The Human RAD5 Homologs, HLTF and SHPRH, Have Separate Functions in DNA Damage Tolerance Dependent on the DNA Lesion Type


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
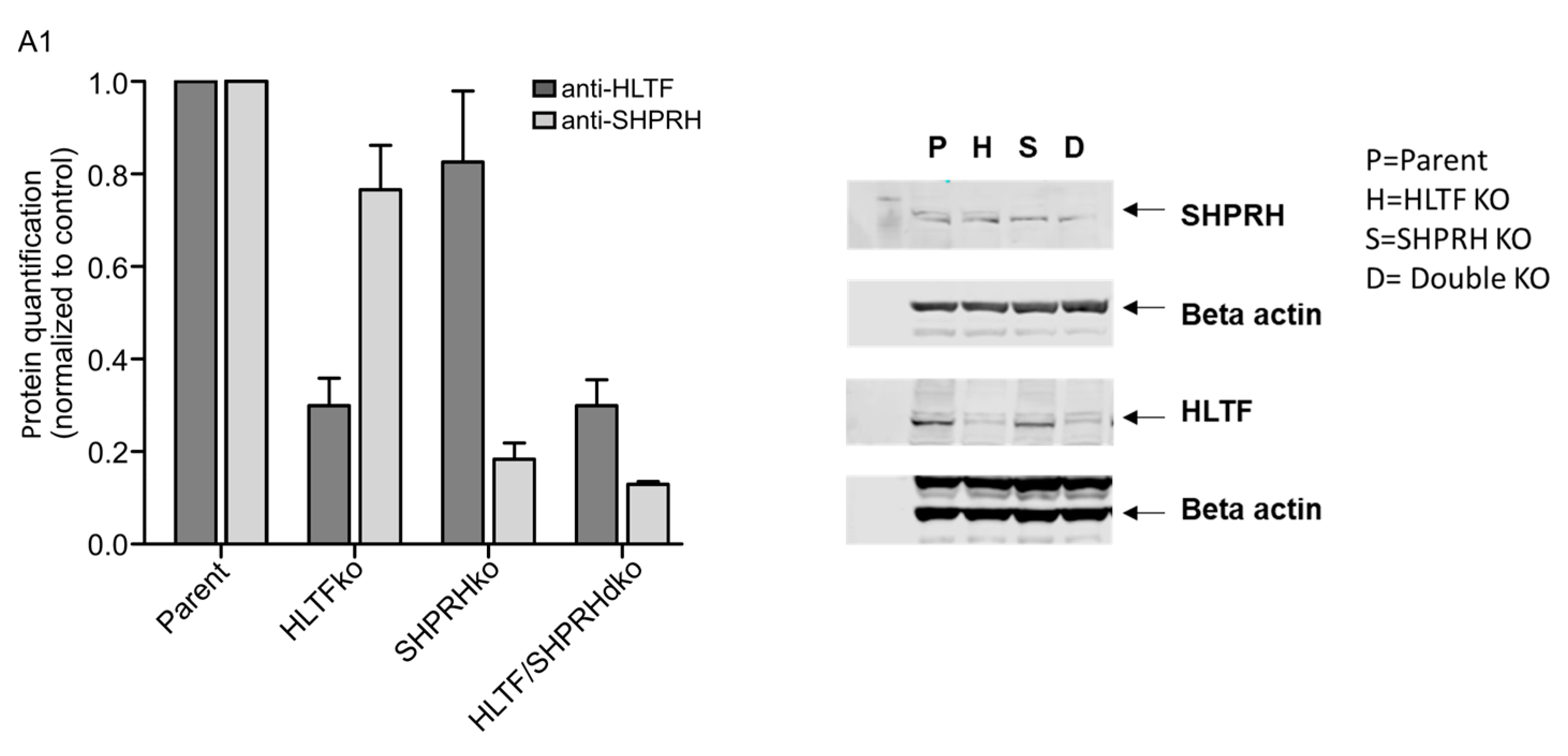
2.1. Cell Lines
2.2. SupF Assay
2.3. Cell Cycle and Western Blot Analysis
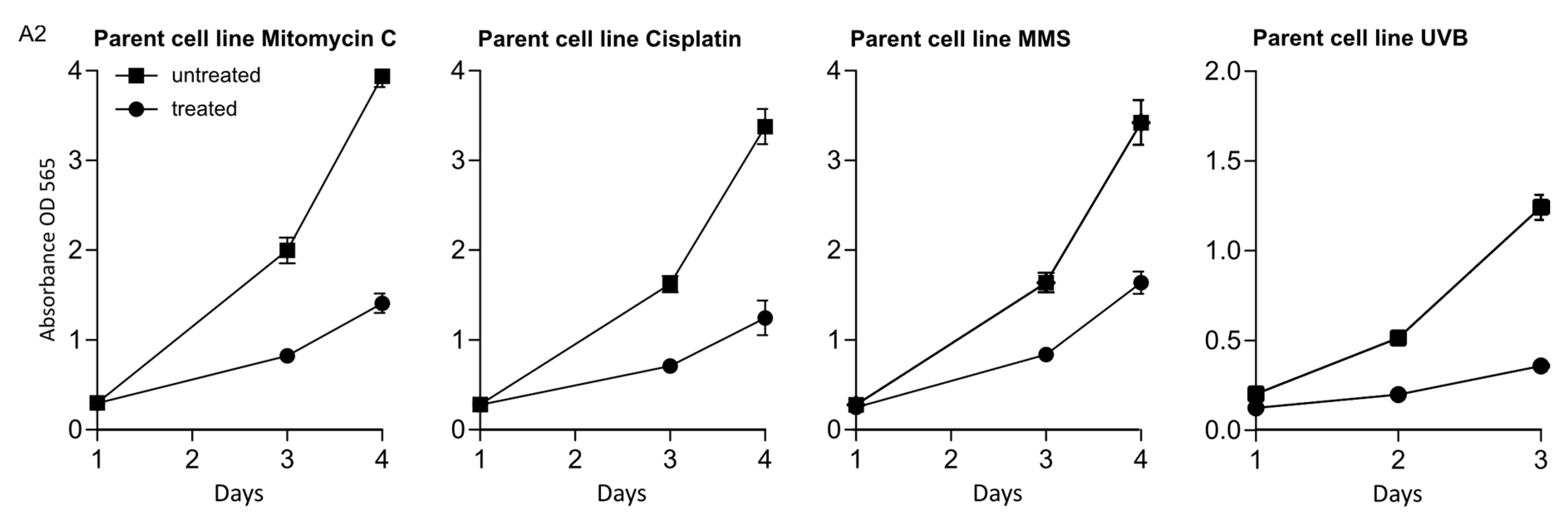
2.4. MTT Assay
3. Results and Discussion
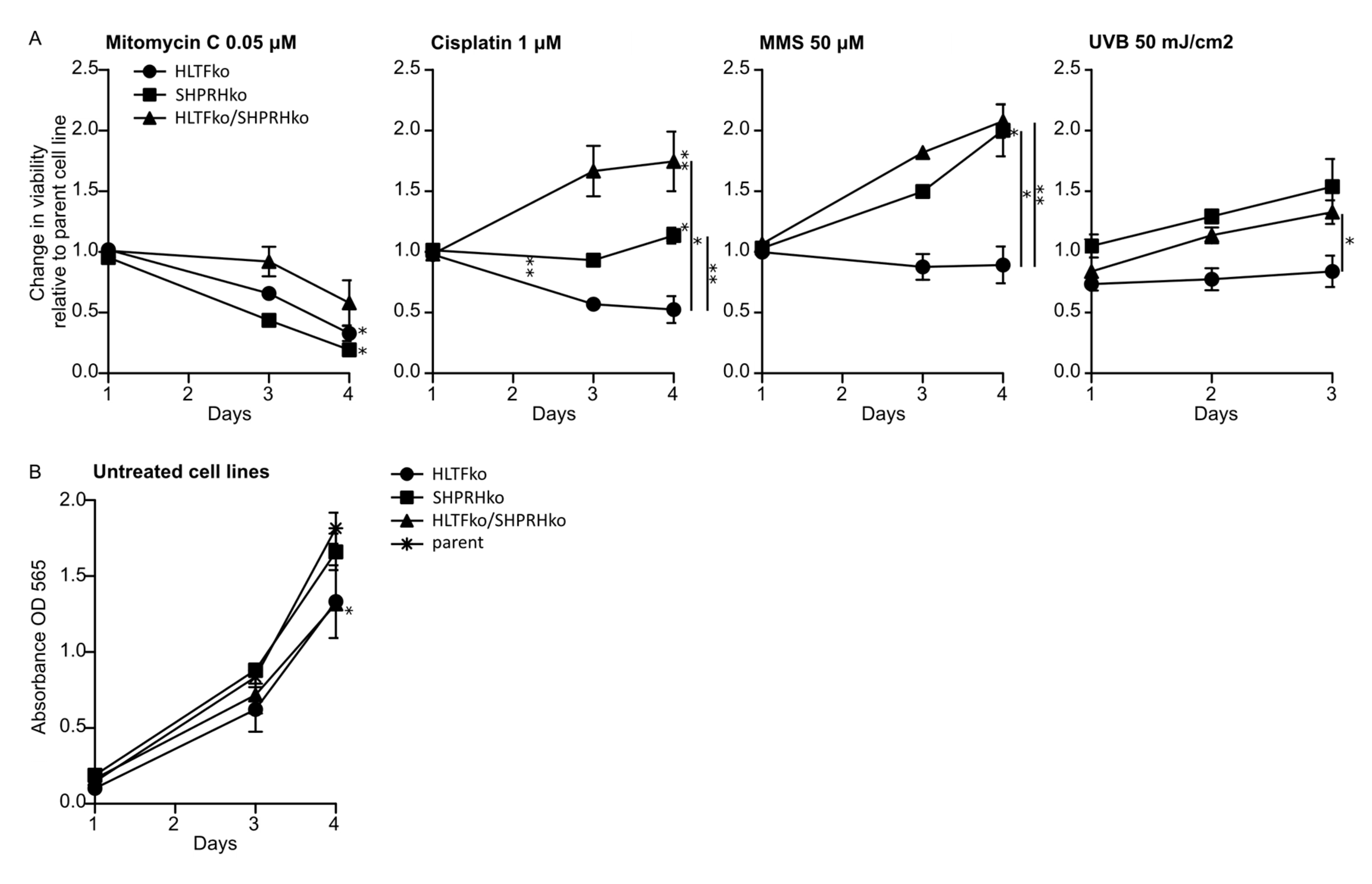
3.1. The Two RAD5 Homologs Have Different Roles in DNA Repair or DNA Damage Tolerance Depending on the DNA Lesion
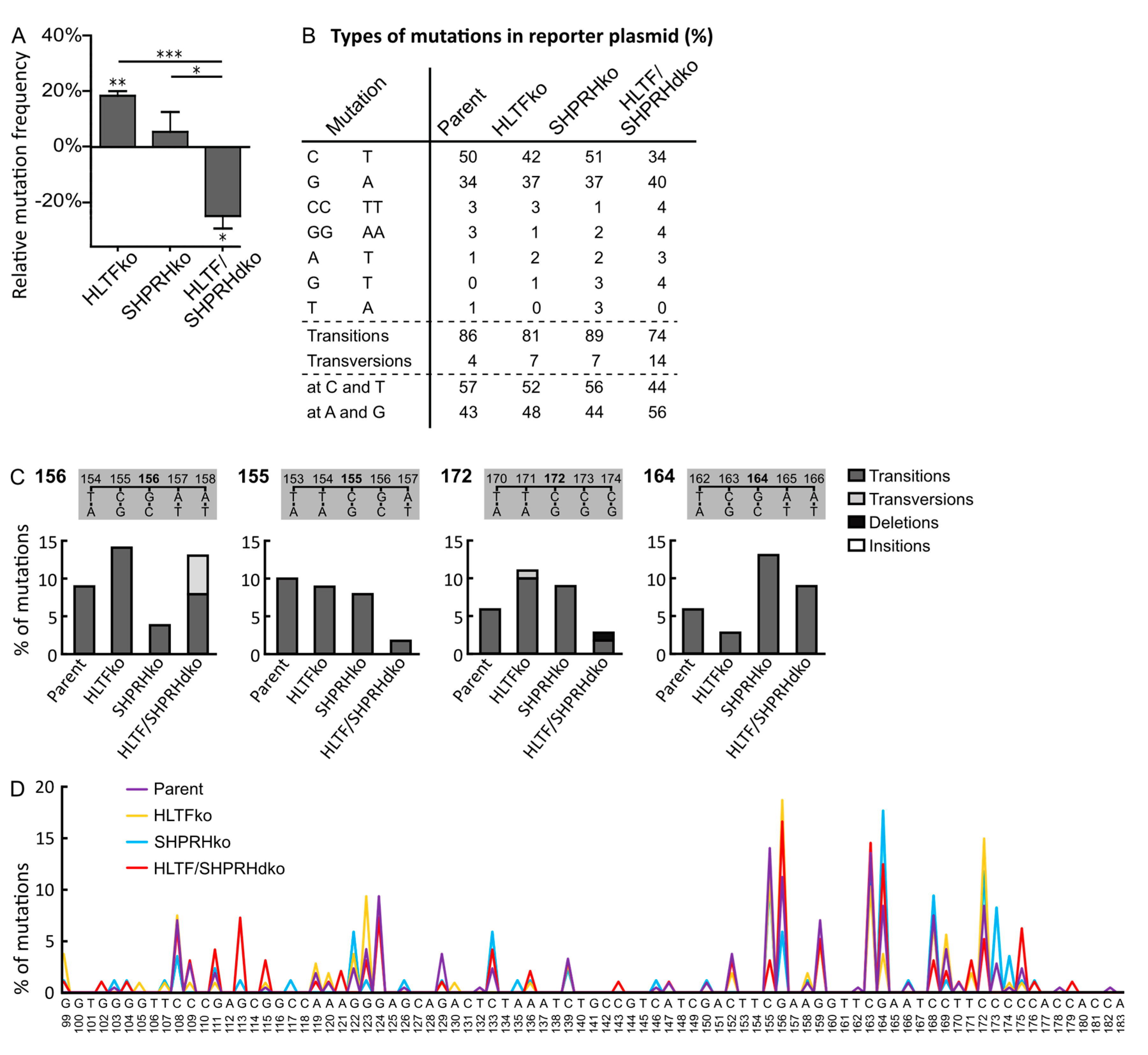
3.2. Absence of HLTF and SHPRH Reduces Error-Prone TLS over UV-induced DNA Lesions
3.3. HLTF and SHPRH Are Interdependent Proteins in Response to UV-Induced DNA Lesions
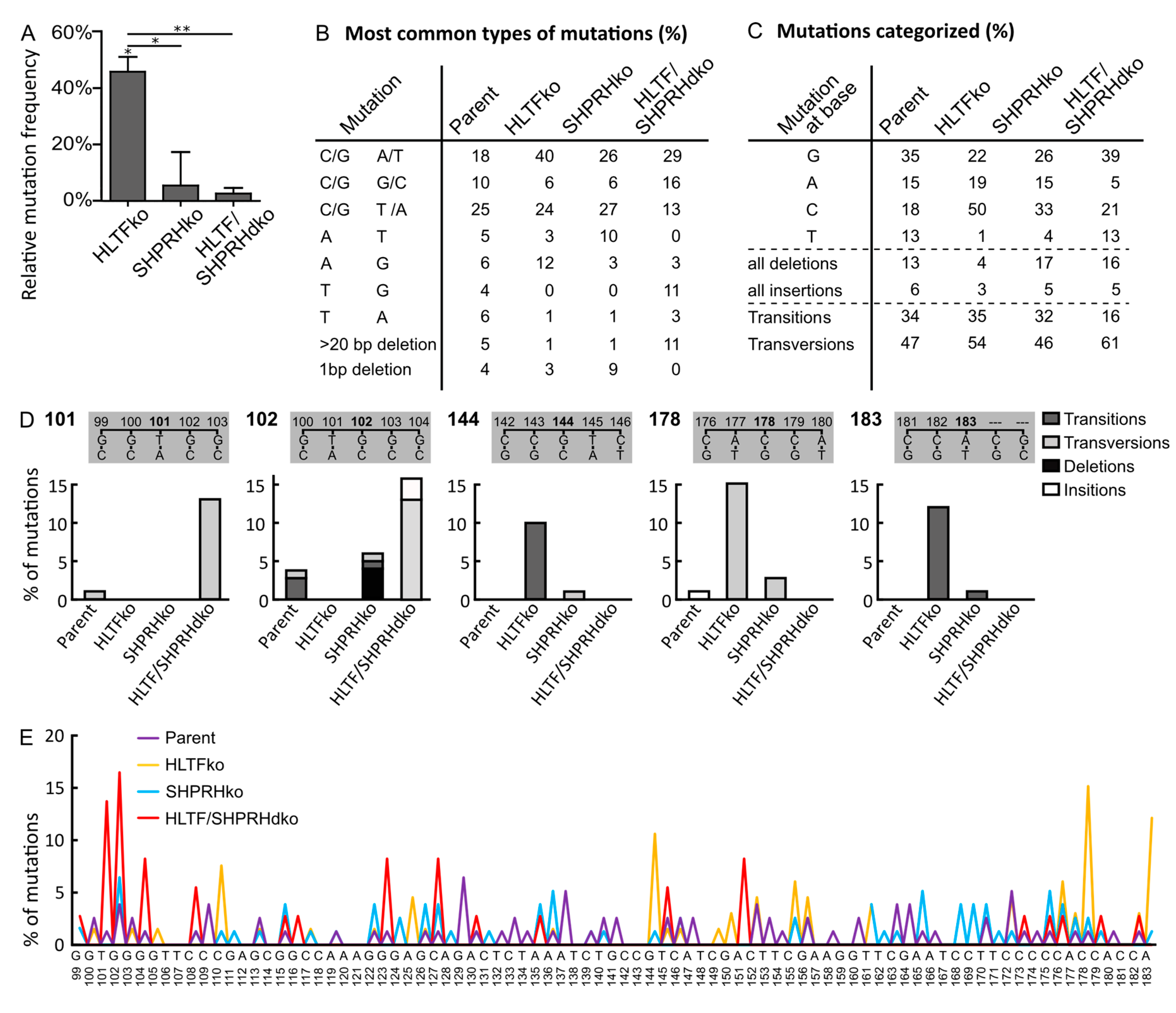
3.4. HLTF Is Important for Correct Bypass and/or Repair of MMS-induced DNA Damage
3.5. SHPRH Has No Central Role in TLS over MMS-induced DNA Lesions, but Is Important for Avoiding DNA Strand Breaks
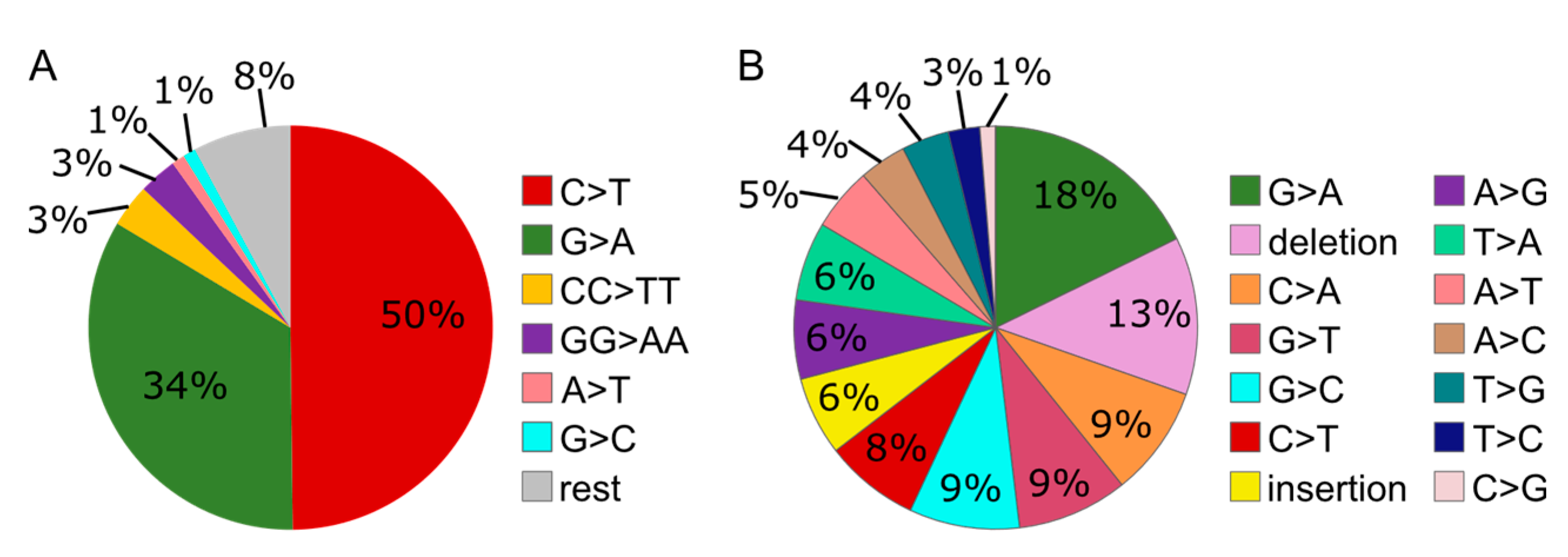
3.6. MMS Results in A More Diverse Mutation Pattern than UV
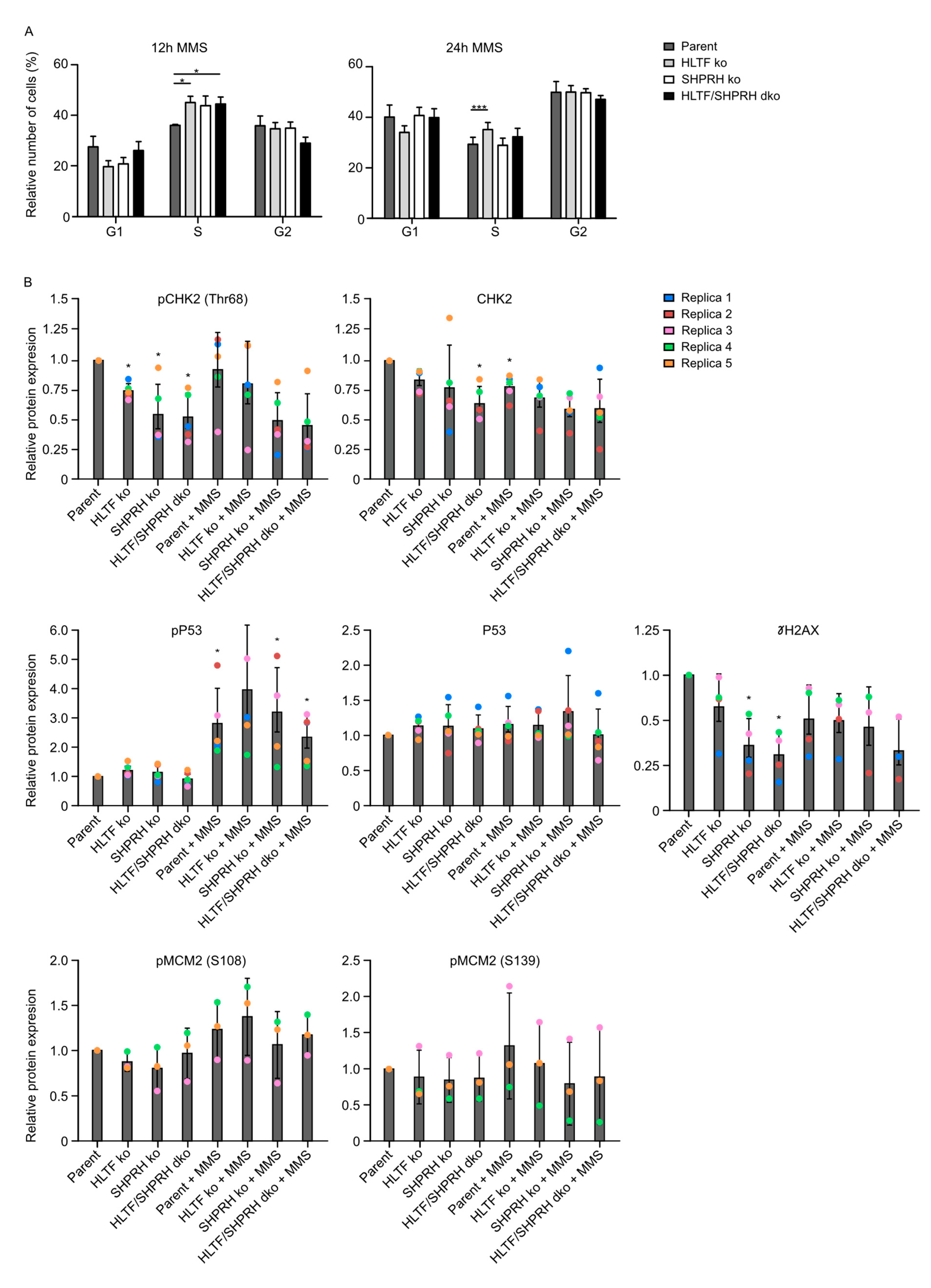
