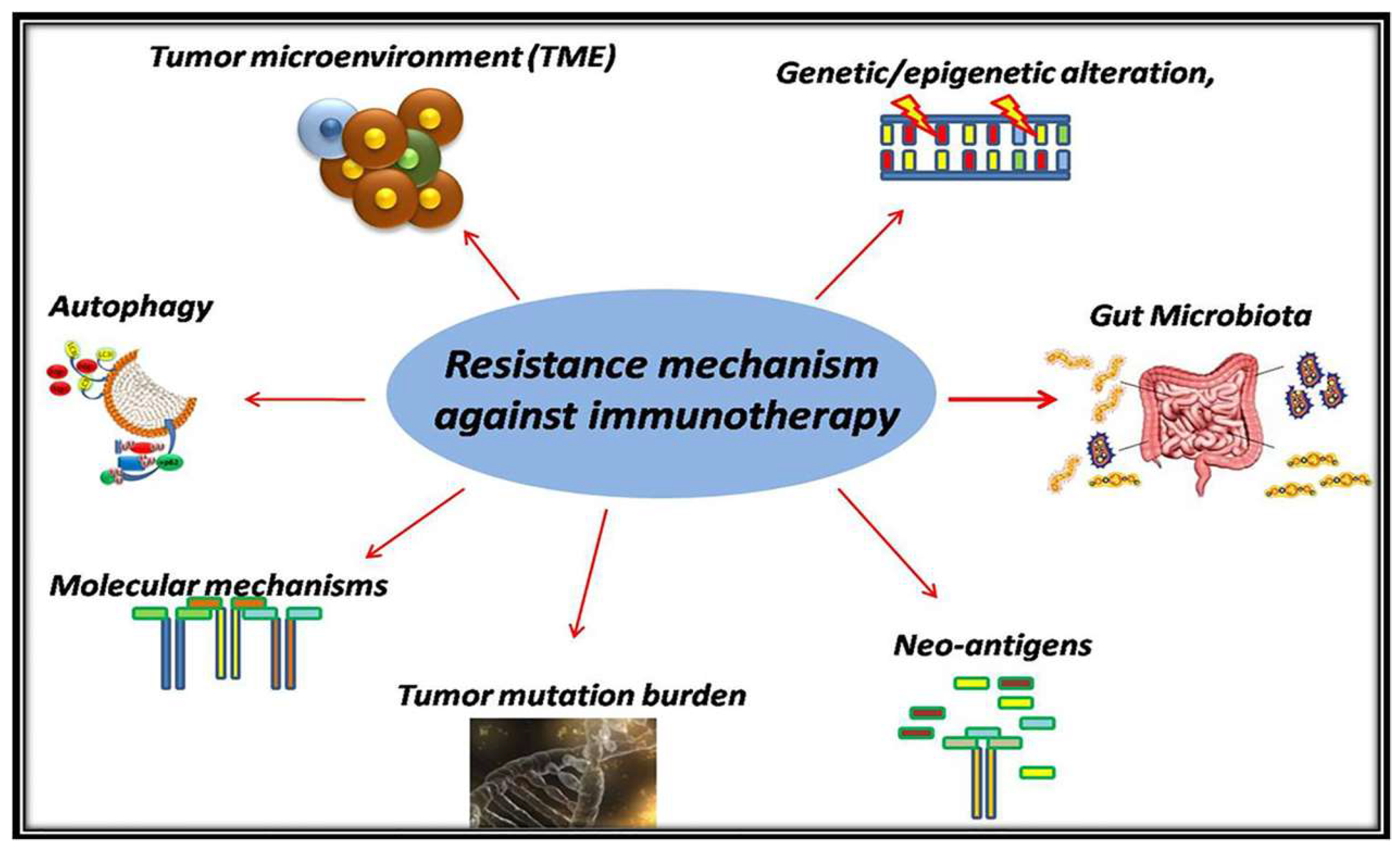
3. Resistance Mechanism
3.1. The Immunosuppressive Mechanism in TME
3.2. Autophagy as a Modulator Mechanism for Antigen Presentation
3.2.1. The Association of Autophagy and the Immune System
3.2.2. The Correlation of Autophagy and Antigen Presenting Cells
3.3. Genetic/Epigenetic Alteration in Cancer
3.4. Tumor Mutational Burden
3.5. Molecular Mechanisms as Immunosuppressive Mechanisms
3.6. The Relation between Gut Microbiota and Immune Response
4. Ways to Overcome the Resistance Mechanism Against Checkpoint Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Decker, W.K.; Da Silva, R.F.; Sanabria, M.H.; Angelo, L.S.; Guimarães, F.; Burt, B.M.; Kheradmand, F.; Paust, S. Cancer Immunotherapy: Historical Perspective of a Clinical Revolution and Emerging Preclinical Animal Models. Front. Immunol. 2017, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2014, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genome Res. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Pio, R.; Ajona, D.; Ortiz-Espinosa, S.; Mantovani, A.; Lambris, J.D. Complementing the Cancer-Immunity Cycle. Front. Immunol. 2019, 10, 774. [Google Scholar] [CrossRef]
- Seliger, B.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Young, K.; Hughes, D.J.; Cunningham, D.; Starling, N. Immunotherapy and pancreatic cancer: Unique challenges and potential opportunities. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816281. [Google Scholar] [CrossRef]
- Fares, C.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Huang, P.-Y.; Guo, S.-S.; Zhang, Y.; Lu, J.-B.; Chen, Q.-Y.; Tang, L.-Q.; Zhang, L.; Liu, L.-T.; Zhang, L.; Mai, H.-Q. Tumor CTLA-4 overexpression predicts poor survival in patients with nasopharyngeal carcinoma. Oncotarget 2016, 7, 13060–13068. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Fontana, V.; Boccardo, S.; Merlo, D.F.; Margallo, E.; Laurent, S.; Morabito, A.; Rijavec, E.; Bello, M.G.D.; Mora, M.; et al. Evaluation of CTLA-4 expression and relevance as a novel prognostic factor in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2012, 61, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H. nab-Paclitaxel as a potential partner with checkpoint inhibitors in solid tumors. OncoTargets Ther. 2016, 10, 101–112. [Google Scholar] [CrossRef]
- Larkin, J.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Caro, R.B.; Zurawski, B.; Kim, S.-W.; Costa, E.C.; Park, K.; Alexandru, A.; Lupinacci, L.; Jimenez, E.D.L.M.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Frontera, O.A.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Powles, T.; Durán, I.; Van Der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castellano, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasić, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, T.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pascual, J.; Ayuso-Sacido, A.; Belda-Iniesta, C. Drug resistance in cancer immunotherapy: New strategies to improve checkpoint inhibitor therapies. Cancer Drug Resist. 2019, 2, 980–993. [Google Scholar] [CrossRef]
