Functional Interactomes of Genes Showing Association with Type-2 Diabetes and Its Intermediate Phenotypic Traits Point towards Adipo-Centric Mechanisms in Its Pathophysiology


Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.Y.; Fernandes, J.D.R.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ali, O. Genetics of type 2 diabetes. World J. Diabetes 2013, 4, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Vig, S.; Datta, M.; Jindel, D.; Mathur, A.K.; Mathur, S.K.; Sharma, A. Systems biology approach reveals genome to phenome correlation in type 2 diabetes. PLoS ONE 2013, 8, e53522. [Google Scholar]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Naji, N.B.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risks. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar]
- Soul, J.; Dunn, S.L.; Hardingham, T.E.; Boot-Handford, R.P.; Schwartz, J.M. Phenome Scape: A cytoscape app to identify differentially regulated subnetworks using known disease associations. Bioinformatics 2016, 32, 3847–3849. [Google Scholar]
- Mishra, A.; Macgregor, S. VEGAS2:Software for More Flexible Gene-Based Testing. Twin Res. Hum. Genet. 2014, 18, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Ideker, T.; Ozier, O.; Schwikowski, B.; Siegel, A.F. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 2002, 18, S233–S240. [Google Scholar]
- Rani., J.; Mittal, I.; PramanikA., A.; Singh, N.; Dube, N.; Sharma, S.; Puniya, B.L.; Raghunandanan, M.V.; Mobeen, A.; Ramachandran, S. T2DiACoD A gene Atlas of Type2 Diabetes Mellitus Associated Complex Disorders. Sci. Rep. 2017, 7, 6892. [Google Scholar]