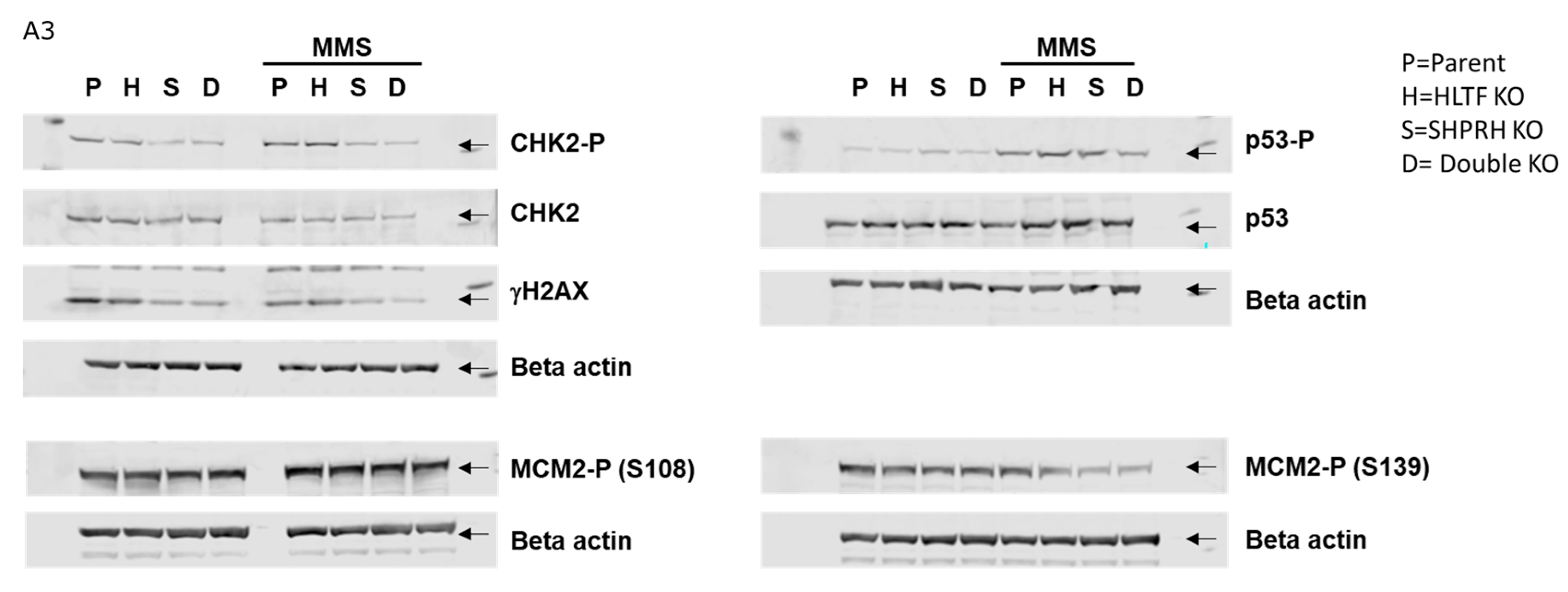
3.7. Knockout of the RAD5 Homologs Affects Cell Cycle Distribution after MMS Treatment
3.8. Reduced Phosphorylation of CHK2 and MCM2 in Cells Lacking SHPRH
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet J. Rare Dis. 2011, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Psakhye, I. DNA damage tolerance. Curr. Opin. Cell Biol. 2016, 40, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenes. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Chang, D.J.; Cimprich, K.A. DNA damage tolerance: When it’s OK to make mistakes. Nat. Chem. Biol. 2009, 5, 82–90. [Google Scholar] [CrossRef]
- Lin, J.R.; Zeman, M.K.; Chen, J.Y.; Yee, M.C.; Cimprich, K.A. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell 2011, 42, 237–249. [Google Scholar] [CrossRef]
- Motegi, A.; Liaw, H.J.; Lee, K.Y.; Roest, H.P.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.D.; Ding, H.; Hoeijmakers, J.H.J.; et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef]
- Chavez, D.A.; Greer, B.H.; Eichman, B.F. The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. J. Biol. Chem. 2018, 293, 8484–8494. [Google Scholar] [CrossRef]
