Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Antibodies and Reagents
2.3. Immunoblotting
2.4. Measurement of Cellular Oxygen Consumption and Extracellular Acidification
2.5. Morphological Analysis by Immunofluorescence Microscopy
2.6. Flow Cytometry
2.7. ATP Assay
2.8. RNA Isolation and qRT-PCR
2.9. Heteroplasmy Determination by RFLP
2.10. Statistical Analysis
3. Results
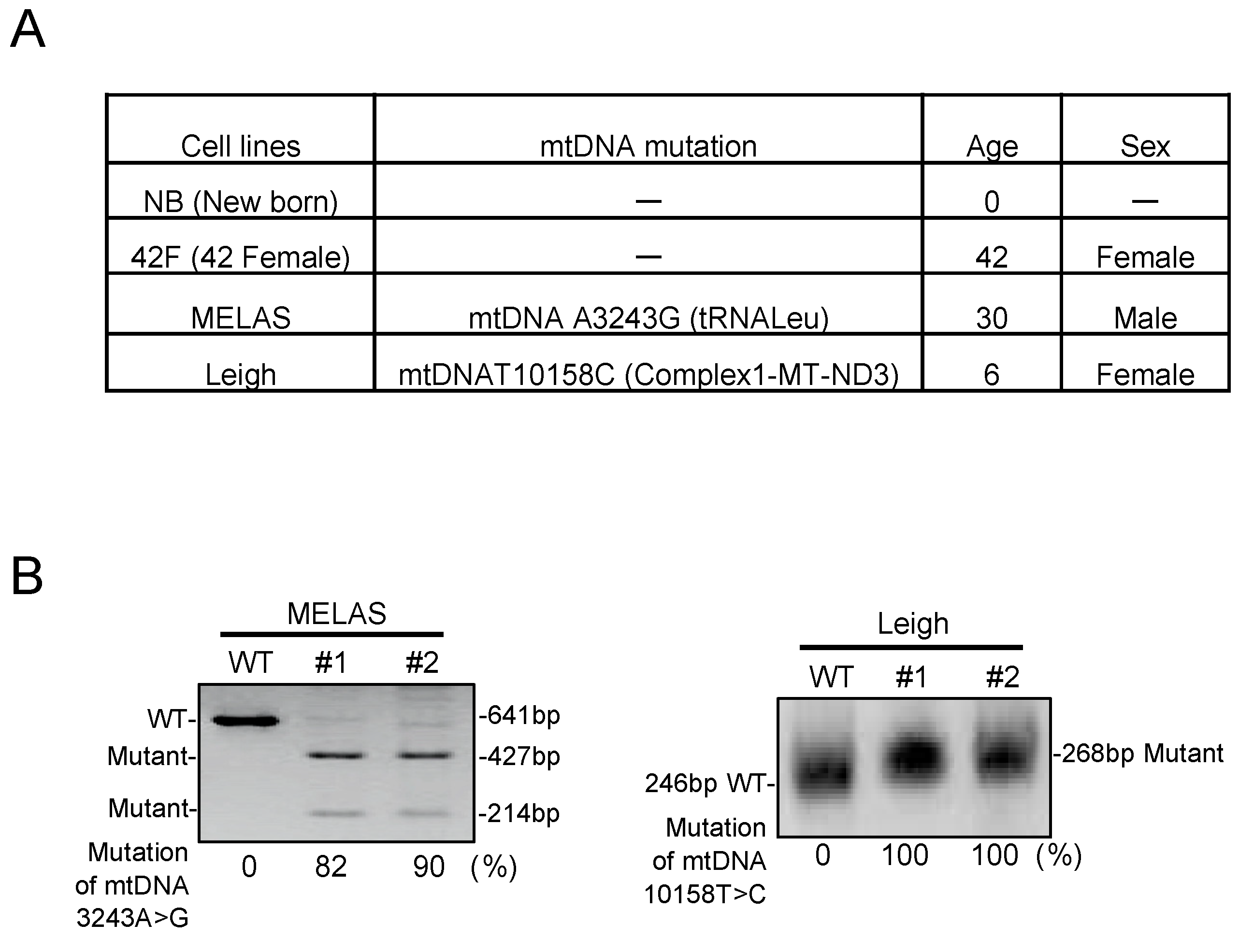
3.1. Characterization in MD Fibroblasts
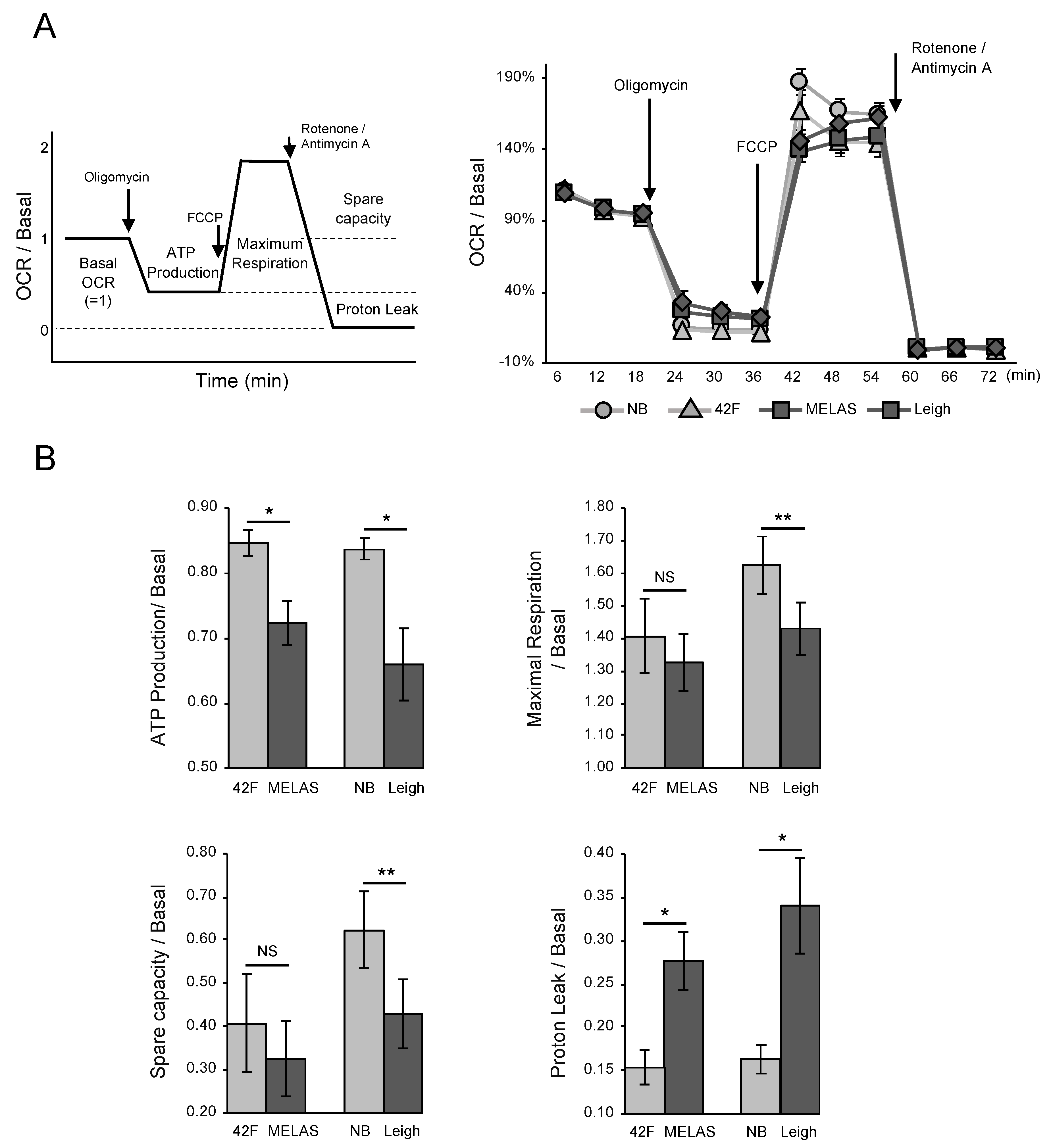
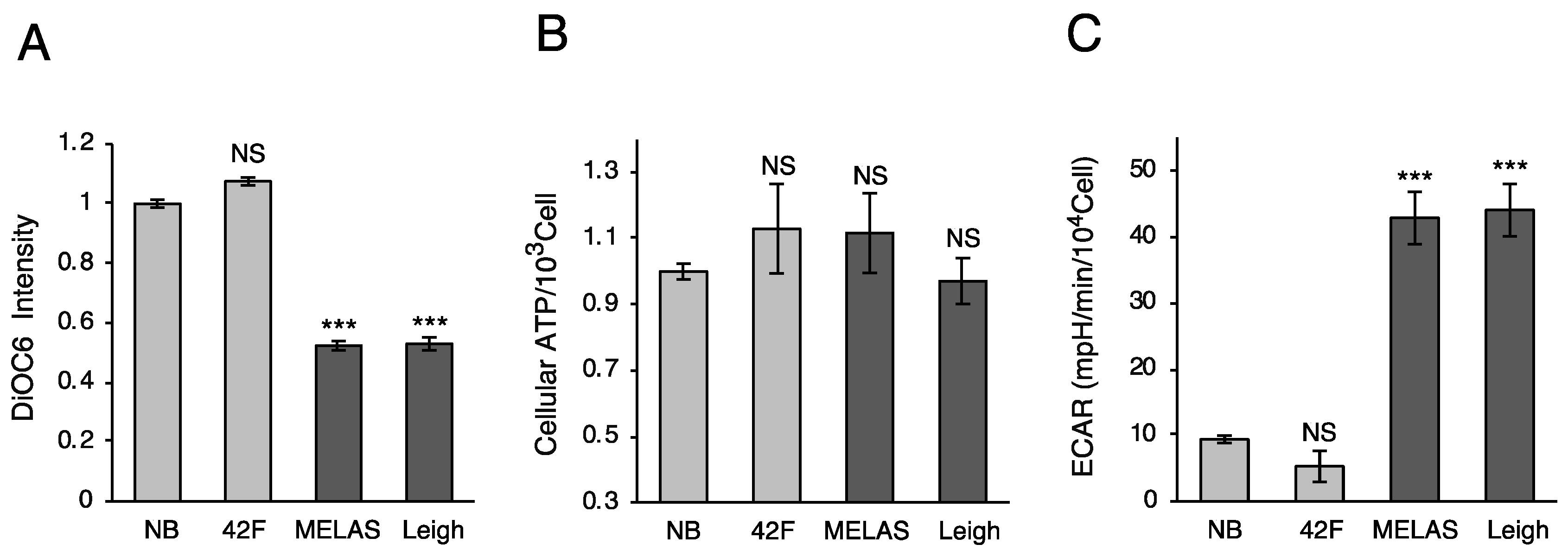
3.2. Oxygen Consumption Rate in MD Fibroblasts
3.3. Mitochondrial Membrane Potential and ATP Production
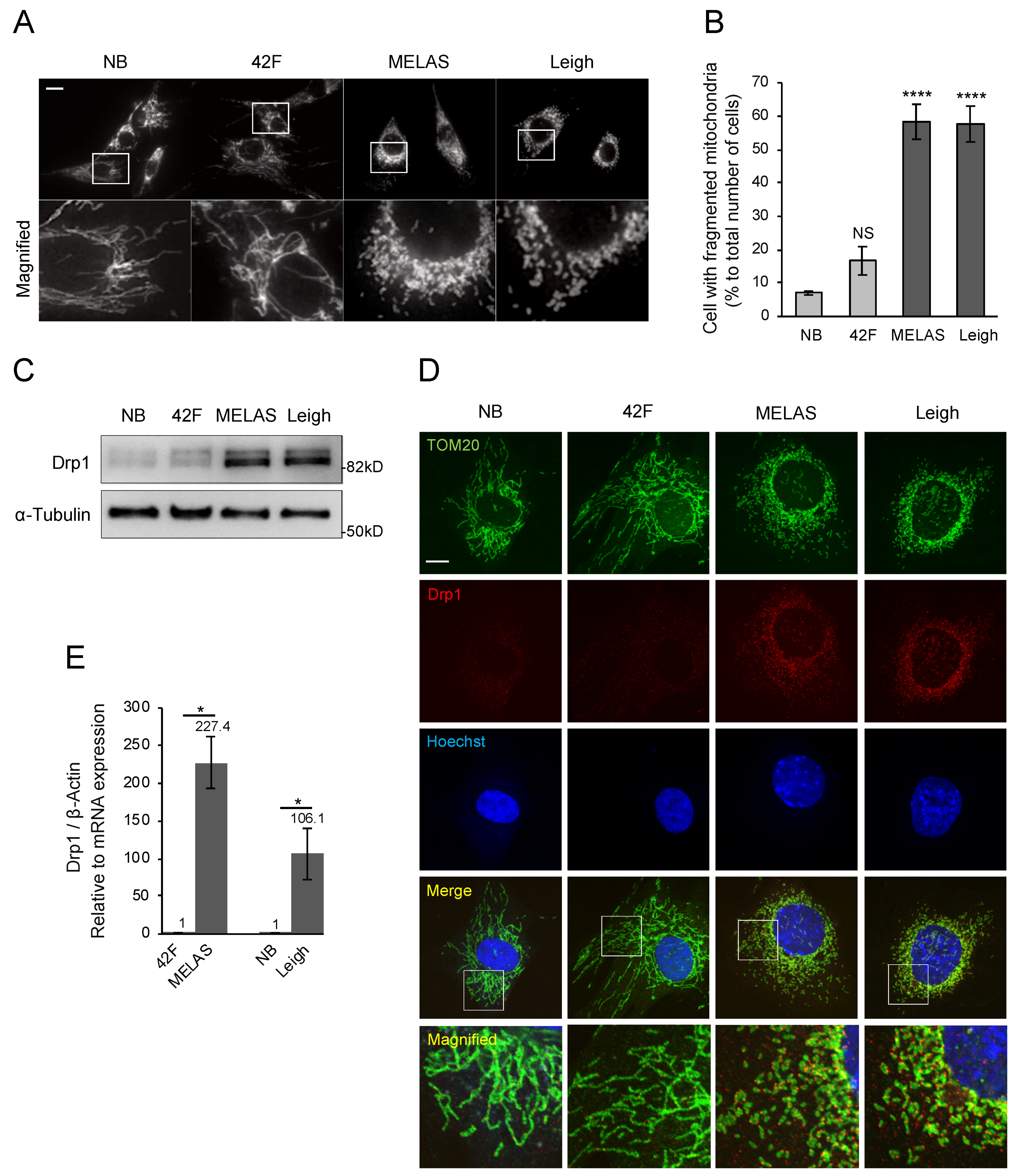
3.4. Promotion of Mitochondrial Fragmentation in MD Fibroblasts
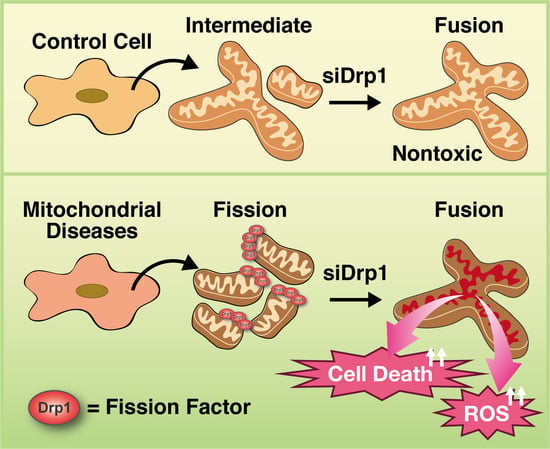
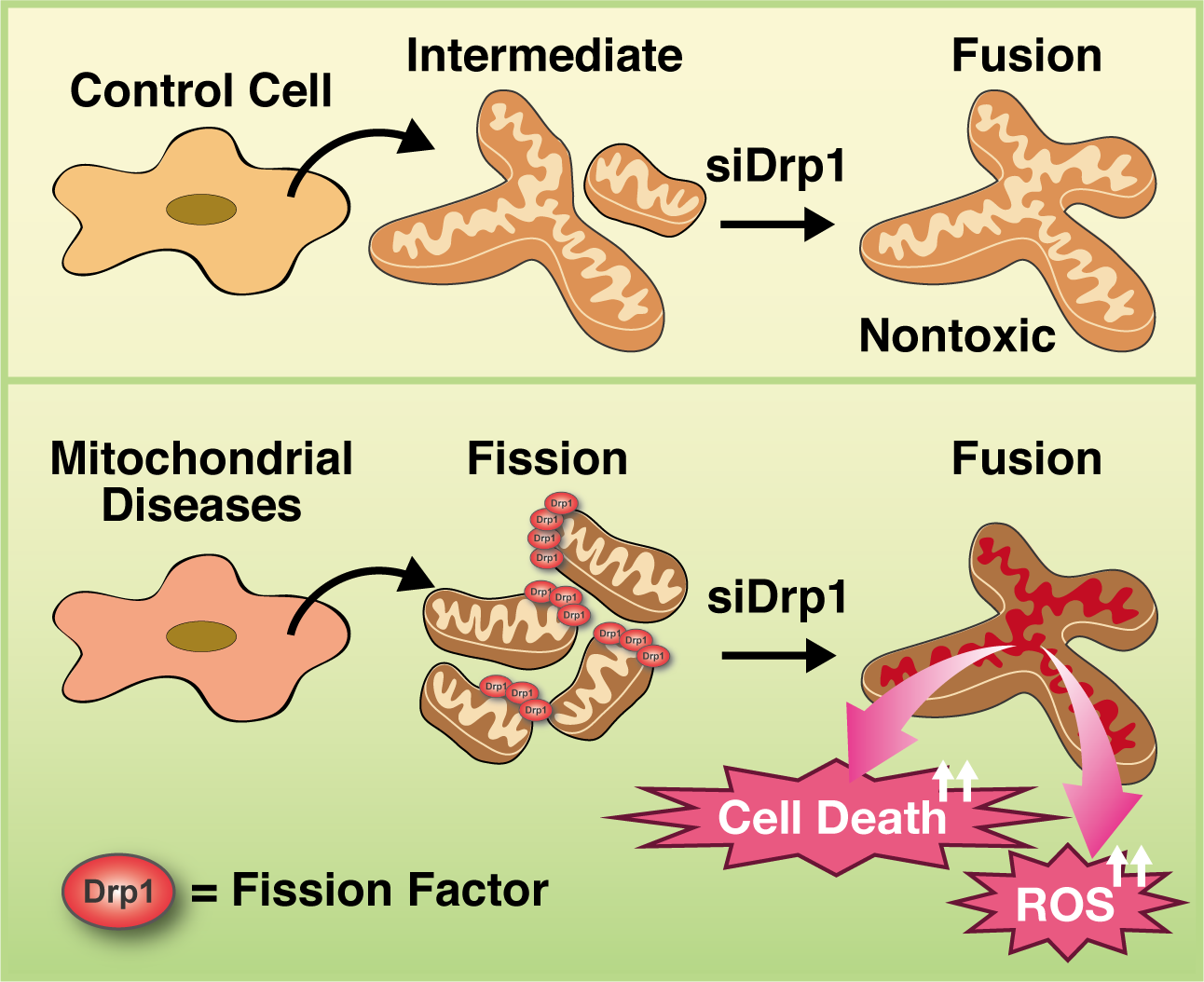
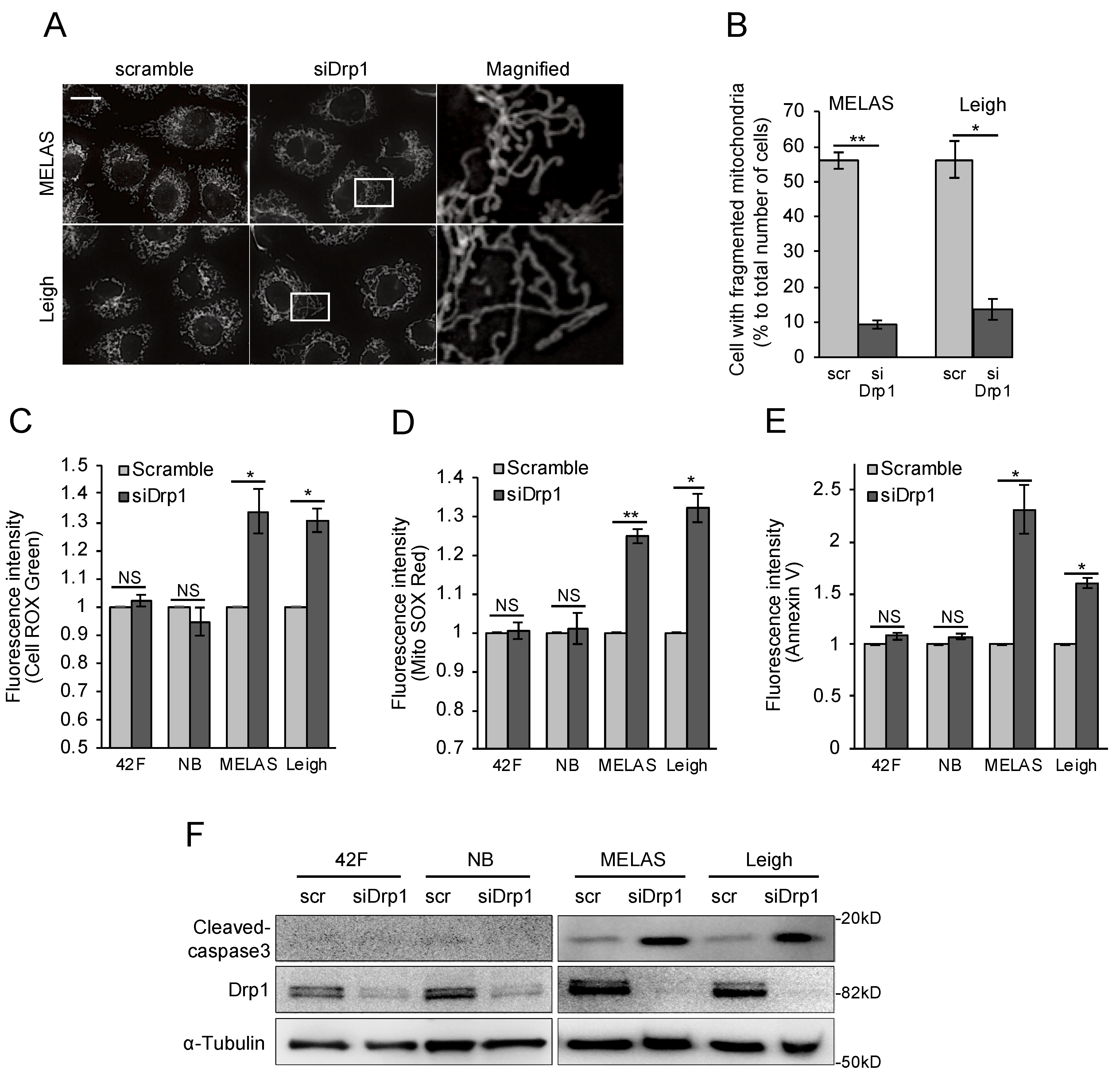
3.5. Inhibition of Mitochondrial Fragmentation Enhances Cellular Toxicity and Cell Death in MD Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sasarman, F.; Antonicka, H.; Shoubridge, E.A. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008, 17, 3697–3707. [Google Scholar] [CrossRef] [PubMed]
