Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging


Abstract
1. Introduction
2. Cellular Senescence
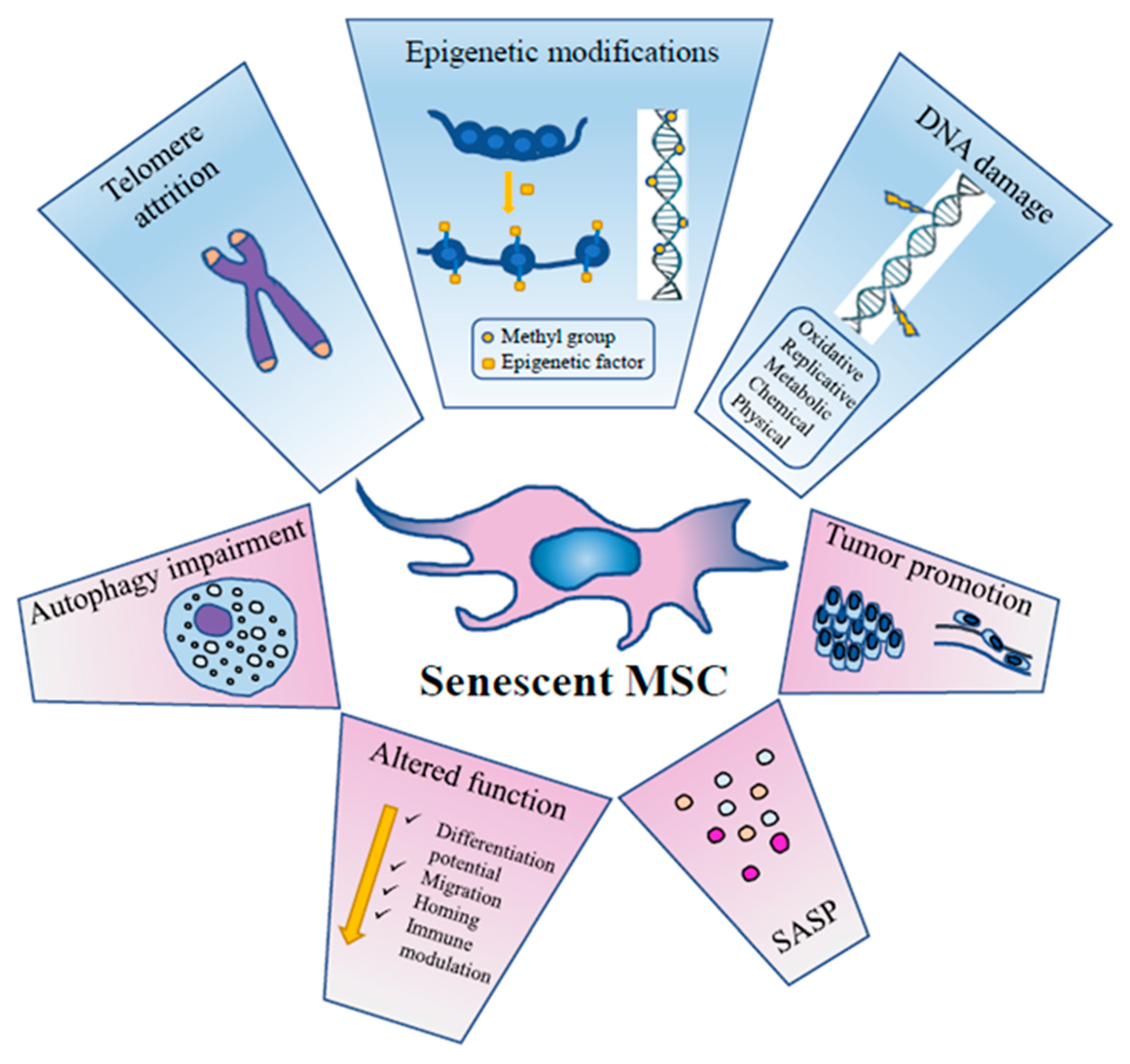
3. MSC Senescence
3.1. DNA Damage
3.2. Chromatin Remodelling
3.3. Epigenetics
3.4. Autophagy
3.5. SASP
4. In Vivo MSC Senescence
5. In Vitro MSC Senescence
5.1. Epigenetics: In Vitro Data
5.2. Autophagy: In Vitro Data
5.3. ROS (Reactive Oxygen Species) Production: In Vitro Data
5.4. Telomeres: In Vitro Data
5.5. SASP: In Vitro Data
6. Detection of Senescent MSCs
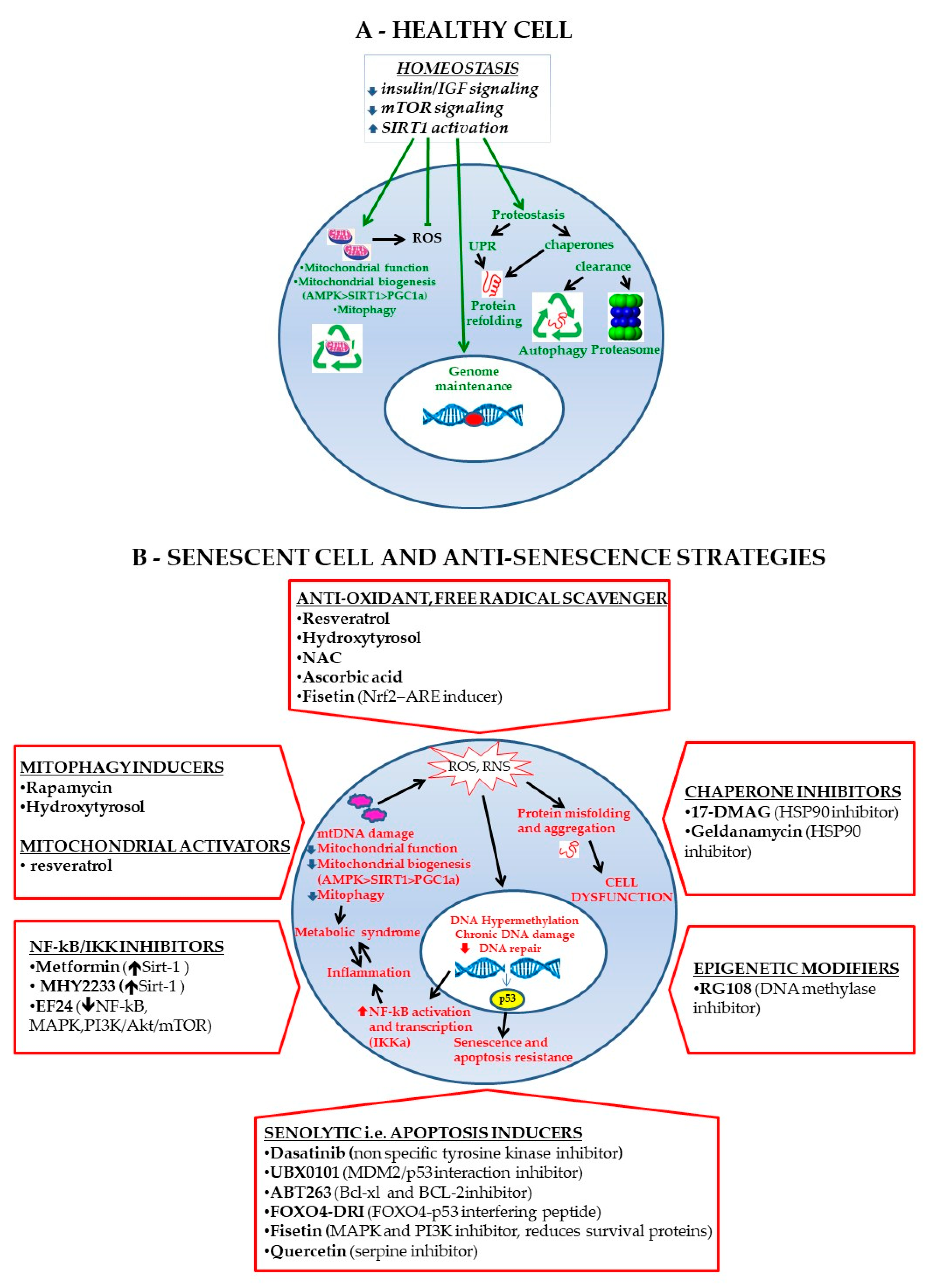
7. MSC Rejuvenating Strategies
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Li, C.Y.; Wu, X.Y.; Tong, J.B.; Yang, X.X.; Zhao, J.L.; Zheng, Q.F.; Zhao, G.B.; Ma, Z.J. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res. Ther. 2015, 6, 55. [Google Scholar] [CrossRef]
- Neri, S. Genetic stability of mesenchymal stromal cells for regenerative medicine applications: A fundamental biosafety aspect. Int J. Mol. Sci. 2019, 20, 2406. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Graf, T. Covering the stem cell explosion at the 2017 isscr conference in boston. Stem Cell Rep. 2017, 9, 1017–1023. [Google Scholar] [CrossRef][Green Version]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef]
- Mastrolia, I.; Foppiani, E.M.; Murgia, A.; Candini, O.; Samarelli, A.V.; Grisendi, G.; Veronesi, E.; Horwitz, E.M.; Dominici, M. Challenges in clinical development of mesenchymal stromal/stem cells: Concise review. Stem Cells Transl. Med. 2019, 8, 1135–1148. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Moretta, L.; Uccelli, A.; Pistoia, V. Mesenchymal stromal cells and immunity: Introductory overview. Immunol. Lett. 2015, 168, 127–128. [Google Scholar] [CrossRef]
- English, K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol. Cell Biol. 2013, 91, 19–26. [Google Scholar] [CrossRef]
- Zhao, K.; Lou, R.; Huang, F.; Peng, Y.; Jiang, Z.; Huang, K.; Wu, X.; Zhang, Y.; Fan, Z.; Zhou, H.; et al. Immunomodulation effects of mesenchymal stromal cells on acute graft-versus-host disease after hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2015, 21, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, N.; Kharlampieva, D.; Loguinova, M.; Butenko, I.; Pobeguts, O.; Efimenko, A.; Ageeva, L.; Sharonov, G.; Ischenko, D.; Alekseev, D.; et al. Characterization of secretomes provides evidence for adipose-derived mesenchymal stromal cells subtypes. Stem Cell Res. Ther. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Pittenger, M.F. Concise review: Msc-derived exosomes for cell-free therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Mun, C.H.; Kang, M.I.; Shin, Y.D.; Kim, Y.; Park, Y.B. The expression of immunomodulation-related cytokines and genes of adipose- and bone marrow-derived human mesenchymal stromal cells from early to late passages. Tissue Eng. Regen. Med. 2018, 15, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.R.; Karnik, S.; Gunderson, Z.J.; Nielsen, J.J.; Fennimore, A.; Promer, H.J.; Lowery, J.W.; Loghmani, M.T.; Low, P.S.; McKinley, T.O.; et al. Dysfunctional stem and progenitor cells impair fracture healing with age. World J. Stem Cells 2019, 11, 281–296. [Google Scholar] [CrossRef]
- Oberbauer, E.; Steffenhagen, C.; Wurzer, C.; Gabriel, C.; Redl, H.; Wolbank, S. Enzymatic and non-enzymatic isolation systems for adipose tissue-derived cells: Current state of the art. Cell Regen. 2015, 4, 7. [Google Scholar] [CrossRef]
- Neri, S.; Bourin, P.; Peyrafitte, J.A.; Cattini, L.; Facchini, A.; Mariani, E. Human adipose stromal cells (asc) for the regeneration of injured cartilage display genetic stability after in vitro culture expansion. PLoS ONE 2013, 8, e77895. [Google Scholar] [CrossRef]
- Neri, S.; Cattini, L.; Facchini, A.; Pawelec, G.; Mariani, E. Microsatellite instability in in vitro ageing of t lymphocyte clones. Exp. Gerontol. 2004, 39, 499–505. [Google Scholar] [CrossRef]
- Neri, S.; Pawelec, G.; Facchini, A.; Ferrari, C.; Mariani, E. Altered expression of mismatch repair proteins associated with acquisition of microsatellite instability in a clonal model of human t lymphocyte aging. Rejuvenation Res. 2008, 11, 565–572. [Google Scholar] [CrossRef]
- Neri, S.; Pawelec, G.; Facchini, A.; Mariani, E. Microsatellite instability and compromised mismatch repair gene expression during in vitro passaging of monoclonal human t lymphocytes. Rejuvenation Res. 2007, 10, 145–156. [Google Scholar] [CrossRef]