- Samstein, R.M.; Arvey, A.; Josefowicz, S.Z.; Peng, X.; Reynolds, A.; Sandstrom, R.; Neph, S.; Sabo, P.; Kim, J.M.; Liao, W.; et al. Foxp3 Exploits a Pre-Existent Enhancer Landscape for Regulatory T Cell Lineage Specification. Cell 2012, 151, 153–166. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, B.S.; Lee, S.-K. Regulatory T Cells in Tumor Microenvironment and Approach for Anticancer Immunotherapy. Immune Netw. 2020, 20, e4. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Mauri, C.; Menon, M. Human regulatory B cells in health and disease: Therapeutic potential. J. Clin. Investig. 2017, 127, 772–779. [Google Scholar] [CrossRef]
- Carter, N.A.; Rosser, E.C.; Mauri, C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res. Ther. 2012, 14, R32. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Matsushita, T.; Tedder, T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 38–57. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Boil. 2019, 27, 30171–30173. [Google Scholar] [CrossRef] [PubMed]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Zin, A.A.M.; Ang, K.C.; Ch’Ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.-M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.-P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Oldford, S.A.; Marshall, J.S. Mast cells as targets for immunotherapy of solid tumors. Mol. Immunol. 2015, 63, 113–124. [Google Scholar] [CrossRef]
- Maciel, T.; Moura, I.C.; Hermine, O. The role of mast cells in cancers. F1000Prime Rep. 2015, 7, 09. [Google Scholar] [CrossRef]
- Koustas, E.; Karamouzis, M.V.; Mihailidou, C.; Schizas, D.; Papavassiliou, A.G. Co-targeting of EGFR and autophagy signaling is an emerging treatment strategy in metastatic colorectal cancer. Cancer Lett. 2017, 396, 94–102. [Google Scholar] [CrossRef]
- Aredia, F.; Guamán-Ortiz, L.M.; Giansanti, V.; Scovassi, A.I. Autophagy and Cancer. Cells 2012, 1, 520–534. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Sarantis, P.; Kyriakopoulou, G.; Papavassiliou, A.G.; Karamouzis, M.V. The Interplay of Autophagy and Tumor Microenvironment in Colorectal Cancer-Ways of Enhancing Immunotherapy Action. Cancers 2019, 11, 533. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M. V Upgraded role of autophagy in colorectal carcinomas. World J. Gastrointest. Oncol. 2018, 10, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Houbaert, D.; Meçe, O.; Agostinis, P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 2019, 26, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Fonteneau, J.-F.; Brilot, F.; Münz, C.; Gannagé, M. The Tumor Antigen NY-ESO-1 Mediates Direct Recognition of Melanoma Cells by CD4+ T Cells after Intercellular Antigen Transfer. J. Immunol. 2015, 196, 64–71. [Google Scholar] [CrossRef]