- Liao, Y.; WangJ., J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt2019: Gene set analysis tool kit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernendez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jeniks, S.L.; Jagodink, K.M.; Lachman, A.; et al. Enrichr: A comprehensive genes etenrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Minadakis, G.; Zachariou, M.; Oulas, A.; Spyrou, G.M. Pathway Connector:Finding complementary pathways to enhance functional analysis. Bioinformatics 2018, 35, 889–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florez, J.C. Newly identified loci highlight beta cell dysfunctionas a key cause of type 2 diabetes: Where are the insulin resistance genes? Diabetologia 2008, 51, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García, G.J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Cernea, S.; Cahn, A.; Raz, I. Pharmacological management of nonalcoholic fatty liver disease in type 2 diabetes. Expert Rev. Clin. Pharmacol. 2017, 10, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum Total Adiponectin Nonalcoholic Fatty Liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. ThePI3K/AKT pathway in obesity and type2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar]
- Vergès, B.; Cariou, B. mTOR inhibitors and diabetes. Diabetes Res. Clin. Pract. 2015, 110, 101–108. [Google Scholar]
- Ozaki, K.I.; Awazu, M.; Tamiya, M.; Iwasaki, Y.; Harada, A.; Kugisaki, S.; Tanimura, S.; Kohno, M. Targeting the ERK signaling pathwayas a potential treatment for insulin resistance and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E643–E651. [Google Scholar]
- Prada, P.O.; Ropelle, E.R.; Mourão, R.H.; de-Souza, C.T.; Pauli, J.R.; Cintra, D.E.; Schenka, A.; Rocco, S.A.; Rittner, R.; Franchini, K.G. EGFR tyrosine kinase inhibitor (PD153035) improves glucose tolerance and insulin action in high-fat diet–fed mice. Diabetes 2009, 58, 2910–2919. [Google Scholar] [CrossRef] [Green Version]