- Shibata, K.R.; Aoyama, T.; Shima, Y.; Fukiage, K.; Otsuka, S.; Furu, M.; Kohno, Y.; Ito, K.; Fujibayashi, S.; Neo, M.; et al. Expression of the p16ink4a gene is associated closely with senescence of human mesenchymal stem cells and is potentially silenced by DNA methylation during in vitro expansion. Stem Cells 2007, 25, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Miao, X.; Li, Y.; Smith, C.; Tsang, K.; Cheng, L.; Wang, Q.F. Whole-genome sequencing identifies genetic variances in culture-expanded human mesenchymal stem cells. Stem Cell Rep. 2014, 3, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Behrens, A.; van Deursen, J.M.; Rudolph, K.L.; Schumacher, B. Impact of genomic damage and ageing on stem cell function. Nat. Cell Biol. 2014, 16, 201–207. [Google Scholar] [CrossRef]
- Bonab, M.M.; Alimoghaddam, K.; Talebian, F.; Ghaffari, S.H.; Ghavamzadeh, A.; Nikbin, B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [PubMed]
- de Witte, S.F.H.; Lambert, E.E.; Merino, A.; Strini, T.; Douben, H.; O’Flynn, L.; Elliman, S.J.; de Klein, A.; Newsome, P.N.; Baan, C.C.; et al. Aging of bone marrow- and umbilical cord-derived mesenchymal stromal cells during expansion. Cytotherapy 2017, 19, 798–807. [Google Scholar] [CrossRef]
- Loisel, S.; Dulong, J.; Menard, C.; Renoud, M.L.; Meziere, N.; Isabelle, B.; Latour, M.; Bescher, N.; Pedeux, R.; Bertheuil, N.; et al. Brief report: Proteasomal indoleamine 2,3-dioxygenase degradation reduces the immunosuppressive potential of clinical grade-mesenchymal stromal cells undergoing replicative senescence. Stem Cells 2017, 35, 1431–1436. [Google Scholar] [CrossRef]
- Ganguly, P.; El-Jawhari, J.J.; Giannoudis, P.V.; Burska, A.N.; Ponchel, F.; Jones, E.A. Age-related changes in bone marrow mesenchymal stromal cells: A potential impact on osteoporosis and osteoarthritis development. Cell Transplant. 2017, 26, 1520–1529. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Wang, Y.; Li, L.; Bu, H.; Bao, J. Senescence of mesenchymal stem cells (review). Int. J. Mol. Med. 2017, 39, 775–782. [Google Scholar] [CrossRef]
- Liu, M.; Lei, H.; Dong, P.; Fu, X.; Yang, Z.; Yang, Y.; Ma, J.; Liu, X.; Cao, Y.; Xiao, R. Adipose-derived mesenchymal stem cells from the elderly exhibit decreased migration and differentiation abilities with senescent properties. Cell Transplant. 2017, 26, 1505–1519. [Google Scholar] [CrossRef]
- Turinetto, V.; Vitale, E.; Giachino, C. Senescence in human mesenchymal stem cells: Functional changes and implications in stem cell-based therapy. Int. J. Mol. Sci. 2016, 17, 1164. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, S.; Alessio, N.; Acar, M.B.; Mert, E.; Omerli, F.; Peluso, G.; Galderisi, U. Unbiased analysis of senescence associated secretory phenotype (sasp) to identify common components following different genotoxic stresses. Aging 2016, 8, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Qiu, L.; Ma, J.; Zhang, H.; Cheng, M.; Li, W.; Zhao, X.; Liu, K. Replicative senescence of human bone marrow and umbilical cord derived mesenchymal stem cells and their differentiation to adipocytes and osteoblasts. Mol. Biol. Rep. 2011, 38, 5161–5168. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowska, A.; Schumacher, A.; Filipowicz, N.; Wardowska, A.; Zielinski, M.; Madanecki, P.; Nowicka, E.; Langa, P.; Deptula, M.; Zielinski, J.; et al. Immunophenotyping and transcriptional profiling of in vitro cultured human adipose tissue derived stem cells. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Szychlinska, M.A.; Stoddart, M.J.; D’Amora, U.; Ambrosio, L.; Alini, M.; Musumeci, G. Mesenchymal stem cell-based cartilage regeneration approach and cell senescence: Can we manipulate cell aging and function? Tissue Eng. Part. B. Rev. 2017, 23, 529–539. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Capasso, S.; Alessio, N.; Squillaro, T.; Di Bernardo, G.; Melone, M.A.; Cipollaro, M.; Peluso, G.; Galderisi, U. Changes in autophagy, proteasome activity and metabolism to determine a specific signature for acute and chronic senescent mesenchymal stromal cells. Oncotarget. 2015, 6, 39457–39468. [Google Scholar] [CrossRef]
- Schmitt, C.A. The persistent dynamic secrets of senescence. Nat. Cell Biol. 2016, 18, 913–915. [Google Scholar] [CrossRef][Green Version]
- Habiballa, L.; Salmonowicz, H.; Passos, J.F. Mitochondria and cellular senescence: Implications for musculoskeletal ageing. Free Radic. Biol. Med. 2019, 132, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, B.; Yuk-Wai Lee, W.; Pong, U.K.; Leung, K.T.; Li, X.; Liu, Z.; Chen, R.; Lin, J.C.; Tsang, L.L.; et al. Kdm3a and kdm4c regulate mesenchymal stromal cell senescence and bone aging via condensin-mediated heterochromatin reorganization. iScience 2019, 21, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Enroth, S.; Gyllensten, U. Continuous aging of the human DNA methylome throughout the human lifespan. PLoS ONE 2013, 8, e67378. [Google Scholar] [CrossRef]
- Bork, S.; Pfister, S.; Witt, H.; Horn, P.; Korn, B.; Ho, A.D.; Wagner, W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 2010, 9, 54–63. [Google Scholar] [CrossRef]