- Page, D.B.; Hulett, T.W.; Hilton, T.; Hu, H.-M.; Urba, W.J.; Fox, B.A. Glimpse into the future: Harnessing autophagy to promote anti-tumor immunity with the DRibbles vaccine. J. Immunother. Cancer 2016, 4, 25. [Google Scholar] [CrossRef]
- Patterson, N.L.; Mintern, J.D. Intersection of autophagy with pathways of antigen presentation. Protein Cell 2012, 3, 911–920. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2018, 39, 517–560. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.M.; Klenerman, P.; et al. Autophagy is a critical regulator of memory CD8+ T cell formation. eLife 2014, 3, e03706. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wen, Z.; Cheng, F.; Ma, J.; Li, W.; Ren, H.; Sheng, Y.; Dong, H.; Lu, L.; Hu, H.-M.; et al. Tumor-released autophagosomes induce IL-10-producing B cells with suppressive activity on T lymphocytes via TLR2-MyD88-NF-κB signal pathway. OncoImmunology 2016, 5, e1180485. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The role of autophagy in the treatment of BRAF mutant colorectal carcinomas differs based on microsatellite instability status. PLoS ONE 2018, 13, e0207227. [Google Scholar] [CrossRef]
- Valečka, J.; Almeida, C.R.; Su, B.; Pierre, P.; Gatti, E. Autophagy and MHC-restricted antigen presentation. Mol. Immunol. 2018, 99, 163–170. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef]
- Parekh, V.V.; Wu, L.; Boyd, K.L.; Williams, J.A.; Gaddy, J.A.; Olivares-Villagómez, D.; Cover, T.L.; Zong, W.-X.; Zhang, J.; Van Kaer, L. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J. Immunol. 2013, 190, 5086–5101. [Google Scholar] [CrossRef]
- Mintern, J.D.; Macri, C.; Chin, W.J.; Panozza, S.E.; Segura, E.; Patterson, N.L.; Zeller, P.; Bourges, R.; Bedoui, S.; McMillan, P.; et al. Differential use of autophagy by primary dendritic cells specialized in cross-presentation. Autophagy 2015, 11, 906–917. [Google Scholar] [CrossRef]
- Ghislat, G.; Lawrence, T. Autophagy in dendritic cells. Cell. Mol. Immunol. 2018, 15, 944–952. [Google Scholar] [CrossRef]
- Thiele, F.; Tao, S.; Zhang, Y.; Muschaweckh, A.; Zollmann, T.; Protzer, U.; Abele, R.; Drexler, I. Modified Vaccinia Virus Ankara-Infected Dendritic Cells Present CD4+ T-Cell Epitopes by Endogenous Major Histocompatibility Complex Class II Presentation Pathways. J. Virol. 2014, 89, 2698–2709. [Google Scholar] [CrossRef]
- Bronietzki, A.W.; Schuster, M.; Schmitz, I. Autophagy in T-cell development, activation and differentiation. Immunol. Cell Boil. 2014, 93, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Durães, F.V.; Niven, J.; Dubrot, J.; Hugues, S.; Gannagé, M. Macroautophagy in Endogenous Processing of Self- and Pathogen-Derived Antigens for MHC Class II Presentation. Front. Immunol. 2015, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Mao, L.; Wei, J.; Jin, S.; Yang, C.; Liu, H.; Zhu, L.; Qian, W. The crosstalk between autophagic and endo-/exosomal pathways in antigen processing for MHC presentation in anticancer T cell immune responses. J. Hematol. Oncol. 2017, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenet. 2017, 9, 59. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef]