- Sawayama, H.; Ishimoto, T.; Watanabe, M.; Yoshida, N.; Sugihara, H.; Kurashige, J.; Harshima, K.; Iwatsuki, M.; Baba, Y.; Oki, E.; et al. Small molecule agonists of PPAR-γ exert therapeutic effects in esophageal cancer. Cancer Res 2013, 74, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, M.; Suraamornkul, S.; Hardies, L.J.; Glass, L.; Musi, N.; Defronzo, R.A. Effects of peroxisomeproliferator-activated receptor(PPAR)-alpha and PPAR-gamma agonists on glucose and lipid metabolism in patient swith type2 diabetes mellitus. Diabetologia 2007, 50, 1723–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Yun, Z. The hypoxia-inducible factor pathway in Adipocytes: The role of HIF-2 in adipose inflammation and hypertrophic cardiomyopathy. Front. Endocrinol. (Lausanne) 2015, 6, 1–7. [Google Scholar]
- Vergoni, B.; Cornejo, P.J.; Gilleron, J.; Djedaini, M.; Ceppo, F.; Gonzalez, T.; Maillet, J.; Dhennin, V.; Verbanck, M.; Froguel, P.; et al. DNA damage and the activation of the p53 pathway mediate alterations in metabolic and secretory functions of adipocytes. Diabetes 2016, 65, 3062–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhtar, S.; Yousif, M.H.M.; Dhaunsi, G.S.; Sarkhouh, F.; Chandrasekhar, B.; Attur, S.; Benter, I.F. Activation of ErbB2 and Downstream Signaling via Rho Kinases and ERK1/2 Contributes to Diabetes-Induced Vascular Dysfunction. PLoS ONE 2013, 8, e67813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, S.K.; Chandra, P.; Mishra, S.; Ajmera, P.; Sharma, P. Type-2 diabetes related intermediate phenotypic traits in north Indian diabetics. Indian J. Clin. Biochem. 2007, 22, 70–73. [Google Scholar]