- Kirino, Y.; Yasukawa, T.; Ohta, S.; Akira, S.; Ishihara, K.; Watanabe, K.; Suzuki, T. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc. Natl. Acad. Sci. USA 2004, 101, 15070–15075. [Google Scholar] [CrossRef] [PubMed]
- Reda, H.M.; Copen, W.A.; Karaa, A.; Oakley, D.H. Case 13-2017. A 41-Year-Old Man with Hearing Loss, Seizures, Weakness, and Cognitive Decline. N. Engl. J. Med. 2017, 376, 1668–1678. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Horai, S.; Matsuoka, T.; Koga, Y.; Nihei, K.; Kobayashi, M.; Nonaka, I. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): A correlative study of the clinical features and mitochondrial DNA mutation. Neurology 1992, 42, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta Neurol. Scand. 2007, 116, 1–14. [Google Scholar] [CrossRef]
- Bannwarth, S.; Procaccio, V.; Lebre, A.S.; Jardel, C.; Chaussenot, A.; Hoarau, C.; Maoulida, H.; Charrier, N.; Gai, X.; Xie, H.M.; et al. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J. Med. Genet. 2013, 50, 704–714. [Google Scholar] [CrossRef]
- Gerards, M.; Kamps, R.; van Oevelen, J.; Boesten, I.; Jongen, E.; de Koning, B.; Scholte, H.R.; de Angst, I.; Schoonderwoerd, K.; Sefiani, A.; et al. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain 2013, 136, 882–890. [Google Scholar] [CrossRef]
- Shrikhande, D.Y.; Kalakoti, P.; Syed, M.M.; Ahya, K.; Singh, G. A rare mitochondrial disorder: Leigh syndrome--a case report. Ital. J. Pediatr. 2010, 36, 62. [Google Scholar] [CrossRef]
- Viscomi, C.; Bottani, E.; Zeviani, M. Emerging concepts in the therapy of mitochondrial disease. Biochem. Biophys. Acta 2015, 1847, 544–557. [Google Scholar] [CrossRef]
- Chinnery, P.F. Mitochondrial disease in adults: what’s old and what’s new? EMBO Mol. Med. 2015, 7, 1503–1512. [Google Scholar] [CrossRef]
- Viscomi, C. Toward a therapy for mitochondrial disease. Biochem. Soc. Trans. 2016, 44, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Dombi, E.; Mortiboys, H.; Poulton, J. Modulating Mitophagy in Mitochondrial Disease. Curr. Med. Chem. 2018, 25, 5597–5612. [Google Scholar] [CrossRef]