- Héninger, E.; Krueger, T.E.; Lang, J.M. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. 2015, 6, 29. [Google Scholar] [CrossRef]
- Shalabi, H.; Kraft, I.L.; Wang, H.-W.; Yuan, C.M.; Yates, B.; Delbrook, C.; Zimbelman, J.D.; Giller, R.; Stetler-Stevenson, M.; Jaffe, E.S.; et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematology 2018, 103, e215–e218. [Google Scholar] [CrossRef]
- Ready, N.E.; Farago, A.F.; De Braud, F.; Atmaca, A.; Hellmann, M.D.; Schneider, J.G.; Spigel, D.R.; Moreno, V.; Chau, I.; Hann, C.L.; et al. Third-Line Nivolumab Monotherapy in Recurrent SCLC: CheckMate 032. J. Thorac. Oncol. 2019, 14, 237–244. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef]
- Leclerc, M.; Mezquita, L.; De Nerville, G.G.; Tihy, I.; Malenica, I.; Chouaib, S.; Mami-Chouaib, F. Recent Advances in Lung Cancer Immunotherapy: Input of T-Cell Epitopes Associated With Impaired Peptide Processing. Front. Immunol. 2019, 10, 1505. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Uduman, M.; Pabla, S.; Stein, R.B.; Buell, J.S. Mutation-Derived Neoantigens for Cancer Immunotherapy. Front. Immunol. 2019, 10, 1856. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Fang, M.J.; Yan, K.; Guo, J. Biological and clinical research progress of MITF in melanoma. Tumors 2013, 33, 1130–1134. [Google Scholar]
- Falletta, P.; Del Campo, L.S.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Al Emran, A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol. 2019, 40, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, L.; Li, Y.; Li, Q. Molecular Mechanisms and Countermeasures of Immunotherapy Resistance in Malignant Tumor. J. Cancer 2019, 10, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Anfossi, S.; Ramapriyan, R.; Menon, H.; Atalar, S.C.; Aliru, M.; Welsh, J.; Calin, G.A. Role of miRNAs in immune responses and immunotherapy in cancer. Genes Chromosom. Cancer 2019, 58, 244–253. [Google Scholar] [CrossRef]
- Giri, B.R.; Cheng, G. MicroRNAs, T-cell immunity and immunotherapy. Future Med. Chem. 2019, 11, 2043–2045. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Zhao, H.; Ren, X.-B. Relationship of VEGF/VEGFR with immune and cancer cells: Staggering or forward? Cancer Boil. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Sarantis, P.; Theoharis, S.; Saetta, A.A.; Chatziandreou, I.; Kyriakopoulou, G.; Giannopoulou, I.; Michelli, M.; Schizas, D.; Papavassiliou, A.G.; et al. Autophagy-related Proteins as a Prognostic Factor of Patients With Colorectal Cancer. Am. J. Clin. Oncol. 2019, 42, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.H.; Turka, L.A. Regulation of T Cell Homeostasis and Responses by Pten. Front. Immunol. 2012, 3, 151. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Chan, T.A. Immunotherapy and Oncogenic Pathways: The PTEN Connection. Cancer Discov. 2016, 6, 128–129. [Google Scholar] [CrossRef]