- Kim, J.J.; Chung, S.W.; Kim, J.H.; Kim, J.W.; Oh, J.S.; Kim, S.; Song, S.Y.; Park, J.; Kim, D.H. Promoter methylation of helicase-like transcription factor is associated with the early stages of gastric cancer with family history. Ann. Oncol. 2006, 17, 657–662. [Google Scholar] [CrossRef]
- Capouillez, A.; Noel, J.C.; Arafa, M.; Arcolia, V.; Mouallif, M.; Guenin, S.; Delvenne, P.; Belayew, A.; Saussez, S. Expression of the helicase-like transcription factor and its variants during carcinogenesis of the uterine cervix: Implications for tumour progression. Histopathology 2011, 58, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H.; et al. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Moinova, H.R.; Chen, W.D.; Shen, L.; Smiraglia, D.; Olechnowicz, J.; Ravi, L.; Kasturi, L.; Myeroff, L.; Plass, C.; Parsons, R.; et al. HLTF gene silencing in human colon cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 4562–4567. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdu, I.; Blastyak, A.; Haracska, L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair 2010, 9, 257–267. [Google Scholar] [CrossRef]
- Seelinger, M.; Otterlei, M. Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). Int. J. Mol. Sci. 2020, 21, 693. [Google Scholar] [CrossRef]
- Scharer, O.D. DNA interstrand crosslinks: Natural and drug-induced DNA adducts that induce unique cellular responses. ChemBioChem 2005, 6, 27–32. [Google Scholar] [CrossRef]
- Warren, A.J.; Maccubbin, A.E.; Hamilton, J.W. Detection of mitomycin C-DNA adducts in vivo by 32P-postlabeling: Time course for formation and removal of adducts and biochemical modulation. Cancer Res. 1998, 58, 453–461. [Google Scholar]
- Kartalou, M.; Essigmann, J.M. Recognition of cisplatin adducts by cellular proteins. Mutat. Res. 2001, 478, 1–21. [Google Scholar] [CrossRef]
- Unk, I.; Hajdu, I.; Fatyol, K.; Hurwitz, J.; Yoon, J.H.; Prakash, L.; Prakash, S.; Haracska, L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. USA 2008, 105, 3768–3773. [Google Scholar] [CrossRef]
- Krijger, P.H.; Lee, K.Y.; Wit, N.; van den Berk, P.C.; Wu, X.; Roest, H.P.; Maas, A.; Ding, H.; Hoeijmakers, J.H.; Myung, K.; et al. HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: Existence of an alternative E3 ligase. DNA Repair 2011, 10, 438–444. [Google Scholar] [CrossRef]