- Dunmore, S.J.; Brown, J.E.P. The role of adipokinesinβ-cell failure of type 2 diabetes. J. Endocrinol. 2013, 216, T37–T45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocot, J.; Dziemidok, P.; Kiełczykowska, M.; Hordyjewska, A.; Szcześniak, G.; Musik, I. Adipokine Profile in Patients with Type 2 Diabetes Dependson Degree of Obesity. Med. Sci. Monit. 2017, 23, 4995–5004. [Google Scholar]
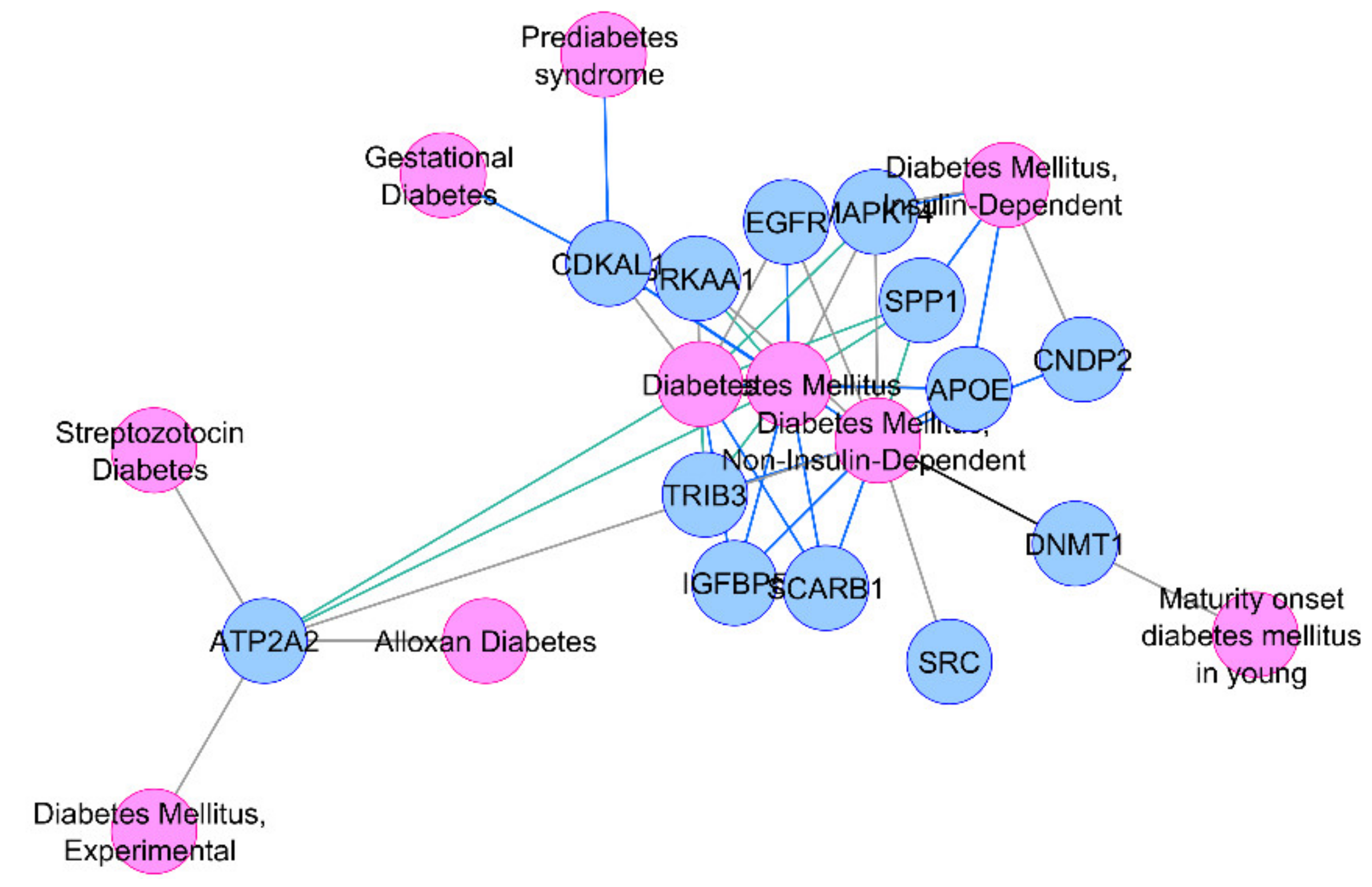

{kind=link}
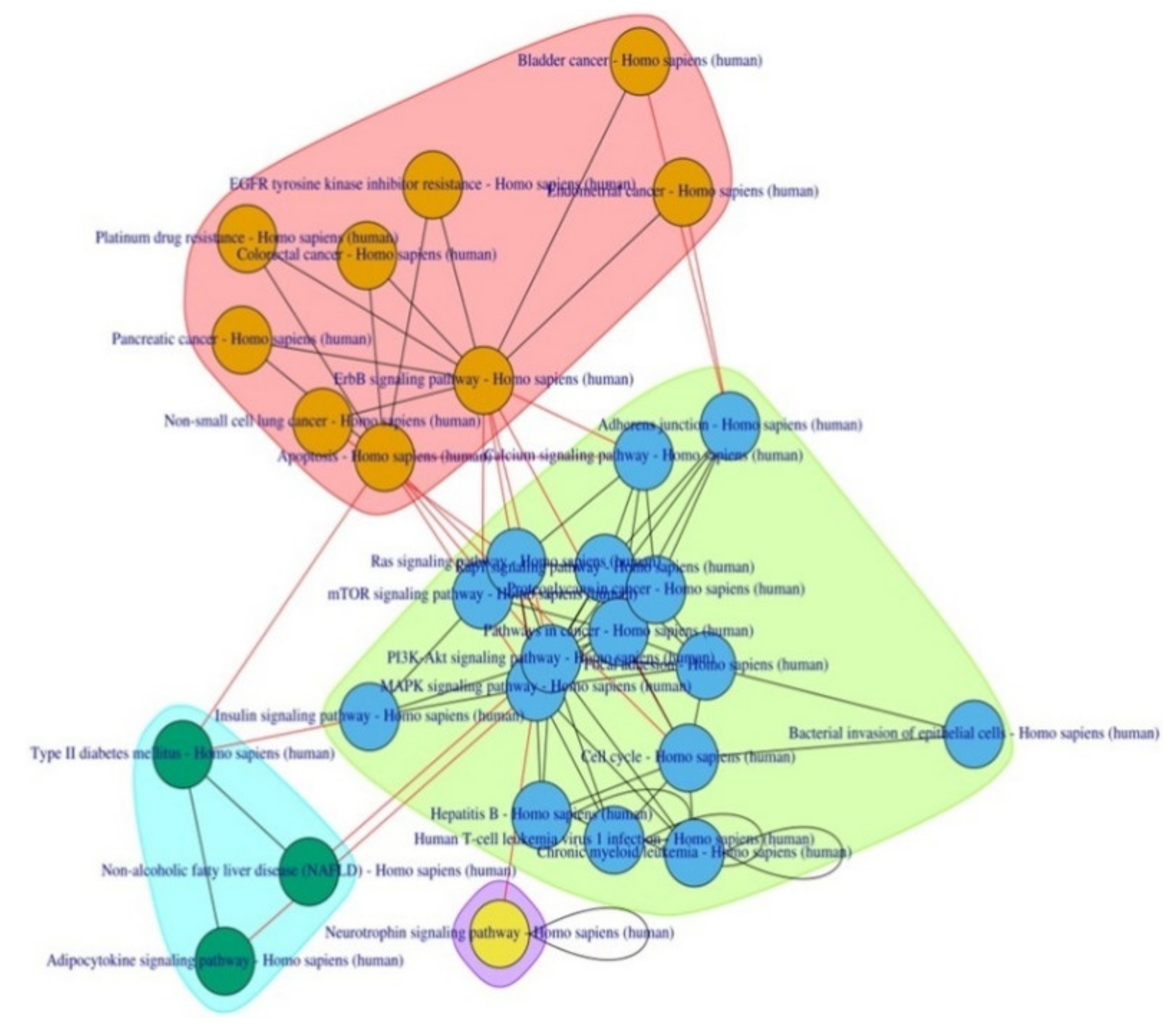
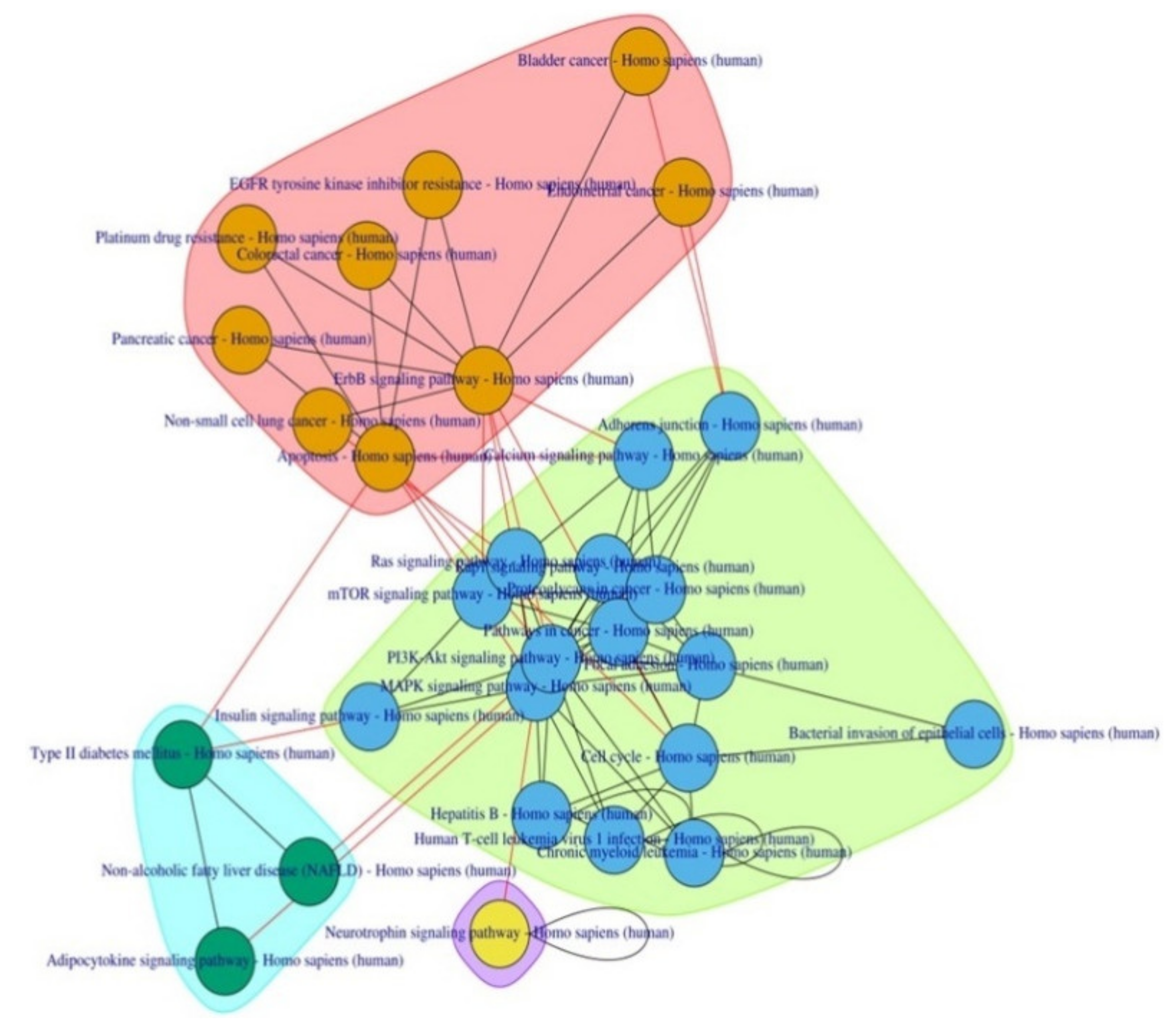{kind=link}
| S. No. | Tissue | # of DEGs Obtained (p < 0.05) | Overlap with HOMA-β | Overlap with HOMA-IR |
|---|---|---|---|---|
| 1 | Pancreas | 295 | 74 (~25%) | 7 |
| 2 | Skeletal | 275 | 63 (~22.7%) | 4 |
| 3 | Adipose | 253 | 55 (~21.7%) | 3 |
| S. No | Pathway | p Value | Common Pathways | Common Genes | Genes Found | Pathway Ratio | Rank |
|---|---|---|---|---|---|---|---|
| 1 | Adherens junction | 4.61653 × 10−26 | 7/4246 | 19/72 | 19/291 | 1.649 × 10−3 | 3 |
| 2 | Type II diabetes mellitus | 1.22599 × 10−13 | 5/4246 | 13/46 | 13/291 | 1.178 × 10−3 | 12 |
| 3 | Chronic myeloid leukemic | 1.35055 × 10−17 | 8/4246 | 18/76 | 18/291 | 1.884 × 10−3 | 4 |
| 4 | Pathways in cancer | 4.61653 × 10−26 | 23/4246 | 52/526 | 52/291 | 5.417 × 10−3 | 1 |
| 5 | ErbB signaling pathway | 7.28619 × 10−15 | 10/4246 | 17/85 | 17/291 | 2.355 × 10−3 | 10 |
| 6 | Bacterial invasion of epithelial cells | 3.71078 × 10−13 | 7/4246 | 15/74 | 15/291 | 1.649 × 10−3 | 15 |
| 7 | Proteoglycans in cancer | 1.21687 × 10−21 | 14/4246 | 30/201 | 30/291 | 3.297 × 10−3 | 2 |
| 8 | Neurotrophin signaling pathway | 4.91599 × 10−17 | 6/4246 | 21/119 | 21/291 | 1.413 × 10−3 | 5 |
| 9 | Pancreatic cancer | 5.48038 × 10−13 | 10/4246 | 14/75 | 14/291 | 2.355 × 10−3 | 16 |