- Sage-Schwaede, A.; Engelstad, K.; Salazar, R.; Curcio, A.; Khandji, A.; Garvin, J.H., Jr.; De Vivo, D.C. Exploring mTOR inhibition as treatment for mitochondrial disease. Ann. Clin. Transl. Neurol. 2019, 6, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Civiletto, G.; Dogan, S.A.; Cerutti, R.; Fagiolari, G.; Moggio, M.; Lamperti, C.; Beninca, C.; Viscomi, C.; Zeviani, M. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef]
- Meeusen, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial fusion intermediates revealed in vitro. Science 2004, 305, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Malka, F.; Guillery, O.; Cifuentes-Diaz, C.; Guillou, E.; Belenguer, P.; Lombes, A.; Rojo, M. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 2005, 6, 853–859. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Yoon, Y.; Pitts, K.R.; McNiven, M.A. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol. Biol. Cell 2001, 12, 2894–2905. [Google Scholar] [CrossRef]
- Shutt, T.E.; McBride, H.M. Staying cool in difficult times: Mitochondrial dynamics, quality control and the stress response. Biochem. Biophys. Acta 2013, 1833, 417–424. [Google Scholar] [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochem. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochem. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Arasaki, K.; Shimizu, H.; Mogari, H.; Nishida, N.; Hirota, N.; Furuno, A.; Kudo, Y.; Baba, M.; Baba, N.; Cheng, J.; et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev. Cell 2015, 32, 304–317. [Google Scholar] [CrossRef]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol 2005, 6, 657–663. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Hirano, M. Pathogenesis and treatment of mitochondrial disorders. Adv. Exp. Med. Biol. 2009, 652, 139–170. [Google Scholar] [PubMed]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef]