- Wagner, W. The link between epigenetic clocks for aging and senescence. Front. Genet. 2019, 10, 303. [Google Scholar] [CrossRef]
- Koch, C.M.; Reck, K.; Shao, K.; Lin, Q.; Joussen, S.; Ziegler, P.; Walenda, G.; Drescher, W.; Opalka, B.; May, T.; et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2013, 23, 248–259. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yu, K.R.; Lee, B.C.; Kang, I.; Kim, J.J.; Jung, E.J.; Kim, H.S.; Seo, Y.; Choi, S.W.; Kang, K.S. Gata4-dependent regulation of the secretory phenotype via mcp-1 underlies lamin a-mediated human mesenchymal stem cell aging. Exp. Mol. Med. 2018, 50, 63. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. Mtor regulates mapkapk2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. Mtor regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting il1a translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. Notch1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Oberacker, T.; Bajorat, J.; Ziola, S.; Schroeder, A.; Roth, D.; Kastl, L.; Edgar, B.A.; Wagner, W.; Gulow, K.; Krammer, P.H. Enhanced expression of thioredoxin-interacting-protein regulates oxidative DNA damage and aging. FEBS Lett. 2018, 592, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Revuelta, M.; Matheu, A. Autophagy in stem cell aging. Aging Cell 2017, 16, 912–915. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Yu, K.R.; Kang, K.S. Aging-related genes in mesenchymal stem cells: A mini-review. Gerontology 2013, 59, 557–563. [Google Scholar] [CrossRef]
- Mani, C.; Reddy, P.H.; Palle, K. DNA repair fidelity in stem cell maintenance, health, and disease. Biochem. Biophys. Acta Mol. Basis Dis. 2019, 1866, 165444. [Google Scholar] [CrossRef]
- Ren, R.; Ocampo, A.; Liu, G.H.; Izpisua Belmonte, J.C. Regulation of stem cell aging by metabolism and epigenetics. Cell Metab. 2017, 26, 460–474. [Google Scholar] [CrossRef]
- Squillaro, T.; Alessio, N.; Capasso, S.; Di Bernardo, G.; Melone, M.A.B.; Peluso, G.; Galderisi, U. Senescence phenomena and metabolic alteration in mesenchymal stromal cells from a mouse model of rett syndrome. Int J. Mol. Sci. 2019, 20, 2508. [Google Scholar] [CrossRef]
- Gharibi, B.; Farzadi, S.; Ghuman, M.; Hughes, F.J. Inhibition of akt/mtor attenuates age-related changes in mesenchymal stem cells. Stem Cells 2014, 32, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.P.; Chiu, F.Y.; Wang, Y.; Yen, M.L.; Kao, S.Y.; Hung, S.C. Rb maintains quiescence and prevents premature senescence through upregulation of dnmt1 in mesenchymal stromal cells. Stem Cell Rep. 2014, 3, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Solis, M.A.; Chen, Y.H.; Huang, L.L. Molecular mechanism of extrinsic factors affecting anti-aging of stem cells. World J. Stem Cells 2015, 7, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Song, L.; Li, Y. Controlling redox status for stem cell survival, expansion, and differentiation. Oxid. Med. Cell Longev. 2015, 2015, 105135. [Google Scholar] [CrossRef]
- Vono, R.; Jover Garcia, E.; Spinetti, G.; Madeddu, P. Oxidative stress in mesenchymal stem cell senescence: Regulation by coding and noncoding rnas. Antioxid. Redox Signal. 2018, 29, 864–879. [Google Scholar] [CrossRef]
- Lee, H.J.; Choi, B.; Kim, Y.; Lee, S.E.; Jin, H.J.; Lee, H.S.; Chang, E.J.; Kim, S.W. The upregulation of toll-like receptor 3 via autocrine ifn-beta signaling drives the senescence of human umbilical cord blood-derived mesenchymal stem cells through jak1. Front. Immunol. 2019, 10, 1659. [Google Scholar] [CrossRef]
- Musavi, M.; Kohram, F.; Abasi, M.; Bolandi, Z.; Ajoudanian, M.; Mohammadi-Yeganeh, S.; Hashemi, S.M.; Sharifi, K.; Fathi, H.R.; Ghanbarian, H. Rn7sk small nuclear rna is involved in cellular senescence. J. Cell Physiol. 2019, 234, 14234–14245. [Google Scholar] [CrossRef]
- Hladik, D.; Hofig, I.; Oestreicher, U.; Beckers, J.; Matjanovski, M.; Bao, X.; Scherthan, H.; Atkinson, M.J.; Rosemann, M. Long-term culture of mesenchymal stem cells impairs atm-dependent recognition of DNA breaks and increases genetic instability. Stem Cell Res. Ther. 2019, 10, 218. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The atm protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. Atr: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef]