- Yoon, J.H.; Prakash, L.; Prakash, S. Error-free replicative bypass of (6-4) photoproducts by DNA polymerase zeta in mouse and human cells. Genes Dev. 2010, 24, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Furrer, A.; van Loon, B. Handling the 3-methylcytosine lesion by six human DNA polymerases members of the B-, X- and Y-families. Nucleic Acids Res. 2014, 42, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Plosky, B.S.; Frank, E.G.; Berry, D.A.; Vennall, G.P.; McDonald, J.P.; Woodgate, R. Eukaryotic Y-family polymerases bypass a 3-methyl-2’-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008, 36, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Sebesta, M.; Balogh, D.; Haracska, L.; Krejci, L. Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res. 2014, 42, 1711–1720. [Google Scholar] [CrossRef]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Nay, S.L.; Lee, D.H.; Bates, S.E.; O’Connor, T.R. Alkbh2 protects against lethality and mutation in primary mouse embryonic fibroblasts. DNA Repair 2012, 11, 502–510. [Google Scholar] [CrossRef]
- Wolfle, W.T.; Washington, M.T.; Prakash, L.; Prakash, S. Human DNA polymerase kappa uses template-primer misalignment as a novel means for extending mispaired termini and for generating single-base deletions. Genes Dev. 2003, 17, 2191–2199. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Feng, Y.; Luo, R. Minichromosome Maintenance Complex is Required for Checkpoint Kinase 2 Chromatin Loading and its Phosphorylation to DNA Damage Response in SCC-4 Cells. Protein Pept. Lett. 2017, 24, 223–228. [Google Scholar] [CrossRef]
- Berger, M.; Stahl, N.; Del Sal, G.; Haupt, Y. Mutations in proline 82 of p53 impair its activation by Pin1 and Chk2 in response to DNA damage. Mol. Cell Biol. 2005, 25, 5380–5388. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.R.; Rhind, N. Replication fork slowing and stalling are distinct, checkpoint-independent consequences of replicating damaged DNA. PLoS Genet. 2017, 13, e1006958. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Preciado, V.; Ufano, S.; Bueno, A. Limiting amounts of budding yeast Rad53 S-phase checkpoint activity results in increased resistance to DNA alkylation damage. Nucleic Acids Res. 2006, 34, 5852–5862. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zegerman, P.; Diffley, J.F. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010, 467, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Limones, C.; Lara-Chica, M.; Jimenez-Jimenez, C.; Perez, M.; Moreno, P.; Munoz, E.; Calzado, M.A. CHK2 stability is regulated by the E3 ubiquitin ligase SIAH2. Oncogene 2016, 35, 4289–4301. [Google Scholar] [CrossRef]
- Bruhl, J.; Trautwein, J.; Schafer, A.; Linne, U.; Bouazoune, K. The DNA repair protein SHPRH is a nucleosome-stimulated ATPase and a nucleosome-E3 ubiquitin ligase. Epigenet. Chromatin 2019, 12, 52. [Google Scholar] [CrossRef]
- Coulombe, P.; Nassar, J.; Peiffer, I.; Stanojcic, S.; Sterkers, Y.; Delamarre, A.; Bocquet, S.; Mechali, M. The ORC ubiquitin ligase OBI1 promotes DNA replication origin firing. Nat. Commun. 2019, 10, 2426. [Google Scholar] [CrossRef]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef]
- Fan, Q.; Xu, X.; Zhao, X.; Wang, Q.; Xiao, W.; Guo, Y.; Fu, Y.V. Rad5 coordinates translesion DNA synthesis pathway by recognizing specific DNA structures in saccharomyces cerevisiae. Curr. Genet. 2018, 64, 889–899. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seelinger, M.; Søgaard, C.K.; Otterlei, M. The Human RAD5 Homologs, HLTF and SHPRH, Have Separate Functions in DNA Damage Tolerance Dependent on the DNA Lesion Type. Biomolecules 2020, 10, 463. https://doi.org/10.3390/biom10030463
Seelinger M, Søgaard CK, Otterlei M. The Human RAD5 Homologs, HLTF and SHPRH, Have Separate Functions in DNA Damage Tolerance Dependent on the DNA Lesion Type. Biomolecules. 2020; 10(3):463. https://doi.org/10.3390/biom10030463
Chicago/Turabian StyleSeelinger, Mareike, Caroline Krogh Søgaard, and Marit Otterlei. 2020. "The Human RAD5 Homologs, HLTF and SHPRH, Have Separate Functions in DNA Damage Tolerance Dependent on the DNA Lesion Type" Biomolecules 10, no. 3: 463. https://doi.org/10.3390/biom10030463
APA StyleSeelinger, M., Søgaard, C. K., & Otterlei, M. (2020). The Human RAD5 Homologs, HLTF and SHPRH, Have Separate Functions in DNA Damage Tolerance Dependent on the DNA Lesion Type. Biomolecules, 10(3), 463. https://doi.org/10.3390/biom10030463