- Shi, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188. [Google Scholar]
- Luo, N.; Formisano, L.; Ericsson, P.G.; Sánchez, V.; Dean, P.T.; Opalenik, S.R.; Sanders, M.E.; Cook, R.S.; Arteaga, C.L.; Johnson, D.B.; et al. Melanoma response to anti-PD-L1 immunotherapy requires JAK1 signaling, but not JAK2. OncoImmunology 2018, 7, e1438106. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Goldsberry, W.; Londoño, A.; Randall, T.; Norian, L.; Arend, R. A Review of the Role of Wnt in Cancer Immunomodulation. Cancers 2019, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Different regulation of MHC Class I antigen processing components in human tumors. J. Immunotoxicol. 2008, 5, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Benus, R.F.J.; Van Der Werf, T.S.; Welling, G.W.; Judd, P.A.; Taylor, M.A.; Harmsen, H.J.M.; Whelan, K. Association between Faecalibacterium prausnitzii and dietary fibre in colonic fermentation in healthy human subjects. Br. J. Nutr. 2010, 104, 693–700. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2017, 359, 97–103. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Ueda, K.; Yonekura, S.; Ogasawara, N.; Matsunaga, Y.; Hoshino, R.; Kurose, H.; Chikui, K.; Uemura, K.; Nakiri, M.; Nishihara, K.; et al. Impact of antibiotics on outcome in patients with metastatic renal cell carcinoma treated with immune checkpoint inhibitors. J. Clin. Oncol. 2017, 35, 462. [Google Scholar]
- Humphries, A.; Daud, A. The gut microbiota and immune checkpoint inhibitors. Hum. Vaccines Immunother. 2018, 14, 2178–2182. [Google Scholar] [CrossRef] [PubMed]
- Chilakapati, S.R.; Ricciuti, J.; Zsiros, E. Microbiome and cancer immunotherapy. Curr. Opin. Biotechnol. 2020, 65, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Deshmukh, S.; Reddy, A.; Lightcap, J.; Hayes, M.; McClellan, S.; Singh, S.; Rabideau, B.D.; Glover, T.G.; Roberts, B.; et al. Cancer Immune Checkpoint Inhibitor Therapy and the Gut Microbiota. Integr. Cancer Ther. 2019, 18, 1534735419846379. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced nonsmall-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Signorelli, D.; Giannatempo, P.; Grazia, G.; Aiello, M.M.; Bertolini, F.; Mirabile, A.; Buti, S.; Vasile, E.; Scotti, V.; Pisapia, P.; et al. Patients Selection for Immunotherapy in Solid Tumors: Overcome the Naïve Vision of a Single Biomarker. BioMed Res. Int. 2019, 2019, 9056417. [Google Scholar] [CrossRef]
- Yu, H.; Boyle, T.A.; Zhou, C.; Rimm, D.L.; Hirsch, F.R. PD-L1 Expression in Lung Cancer. J. Thorac. Oncol. 2016, 11, 964–975. [Google Scholar] [CrossRef]
- Da Silva, C.; Camps, M.G.; Li, T.M.; Zerrillo, L.; Löwik, C.W.; Ossendorp, F.; Cruz, L.J. Effective chemoimmunotherapy by co-delivery of doxorubicin and immune adjuvants in biodegradable nanoparticles. Theranostics 2019, 9, 6485–6500. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef] [PubMed]