- Li, H.; Ghazanfari, R.; Zacharaki, D.; Lim, H.C.; Scheding, S. Isolation and characterization of primary bone marrow mesenchymal stromal cells. Ann. N. Y. Acad. Sci. 2016, 1370, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Larson, K.; Yan, S.J.; Tsurumi, A.; Liu, J.; Zhou, J.; Gaur, K.; Guo, D.; Eickbush, T.H.; Li, W.X. Heterochromatin formation promotes longevity and represses ribosomal rna synthesis. PLoS Genet. 2012, 8, e1002473. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ren, R.; Liu, Z.; Song, M.; Li, J.; Wu, Z.; Ren, X.; Fu, L.; Li, W.; Zhang, W.; et al. Stabilizing heterochromatin by dgcr8 alleviates senescence and osteoarthritis. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Severino, V.; Alessio, N.; Farina, A.; Di Bernardo, G.; Cipollaro, M.; Peluso, G.; Chambery, A.; Galderisi, U. De-regulated expression of the brg1 chromatin remodeling factor in bone marrow mesenchymal stromal cells induces senescence associated with the silencing of nanog and changes in the levels of chromatin proteins. Cell Cycle 2015, 14, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Ozkul, Y.; Galderisi, U. The impact of epigenetics on mesenchymal stem cell biology. J. Cell Physiol. 2016, 231, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Cakouros, D.; Gronthos, S. Epigenetic regulation of bone marrow stem cell aging: Revealing epigenetic signatures associated with hematopoietic and mesenchymal stem cell aging. Aging Dis. 2019, 10, 174–189. [Google Scholar] [CrossRef]
- Li, Z.; Liu, C.; Xie, Z.; Song, P.; Zhao, R.C.; Guo, L.; Liu, Z.; Wu, Y. Epigenetic dysregulation in mesenchymal stem cell aging and spontaneous differentiation. PLoS ONE 2011, 6, e20526. [Google Scholar] [CrossRef]
- Lu, Y.; Qu, H.; Qi, D.; Xu, W.; Liu, S.; Jin, X.; Song, P.; Guo, Y.; Jia, Y.; Wang, X.; et al. Oct4 maintains self-renewal and reverses senescence in human hair follicle mesenchymal stem cells through the downregulation of p21 by DNA methyltransferases. Stem Cell Res. Ther. 2019, 10, 28. [Google Scholar] [CrossRef]
- Li, H.; Liu, P.; Xu, S.; Li, Y.; Dekker, J.D.; Li, B.; Fan, Y.; Zhang, Z.; Hong, Y.; Yang, G.; et al. Foxp1 controls mesenchymal stem cell commitment and senescence during skeletal aging. J. Clin. Invest. 2017, 127, 1241–1253. [Google Scholar] [CrossRef]
- So, A.Y.; Jung, J.W.; Lee, S.; Kim, H.S.; Kang, K.S. DNA methyltransferase controls stem cell aging by regulating bmi1 and ezh2 through micrornas. PLoS ONE 2011, 6, e19503. [Google Scholar] [CrossRef]
- Koch, C.M.; Joussen, S.; Schellenberg, A.; Lin, Q.; Zenke, M.; Wagner, W. Monitoring of cellular senescence by DNA-methylation at specific cpg sites. Aging Cell 2012, 11, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, A.; Lin, Q.; Schuler, H.; Koch, C.M.; Joussen, S.; Denecke, B.; Walenda, G.; Pallua, N.; Suschek, C.V.; Zenke, M.; et al. Replicative senescence of mesenchymal stem cells causes DNA-methylation changes which correlate with repressive histone marks. Aging 2011, 3, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W. Epigenetic aging clocks in mice and men. Genome Biol. 2017, 18, 107. [Google Scholar] [CrossRef] [PubMed]
- Bentivegna, A.; Miloso, M.; Riva, G.; Foudah, D.; Butta, V.; Dalpra, L.; Tredici, G. DNA methylation changes during in vitro propagation of human mesenchymal stem cells: Implications for their genomic stability? Stem Cells Int. 2013, 2013, 192425. [Google Scholar] [CrossRef]
- Torano, E.G.; Bayon, G.F.; Del Real, A.; Sierra, M.I.; Garcia, M.G.; Carella, A.; Belmonte, T.; Urdinguio, R.G.; Cubillo, I.; Garcia-Castro, J.; et al. Age-associated hydroxymethylation in human bone-marrow mesenchymal stem cells. J. Transl. Med. 2016, 14, 207. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—Partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef]
- D’Arrigo, D.; Roffi, A.; Cucchiarini, M.; Moretti, M.; Candrian, C.; Filardo, G. Secretome and extracellular vesicles as new biological therapies for knee osteoarthritis: A systematic review. J. Clin. Med. 2019, 8, 1867. [Google Scholar] [CrossRef]
- Liu, L.; Jin, X.; Hu, C.F.; Li, R.; Zhou, Z.; Shen, C.X. Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy via ampk and akt pathways. Cell Physiol. Biochem. 2017, 43, 52–68. [Google Scholar] [CrossRef]
- Ma, Y.; Qi, M.; An, Y.; Zhang, L.; Yang, R.; Doro, D.H.; Liu, W.; Jin, Y. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 2018, 17, e12709. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Tong, F. Autophagy induction is a survival response against oxidative stress in bone marrow-derived mesenchymal stromal cells. Cytotherapy 2014, 16, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Hu, C.J.; Zhuo, R.H.; Lei, Y.S.; Han, N.N.; He, L. Inhibition of autophagy alleviates the senescent state of rat mesenchymal stem cells during long-term culture. Mol. Med. Rep. 2014, 10, 3003–3008. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Bouhtit, F.; Melki, R.; Afif, H.; Hamal, A.; Fahmi, H.; Merimi, M.; Lagneaux, L. Mesenchymal stromal cell-based therapy: New perspectives and challenges. J. Clin. Med. 2019, 8, 626. [Google Scholar] [CrossRef]