| 10 | Colorectal cancer | 2.16852 × 10−13 | 7/4246 | 19/72 | 19/291 | 1.649 × 10−3 | 3 |
| 11 | Insulin signaling pathway | 1.09957 × 10−15 | 5/4246 | 13/46 | 13/291 | 1.178 × 10−3 | 12 |
| 12 | Focal adhesion | 2.55996 × 10−15 | 8/4246 | 18/76 | 18/291 | 1.884 × 10−3 | 4 |
| 13 | Hepatitis B | 2.3797 × 10−16 | 23/4246 | 52/526 | 52/291 | 5.417 × 10−3 | 1 |
| 14 | Human T-cell leukemic virus 1 infection | 9.37 × 10−15 | 10/4246 | 17/85 | 17/291 | 2.355 × 10−3 | 10 |
| 15 | Rap1 signaling pathway | 6.90707 × 10−15 | 7/4246 | 15/74 | 15/291 | 1.649 × 10−3 | 15 |
| 16 | EGFR tyrosine kinase inhibitor resistance | - | 14/4246 | 30/201 | 30/291 | 3.297 × 10−3 | New |
| 17 | Ras signaling pathway | 3.086743 × 10−13 | 6/4246 | 21/119 | 21/291 | 1.413 × 10−3 | 5 |
| 18 | Non-small cell lung cancer | 1.41 × 10−6 | 10/4246 | 14/75 | 14/291 | 2.355 × 10−3 | 16 |
| 19 | Endometrial cancer | 7.88 × 10−7 | 10/4246 | 15/86 | 15/291 | 2.355 × 10−3 | 13 |
| 20 | MAPK signaling pathway | 1.13 × 10−9 | 7/4246 | 21/137 | 21/291 | 1.649 × 10−3 | 7 |
| 21 | Adipocytokine signaling pathway | 7.72 × 10−7 | 9/4246 | 24/199 | 24/291 | 2.120 × 10−3 | 8 |
| 22 | PI3K-Akt signaling pathway | 6.34 × 10−9 | 11/4246 | 21/163 | 21/291 | 2.591 × 10−3 | 6 |
| 23 | Apoptosis | 1.38 × 10−7 | 12/4246 | 25/219 | 25/291 | 2.826 × 10−3 | 11 |
| 24 | Cell cycle | 2.02 × 10−6 | 9/4246 | 24/206 | 24/291 | 2.120 × 10−3 | 9 |
| 25 | Platinum drug resistance | - | 10/4246 | 12/79 | 12/291 | 2.355 × 10−3 | New |
| 26 | Non-alcoholic fatty liver disease (NAFLD) | 1.36 × 10−5 | 9/4246 | 24/232 | 24/291 | 2.120 × 10−3 | 14 |
| 27 | Bladder cancer | 2.91 × 10−3 | 8/4246 | 10/66 | 10/291 | 1.884 × 10−3 | 69 |
| 28 | mTOR signaling pathway | 2.33 × 10−4 | 9/4246 | 8/58 | 8/291 | 2.120 × 10−3 | 64 |