- Bokas, A.; Papakotoulas, P.; Sarantis, P.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Mechanisms of the Antitumor Activity of Low Molecular Weight Heparins in Pancreatic Adenocarcinomas. Cancers 2020, 12, 432. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Le, T.Q.; Massarelli, E.; Hendifar, A.E.; Tuli, R. Radiation therapy and PD-1/PD-L1 blockade: The clinical development of an evolving anticancer combination. J. Immunother. Cancer 2018, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.-G.; Liu, X.; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2016, 77, 839–850. [Google Scholar] [CrossRef]
- Ciciola, P.; Cascetta, P.; Bianco, C.; Formisano, L.; Bianco, R. Combining Immune Checkpoint Inhibitors with Anti-Angiogenic Agents. J. Clin. Med. 2020, 9, 675. [Google Scholar] [CrossRef]
- Yaghoubi, N.; Soltani, A.; Ghazvini, K.; Hassanian, S.M.; Hashemy, S.I. PD-1/PD-L1 blockade as a novel treatment for colorectal cancer. Biomed. Pharmacother. 2019, 110, 312–318. [Google Scholar] [CrossRef]
- Della Gravara, L.; Battiloro, C.; Cantile, R.; Letizia, A.; Vitiello, F.; Montesarchio, V.; Rocco, D. Chemotherapy and/or immune checkpoint inhibitors in NSCLC first-line setting: What is the best approach? Lung Cancer Manag. 2020, 9, LMT22. [Google Scholar] [CrossRef]
- Liang, X.; De Vera, M.E.; Buchser, W.; Chavez, A.R.D.V.; Loughran, P.; Stolz, N.B.; Basse, P.; Wang, T.; Van Houten, B.; Zeh, H.J.; et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res. 2012, 72, 2791–2801. [Google Scholar] [CrossRef]
- Lotze, M.T.; Buchser, W.; Liang, X. Blocking the interleukin 2 (IL2)-induced systemic autophagic syndrome promotes profound antitumor effects and limits toxicity. Autophagy 2012, 8, 1264–1266. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Wang, W.; Piao, S.; Zhou, J.; Saiyin, W.; Zheng, C.; Sun, H.; Li, Y. Inhibition of autophagy by 3-MA enhances IL-24-induced apoptosis in human oral squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. 2015, 34, 97. [Google Scholar] [CrossRef]
- Li, W.; Deng, Y.; Chu, Q.; Zhang, P. Gut microbiome and cancer immunotherapy. Cancer Lett. 2019, 447, 41–47. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Number of Study. | Type of Cancer | Phase | Agent/Compound |
|---|---|---|---|
| NCT04069273 | Adenocarcinomas of the esophagogastric junction | II | Ramucirumab + pembrolizumab + paclitaxel |
| NCT02501096 | Advanced solid tumors | I/II | Lenvatinib + pembrolizumab |
| NCT02646748 | Advanced solid tumors | Ι | Pembrolizumab+INCB combinations |
| NCT04317105 | Advanced malignant solid neoplasm | I/II | Copanlisib, ipilimumab, nivolumab |
| NCT03525795 | Advanced solid tumors | I/II | CPI-1205, ipilimumab |
| NCT02617589 | Brain cancer | III | Nivolumab, temozolomide |
| NCT02684006 | Clear cell | III | Avelumab + axitinib vs. sunitinib |
| NCT02853331 | Clear cell | III | Pembrolizumab + axitinib vs. sunitinib |