- Fafian-Labora, J.; Lesende-Rodriguez, I.; Fernandez-Pernas, P.; Sangiao-Alvarellos, S.; Monserrat, L.; Arntz, O.J.; van de Loo, F.J.; Mateos, J.; Arufe, M.C. Effect of age on pro-inflammatory mirnas contained in mesenchymal stem cell-derived extracellular vesicles. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Kulkarni, R.; Bajaj, M.; Ghode, S.; Jalnapurkar, S.; Limaye, L.; Kale, V.P. Intercellular transfer of microvesicles from young mesenchymal stromal cells rejuvenates aged murine hematopoietic stem cells. Stem Cells 2018, 36, 420–433. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal stem cells: Time to change the name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B.; Paganelli, R. Aging, obesity, and inflammatory age-related diseases. Front. Immunol. 2017, 8, 1745. [Google Scholar] [CrossRef]
- Philipot, D.; Guerit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.M.; Piette, J.; Borzi, R.M.; et al. P16ink4a and its regulator mir-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef]
- Mazzotti, E.; Teti, G.; Falconi, M.; Chiarini, F.; Barboni, B.; Mazzotti, A.; Muttini, A. Age-related alterations affecting the chondrogenic differentiation of synovial fluid mesenchymal stromal cells in an equine model. Cells 2019, 8, 1116. [Google Scholar] [CrossRef]
- Perez, L.M.; Bernal, A.; de Lucas, B.; San Martin, N.; Mastrangelo, A.; Garcia, A.; Barbas, C.; Galvez, B.G. Altered metabolic and stemness capacity of adipose tissue-derived stem cells from obese mouse and human. PLoS ONE 2015, 10, e0123397. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Pan, Z.Y.; Zou, Y.; He, Y.; Yang, P.Y.; Tang, Q.Q.; Yin, F. A comparative assessment of adipose-derived stem cells from subcutaneous and visceral fat as a potential cell source for knee osteoarthritis treatment. J. Cell Mol. Med. 2017, 21, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Kalupahana, N.S.; Claycombe, K.J.; Moustaid-Moussa, N. (n-3) fatty acids alleviate adipose tissue inflammation and insulin resistance: Mechanistic insights. Adv. Nutr. 2011, 2, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Karolina, D.S.; Tavintharan, S.; Armugam, A.; Sepramaniam, S.; Pek, S.L.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. Circulating mirna profiles in patients with metabolic syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E2271–E2276. [Google Scholar] [CrossRef] [PubMed]
- Roldan, M.; Macias-Gonzalez, M.; Garcia, R.; Tinahones, F.J.; Martin, M. Obesity short-circuits stemness gene network in human adipose multipotent stem cells. FASEB J. 2011, 25, 4111–4126. [Google Scholar] [CrossRef] [PubMed]
- Ejarque, M.; Ceperuelo-Mallafre, V.; Serena, C.; Maymo-Masip, E.; Duran, X.; Diaz-Ramos, A.; Millan-Scheiding, M.; Nunez-Alvarez, Y.; Nunez-Roa, C.; Gama, P.; et al. Adipose tissue mitochondrial dysfunction in human obesity is linked to a specific DNA methylation signature in adipose-derived stem cells. Int. J. Obes. 2019, 43, 1256–1268. [Google Scholar] [CrossRef]
- Minguzzi, M.; Guidotti, S.; Platano, D.; D’Adamo, S.; Cetrullo, S.; Assirelli, E.; Santi, S.; Mariani, E.; Trisolino, G.; Filardo, G.; et al. Polyamine supplementation reduces DNA damage in adipose stem cells cultured in 3-d. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Gurkar, A.; Niedernhofer, L.J.; Robbins, P.D. Methods to quantify the nf-kappab pathway during senescence. Methods Mol. Biol. 2019, 1896, 231–250. [Google Scholar]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Birch, J.; Passos, J.F. Targeting the sasp to combat ageing: Mitochondria as possible intracellular allies? Bioessays 2017, 39, 1600235. [Google Scholar] [CrossRef]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The nf-kappab family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Ichim, G.; Tait, S.W.; Passos, J.F. Depletion of mitochondria in mammalian cells through enforced mitophagy. Nat. Protoc. 2017, 12, 183–194. [Google Scholar] [CrossRef]
- Ghanta, S.; Tsoyi, K.; Liu, X.; Nakahira, K.; Ith, B.; Coronata, A.A.; Fredenburgh, L.E.; Englert, J.A.; Piantadosi, C.A.; Choi, A.M.; et al. Mesenchymal stromal cells deficient in autophagy proteins are susceptible to oxidative injury and mitochondrial dysfunction. Am. J. Respir. Cell Mol. Biol. 2017, 56, 300–309. [Google Scholar] [CrossRef]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle micrornas. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A.; et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef]
- Fan, J.; An, X.; Yang, Y.; Xu, H.; Fan, L.; Deng, L.; Li, T.; Weng, X.; Zhang, J.; Chunhua Zhao, R. Mir-1292 targets fzd4 to regulate senescence and osteogenic differentiation of stem cells in te/sj/mesenchymal tissue system via the wnt/beta-catenin pathway. Aging Dis. 2018, 9, 1103–1121. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Qi, M.; Konermann, A.; Zhang, L.; Jin, F.; Jin, Y. The p53/mir-17/smurf1 pathway mediates skeletal deformities in an age-related model via inhibiting the function of mesenchymal stem cells. Aging 2015, 7, 205–218. [Google Scholar] [CrossRef]