- Kuruva, C.S.; Manczak, M.; Yin, X.; Ogunmokun, G.; Reddy, A.P.; Reddy, P.H. Aqua-soluble DDQ reduces the levels of Drp1 and Abeta and inhibits abnormal interactions between Abeta and Drp1 and protects Alzheimer’s disease neurons from Abeta- and Drp1-induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 2017, 26, 3375–3395. [Google Scholar] [CrossRef]
- De Vos, K.J.; Allan, V.J.; Grierson, A.J.; Sheetz, M.P. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr. Biol. 2005, 15, 678–683. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef]
- Sauvanet, C.; Duvezin-Caubet, S.; di Rago, J.P.; Rojo, M. Energetic requirements and bioenergetic modulation of mitochondrial morphology and dynamics. Semin Cell Dev. Biol. 2010, 21, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria-Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef]
- Fernandez-Mosquera, L.; Yambire, K.F.; Couto, R.; Pereyra, L.; Pabis, K.; Ponsford, A.H.; Diogo, C.V.; Stagi, M.; Milosevic, I.; Raimundo, N. Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy 2019, 15, 1572–1591. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef]
- Malena, A.; Loro, E.; Di Re, M.; Holt, I.J.; Vergani, L. Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum. Mol. Genet. 2009, 18, 3407–3416. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020, 10, 450. https://doi.org/10.3390/biom10030450
Tokuyama T, Hirai A, Shiiba I, Ito N, Matsuno K, Takeda K, Saito K, Mii K, Matsushita N, Fukuda T, et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules. 2020; 10(3):450. https://doi.org/10.3390/biom10030450
Chicago/Turabian StyleTokuyama, Takeshi, Asei Hirai, Isshin Shiiba, Naoki Ito, Keigo Matsuno, Keisuke Takeda, Kanata Saito, Koki Mii, Nobuko Matsushita, Toshifumi Fukuda, and et al. 2020. "Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients" Biomolecules 10, no. 3: 450. https://doi.org/10.3390/biom10030450
APA StyleTokuyama, T., Hirai, A., Shiiba, I., Ito, N., Matsuno, K., Takeda, K., Saito, K., Mii, K., Matsushita, N., Fukuda, T., Inatome, R., & Yanagi, S. (2020). Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules, 10(3), 450. https://doi.org/10.3390/biom10030450