| NCT01472081 | Clear cell/non-clear cell | I | Nivolumab + sunitib/pazopanib |
| NCT02420821 | Clear cell, sarcomatoid | III | Atezolizumab+bevacizumab vs. sunitib |
| NCT03202758 | Colorectal cancer | I/II | Durvalumab, tremelimumab and FOLFOX |
| NCT02981524 | Colorectal cancer | II | Cyclophosphamide followed by Pembrolizumab |
| NCT03657641 | Colorectal cancer | I/II | Pembrolizumab + vicriviroc |
| NCT02375672 | Colorectal cancer | II | Pembrolizumab + FOLFOX |
| NCT03711058 | Colorectal cancer | I/II | Nivolumab + copanlisi, nivolumab |
| NCT02327078 | Colorectal cancer | VII | Nivolumab + epacadostat |
| NCT03832621 | Colorectal cancer | II | Nivolumab, ipilimumab, temozolomide |
| NCT02675946 | Gastrointestinal cancers | I | CGX1321+pembrolizumab |
| NCT02496208 | Genitourinary tumors | I | Cabozantinib + nivolumab/ipilimumab |
| NCT02997228 | mCRC | III | Atezolizumab + bevacizumab + mFOLFOX6 |
| NCT01950390 | Melanoma | II | Ipilimumab + bevacizumab |
| NCT02802098 | Metastatic breast cancer | I | Durvalumab+ bevacizumab, taxane+ bevacizumab |
| NCT00790010 | Metastatic melanoma | I | Ipilimumab, bevacizumab |
| NCT02959554 | Metastatic renal cell carcinoma | II | Nivolumab after sunitinib/pazopanib |
| NCT03149822 | Metastatic renal cell carcinoma | I/II | Pembrolizumab + cabozantinib |
| NCT02681549 | mNSCLC | II | Pembrolizumab + bevacizumab |
| NCT03976375 | mNSCLC | III | Pembrolizumab, lenvatinib, docetaxel |
| NCT03838159 | NSCLC | II | Paclitaxel, carboplatin, nivolumab |
| NCT03425006 | NSCLC | II | Itacitinib, Pembrolizumab |
| NCT02492568 | NSCLC | II | SBRT, pembrolizumab |
| NCT02443324 | NSCLC, Biliary tract carcinoma, Urothelial carcinoma | I | Ramucirumab + pembrolizumab |
| NCT03153410 | Pancreatic cancer | I | Pembrolizumab, GVAX, cyclophosphamide, IMC-CS4 |
| NCT02648282 | Pancreatic cancer | II | Pembrolizumab, GVAX, cyclophosphamide |
| NCT03563248 | Pancreatic cancer | II | Nivolumab, losartan, FOLFIRINOX |
| NCT03829111 | Renal Cell carcinoma | I | Nivolumab, Ipilimumab, Clostridium butyricum CBM 588 probiotic strain |
| NCT02888665 | Sarcoma | I/II | Doxorubicin+ pembrolizumab |
| NCT03898180 | Urothelial carcinoma | III | Lenvatinib + pembrolizumab |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors. Biomolecules 2020, 10, 666. https://doi.org/10.3390/biom10050666
Koustas E, Sarantis P, Papavassiliou AG, Karamouzis MV. The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors. Biomolecules. 2020; 10(5):666. https://doi.org/10.3390/biom10050666
Chicago/Turabian StyleKoustas, Evangelos, Panagiotis Sarantis, Athanasios G. Papavassiliou, and Michalis V. Karamouzis. 2020. "The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors" Biomolecules 10, no. 5: 666. https://doi.org/10.3390/biom10050666
APA StyleKoustas, E., Sarantis, P., Papavassiliou, A. G., & Karamouzis, M. V. (2020). The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors. Biomolecules, 10(5), 666. https://doi.org/10.3390/biom10050666