| S. No | Pathway | p Value | Common Pathways | Common Genes | Genes Found | Pathway Ratio | Rank |
|---|---|---|---|---|---|---|---|
| 1 | Proteoglycans in cancer | 4.932838 × 10−13 | 14/4246 | 10/201 | 10/32 | 3.297 × 10−3 | 1 |
| 2 | Glioma | 8.665005 × 10−12 | 8/4246 | 7/75 | 7/32 | 1.884 × 10−3 | 2 |
| 3 | Pathways in cancer | 1.556996 × 10−7 | 23/4246 | 9/526 | 9/32 | 5.417 × 10−3 | 10 |
| 4 | MicroRNAs in cancer | 1.661089 × 10−8 | 7/4246 | 8/299 | 8/32 | 1.649 × 10−3 | 5 |
| 5 | FoxO signaling pathway | 1.437119 × 10− 9 | 13/4246 | 7/132 | 7/32 | 3.062 × 10−3 | 3 |
| 6 | mTOR signaling pathway | 2.470845 × 10−6 | 8/4246 | 7/153 | 7/32 | 1.884 × 10−3 | 18 |
| 7 | HIF-1 signaling pathway | 1.30885 × 10−8 | 9/4246 | 6/100 | 6/32 | 2.120 × 10−3 | 4 |
| 8 | PI3K-Akt signaling pathway | 9.186771 × 10−7 | 17/4246 | 7/354 | 7/32 | 4.004 × 10−3 | 14 |
| 9 | Neurotrophin signaling pathway | 3.280781 × 10−8 | 6/4246 | 6/119 | 6/32 | 1.413 × 10−3 | 6 |
| 10 | Longevity regulating pathway | 3.751219 × 10−7 | 8/4246 | 5/62 | 5/32 | 1.884 × 10−3 | 7 |
| 11 | Melanoma | 9.131152 × 10−8 | 7/4246 | 5/72 | 5/32 | 1.649 × 10−13 | 8 |
| 12 | Bacterial invasion of epithelial cells | 1.468947 × 10−7 | 7/4246 | 5/74 | 5/32 | 1.649 × 10−3 | 9 |
| 13 | EGFR tyrosine kinase inhibitor resistance | - | 10/4246 | 5/79 | 5/32 | 2.355 × 10−3 | new |
| 14 | Focal adhesion | 7.174111 × 10−7 | 9/4246 | 6/199 | 6/32 | 2.120 × 10−3 | 13 |
| 15 | Longevity regulating pathway | 5.389533 × 10−8 | 9/4246 | 5/89 | 5/32 | 2.120 × 10−3 | 12 |
| 16 | Prostate cancer | 2.852625 × 10−7 | 9/4246 | 5/97 | 5/32 | 2.120 × 10−3 | 11 |
| 17 | Leukocyte transendothelial migration | 1.163798 × 10−6 | 3/4246 | 5/112 | 5/32 | 7.065 × 10−4 | 15 |
| 18 | ErbB signaling pathway | 1.1 × 10−5 | 10/4246 | 4/85 | 4/32 | 2.355 × 10−3 | 20 |