- Gnani, D.; Crippa, S.; Della Volpe, L.; Rossella, V.; Conti, A.; Lettera, E.; Rivis, S.; Ometti, M.; Fraschini, G.; Bernardo, M.E.; et al. An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell 2019, 18, e12933. [Google Scholar] [CrossRef] [PubMed]
- Malaise, O.; Tachikart, Y.; Constantinides, M.; Mumme, M.; Ferreira-Lopez, R.; Noack, S.; Krettek, C.; Noel, D.; Wang, J.; Jorgensen, C.; et al. Mesenchymal stem cell senescence alleviates their intrinsic and seno-suppressive paracrine properties contributing to osteoarthritis development. Aging 2019, 11, 9128–9146. [Google Scholar] [CrossRef] [PubMed]
- Stolzing, A.; Jones, E.; McGonagle, D.; Scutt, A. Age-related changes in human bone marrow-derived mesenchymal stem cells: Consequences for cell therapies. Mech. Ageing Dev. 2008, 129, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Choudhery, M.S.; Badowski, M.; Muise, A.; Pierce, J.; Harris, D.T. Donor age negatively impacts adipose tissue-derived mesenchymal stem cell expansion and differentiation. J. Transl. Med. 2014, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Zaim, M.; Karaman, S.; Cetin, G.; Isik, S. Donor age and long-term culture affect differentiation and proliferation of human bone marrow mesenchymal stem cells. Ann. Hematol. 2012, 91, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; El-Jawhari, J.J.; Burska, A.N.; Ponchel, F.; Giannoudis, P.V.; Jones, E.A. The analysis of in vivo aging in human bone marrow mesenchymal stromal cells using colony-forming unit-fibroblast assay and the cd45(low)cd271(+) phenotype. Stem Cells Int. 2019, 2019, 5197983. [Google Scholar] [CrossRef] [PubMed]
- Choumerianou, D.M.; Dimitriou, H.; Perdikogianni, C.; Martimianaki, G.; Riminucci, M.; Kalmanti, M. Study of oncogenic transformation in ex vivo expanded mesenchymal cells, from paediatric bone marrow. Cell Prolif. 2008, 41, 909–922. [Google Scholar] [CrossRef]
- Pipino, C.; Pierdomenico, L.; Di Tomo, P.; Di Giuseppe, F.; Cianci, E.; D’Alimonte, I.; Morabito, C.; Centurione, L.; Antonucci, I.; Mariggio, M.A.; et al. Molecular and phenotypic characterization of human amniotic fluid-derived cells: A morphological and proteomic approach. Stem Cells Dev. 2015, 24, 1415–1428. [Google Scholar] [CrossRef]
- Alessio, N.; Squillaro, T.; Ozcan, S.; Di Bernardo, G.; Venditti, M.; Melone, M.; Peluso, G.; Galderisi, U. Stress and stem cells: Adult muse cells tolerate extensive genotoxic stimuli better than mesenchymal stromal cells. Oncotarget. 2018, 9, 19328–19341. [Google Scholar] [CrossRef] [PubMed]
- Kornicka, K.; Marycz, K.; Maredziak, M.; Tomaszewski, K.A.; Nicpon, J. The effects of the DNA methyltranfserases inhibitor 5-azacitidine on ageing, oxidative stress and DNA methylation of adipose derived stem cells. J. Cell Mol. Med. 2017, 21, 387–401. [Google Scholar] [CrossRef]
- Abolhasani, M.; Rezaee, M.A.; Mohammadi, M.; Ghadimi, T.; Rahmani, M.R. Immunomodulatory properties of umbilical cord vein mesenchymal stromal cells influenced by gestational age and in vitro expansion. Immunol. Lett. 2018, 194, 62–68. [Google Scholar] [CrossRef]
- Kim, M.K.; Lee, W.; Yoon, G.H.; Chang, E.J.; Choi, S.C.; Kim, S.W. Links between accelerated replicative cellular senescence and down-regulation of sphk1 transcription. BMB Rep. 2019, 52, 220–225. [Google Scholar] [CrossRef]
- Kilpinen, L.; Tigistu-Sahle, F.; Oja, S.; Greco, D.; Parmar, A.; Saavalainen, P.; Nikkila, J.; Korhonen, M.; Lehenkari, P.; Kakela, R.; et al. Aging bone marrow mesenchymal stromal cells have altered membrane glycerophospholipid composition and functionality. J. Lipid Res. 2013, 54, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Li, P.; Cui, D.C.; Dang, R.J.; Zhang, L.; Wen, N.; Jiang, X.X. Effect of aged bone marrow microenvironment on mesenchymal stem cell migration. Age 2015, 37, 16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.; Kim, C.; Choi, Y.S.; Park, C.; Suh, Y. Age-related alterations in mesenchymal stem cells related to shift in differentiation from osteogenic to adipogenic potential: Implication to age-associated bone diseases and defects. Mech. Ageing Dev. 2012, 133, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Boyette, L.B.; Tuan, R.S. Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone 2015, 70, 37–47. [Google Scholar] [CrossRef]