| 19 | Small cell lung cancer | 0.000348 | 7/4246 | 4/93 | 4/32 | 1.649 × 10−3 | 37 |
| 20 | Non-small cell lung cancer | 0.003619 | 8/4246 | 3/66 | 3/32 | 1.884 × 10−3 | 57 |
| 21 | MAPK signaling pathway | 0.062537 | 7/4246 | 4/295 | 4/32 | 1.649 × 10−3 | 96 |
| 22 | p53 signaling pathway | 0.000181 | 4/4246 | 3/72 | 3/32 | 9.421 × 10−4 | 31 |
| 23 | Colorectal cancer | 0.004417 | 10/4246 | 3/86 | 3/32 | 2.355 × 10−3 | 61 |
| 24 | Fluid shear stress and atherosclerosis | 9/4246 | 3/139 | 3/32 | 2.120 × 10−3 | new | |
| 25 | Cell cycle | 0.016751 | 4/4246 | 2/124 | 2/32 | 9.421 × 10−4 | 81 |
| 26 | Apoptosis | 0.021037 | 9/4246 | 2/136 | 2/32 | 2.120 × 10−3 | 87 |
| 27 | Chemokine signaling pathway | 0.035898 | 7/4246 | 2/190 | 2/32 | 1.649 × 10−3 | 94 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saxena, A.; Wahi, N.; Kumar, A.; Mathur, S.K. Functional Interactomes of Genes Showing Association with Type-2 Diabetes and Its Intermediate Phenotypic Traits Point towards Adipo-Centric Mechanisms in Its Pathophysiology. Biomolecules 2020, 10, 601. https://doi.org/10.3390/biom10040601
Saxena A, Wahi N, Kumar A, Mathur SK. Functional Interactomes of Genes Showing Association with Type-2 Diabetes and Its Intermediate Phenotypic Traits Point towards Adipo-Centric Mechanisms in Its Pathophysiology. Biomolecules. 2020; 10(4):601. https://doi.org/10.3390/biom10040601
Chicago/Turabian StyleSaxena, Aditya, Nitin Wahi, Anshul Kumar, and Sandeep Kumar Mathur. 2020. "Functional Interactomes of Genes Showing Association with Type-2 Diabetes and Its Intermediate Phenotypic Traits Point towards Adipo-Centric Mechanisms in Its Pathophysiology" Biomolecules 10, no. 4: 601. https://doi.org/10.3390/biom10040601