- Wiese, D.M.; Ruttan, C.C.; Wood, C.A.; Ford, B.N.; Braid, L.R. Accumulating transcriptome drift precedes cell aging in human umbilical cord-derived mesenchymal stromal cells serially cultured to replicative senescence. Stem Cells Transl. Med. 2019, 8, 945–958. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, W.; Shen, F.; Yu, Y.; Wang, Y.; Xiang, C. Histone arginine methylation-mediated epigenetic regulation of discoidin domain receptor 2 controls the senescence of human bone marrow mesenchymal stem cells. Stem Cells Int. 2019, 2019, 7670316. [Google Scholar] [CrossRef]
- Neri, S.; Guidotti, S.; Lilli, N.L.; Cattini, L.; Mariani, E. Infrapatellar fat pad-derived mesenchymal stromal cells from osteoarthritis patients: In vitro genetic stability and replicative senescence. J. Orthop. Res. 2017, 35, 1029–1037. [Google Scholar] [CrossRef]
- Pasumarthy, K.K.; Doni Jayavelu, N.; Kilpinen, L.; Andrus, C.; Battle, S.L.; Korhonen, M.; Lehenkari, P.; Lund, R.; Laitinen, S.; Hawkins, R.D. Methylome analysis of human bone marrow mscs reveals extensive age- and culture-induced changes at distal regulatory elements. Stem Cell Reports 2017, 9, 999–1015. [Google Scholar] [CrossRef]
- Choi, M.R.; In, Y.H.; Park, J.; Park, T.; Jung, K.H.; Chai, J.C.; Chung, M.K.; Lee, Y.S.; Chai, Y.G. Genome-scale DNA methylation pattern profiling of human bone marrow mesenchymal stem cells in long-term culture. Exp. Mol. Med. 2012, 44, 503–512. [Google Scholar] [CrossRef]
- Hon, G.C.; Rajagopal, N.; Shen, Y.; McCleary, D.F.; Yue, F.; Dang, M.D.; Ren, B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 2013, 45, 1198–1206. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Phermthai, T.; Suksompong, S.; Tirawanchai, N.; Issaragrisil, S.; Julavijitphong, S.; Wichitwiengrat, S.; Silpsorn, D.; Pokathikorn, P. Epigenetic analysis and suitability of amniotic fluid stem cells for research and therapeutic purposes. Stem Cells Dev. 2013, 22, 1319–1328. [Google Scholar] [CrossRef]
- Zhu, Y.; Song, X.; Han, F.; Li, Y.; Wei, J.; Liu, X. Alteration of histone acetylation pattern during long-term serum-free culture conditions of human fetal placental mesenchymal stem cells. PLoS ONE 2015, 10, e0117068. [Google Scholar] [CrossRef]
- Redaelli, S.; Bentivegna, A.; Foudah, D.; Miloso, M.; Redondo, J.; Riva, G.; Baronchelli, S.; Dalpra, L.; Tredici, G. From cytogenomic to epigenomic profiles: Monitoring the biologic behavior of in vitro cultured human bone marrow mesenchymal stem cells. Stem Cell Res. Ther. 2012, 3, 47. [Google Scholar] [CrossRef]
- Schellenberg, A.; Mauen, S.; Koch, C.M.; Jans, R.; de Waele, P.; Wagner, W. Proof of principle: Quality control of therapeutic cell preparations using senescence-associated DNA-methylation changes. BMC Res. Notes 2014, 7, 254. [Google Scholar] [CrossRef]
- Franzen, J.; Zirkel, A.; Blake, J.; Rath, B.; Benes, V.; Papantonis, A.; Wagner, W. Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell 2017, 16, 183–191. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, H.; Wu, M.; Tan, B.; Zhao, L.; Yi, Q.; Xu, X.; Pan, H.; Bi, Y.; Yang, K. Decline of p300 contributes to cell senescence and growth inhibition of huc-mscs through p53/p21 signaling pathway. Biochem. Biophys. Res. Commun. 2019, 515, 24–30. [Google Scholar] [CrossRef]
- Zheng, Y.; Lei, Y.; Hu, C. P53 regulates autophagic activity in senescent rat mesenchymal stromal cells. Exp. Gerontol. 2016, 75, 64–71. [Google Scholar] [CrossRef]
- Chang, T.C.; Hsu, M.F.; Wu, K.K. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PLoS ONE 2015, 10, e0126537. [Google Scholar] [CrossRef]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Grukova, A.A.; Nikolsky, N.N.; Burova, E.B. Tetraploidization or autophagy: The ultimate fate of senescent human endometrial stem cells under atm or p53 inhibition. Cell Cycle 2016, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Alessio, N.; Del Gaudio, S.; Capasso, S.; Di Bernardo, G.; Cappabianca, S.; Cipollaro, M.; Peluso, G.; Galderisi, U. Low dose radiation induced senescence of human mesenchymal stromal cells and impaired the autophagy process. Oncotarget. 2015, 6, 8155–8166. [Google Scholar] [CrossRef] [PubMed]
- Fafian-Labora, J.A.; Morente-Lopez, M.; Arufe, M.C. Effect of aging on behaviour of mesenchymal stem cells. World J. Stem Cells 2019, 11, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.G.; Cho, G.W. Endogenous ros levels are increased in replicative senescence in human bone marrow mesenchymal stromal cells. Biochem. Biophys. Res. Commun. 2015, 460, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Chen, P.K.; Sun, L.Y.; Pang, C.Y. Enhancement of mitochondrial transfer by antioxidants in human mesenchymal stem cells. Oxid. Med. Cell Longev. 2017, 2017, 8510805. [Google Scholar] [CrossRef]
- Borodkina, A.; Shatrova, A.; Abushik, P.; Nikolsky, N.; Burova, E. Interaction between ros dependent DNA damage, mitochondria and p38 mapk underlies senescence of human adult stem cells. Aging 2014, 6, 481–495. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yu, K.R.; Kim, H.S.; Kang, I.; Kim, J.J.; Lee, B.C.; Choi, S.W.; Shin, J.H.; Seo, Y.; Kang, K.S. Bmi1 inhibits senescence and enhances the immunomodulatory properties of human mesenchymal stem cells via the direct suppression of mkp-1/dusp1. Aging 2016, 8, 1670–1689. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.; Yu, S.; Sun, W.; Miao, D. Bmi1 overexpression in mesenchymal stem cells exerts antiaging and antiosteoporosis effects by inactivating p16/p19 signaling and inhibiting oxidative stress. Stem Cells 2019, 37, 1200–1211. [Google Scholar] [CrossRef]
- Bertolo, A.; Capossela, S.; Frankl, G.; Baur, M.; Potzel, T.; Stoyanov, J. Oxidative status predicts quality in human mesenchymal stem cells. Stem Cell Res. Ther. 2017, 8, 3. [Google Scholar] [CrossRef]
- Fernandez-Rebollo, E.; Franzen, J.; Hollmann, J.; Ostrowska, A.; Oliverio, M.; Sieben, T.; Rath, B.; Kornfeld, J.-W.; Wagner, W. Senescence-associated metabolomic phenotype in primary and ipsc-derived mesenchymal stromal cells. BioRxiv 2019, 14, 201–209. [Google Scholar] [CrossRef]
- Denu, R.A.; Hematti, P. Effects of oxidative stress on mesenchymal stem cell biology. Oxid. Med. Cell Longev. 2016, 2016, 2989076. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Lee, H.J.; Kim, J.S.; Lee, S.J.; Han, H.J. Ephb2 signaling-mediated sirt3 expression reduces msc senescence by maintaining mitochondrial ros homeostasis. Free Radic. Biol. Med. 2017, 110, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Shao, Y.; Ma, C.Y.; Chen, W.; Sun, L.; Liu, W.; Zhang, D.Y.; Fu, B.C.; Liu, K.Y.; Jia, Z.B.; et al. Decreased sirt3 in aged human mesenchymal stromal/stem cells increases cellular susceptibility to oxidative stress. J. Cell Mol. Med. 2014, 18, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Denu, R.A. Sirt3 enhances mesenchymal stem cell longevity and differentiation. Oxid. Med. Cell Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Yuan, H.F.; Zhai, C.; Yan, X.L.; Zhao, D.D.; Wang, J.X.; Zeng, Q.; Chen, L.; Nan, X.; He, L.J.; Li, S.T.; et al. Sirt1 is required for long-term growth of human mesenchymal stem cells. J. Mol. Med. 2012, 90, 389–400. [Google Scholar] [CrossRef]
- Khanh, V.C.; Zulkifli, A.F.; Tokunaga, C.; Yamashita, T.; Hiramatsu, Y.; Ohneda, O. Aging impairs beige adipocyte differentiation of mesenchymal stem cells via the reduced expression of sirtuin 1. Biochem. Biophys. Res. Commun. 2018, 500, 682–690. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, P.; Liu, Y.; Zhou, J.; Shi, Z.; Cheng, K.; Huang, T.; Wang, X.; Yang, G.L.; Yang, B.; et al. Overexpression of foxq1 enhances anti-senescence and migration effects of human umbilical cord mesenchymal stem cells in vitro and in vivo. Cell Tissue Res. 2018, 373, 379–393. [Google Scholar] [CrossRef]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The longest telomeres: A general signature of adult stem cell compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef]
- Kim, J.A.; Im, K.O.; Park, S.N.; Kwon, J.S.; Kim, S.Y.; Oh, K.; Lee, D.S.; Kim, M.K.; Kim, S.W.; Jang, M.; et al. Cytogenetic heterogeneity and their serial dynamic changes during acquisition of cytogenetic aberrations in cultured mesenchymal stem cells. Mutat. Res. 2015, 777, 60–68. [Google Scholar] [CrossRef]
- Baxter, M.A.; Wynn, R.F.; Jowitt, S.N.; Wraith, J.E.; Fairbairn, L.J.; Bellantuono, I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004, 22, 675–682. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, T.; Yan, H.; Qi, H.; Deng, C.; Ye, T.; Zhou, S.; Li, F.R. Role of histone deacetylase inhibitors in the aging of human umbilical cord mesenchymal stem cells. J. Cell Biochem. 2013, 114, 2231–2239. [Google Scholar] [CrossRef]
- Twine, N.A.; Harkness, L.; Adjaye, J.; Aldahmash, A.; Wilkins, M.R.; Kassem, M. Molecular phenotyping of telomerized human bone marrow skeletal stem cells reveals a genetic program of enhanced proliferation and maintenance of differentiation responses. JBMR Plus 2018, 2, 257–267. [Google Scholar] [CrossRef]
- Trachana, V.; Petrakis, S.; Fotiadis, Z.; Siska, E.K.; Balis, V.; Gonos, E.S.; Kaloyianni, M.; Koliakos, G. Human mesenchymal stem cells with enhanced telomerase activity acquire resistance against oxidative stress-induced genomic damage. Cytotherapy 2017, 19, 808–820. [Google Scholar] [CrossRef]
- Lei, Q.; Liu, T.; Gao, F.; Xie, H.; Sun, L.; Zhao, A.; Ren, W.; Guo, H.; Zhang, L.; Wang, H.; et al. Microvesicles as potential biomarkers for the identification of senescence in human mesenchymal stem cells. Theranostics 2017, 7, 2673–2689. [Google Scholar] [CrossRef]
- Hisamatsu, D.; Ohno-Oishi, M.; Nakamura, S.; Mabuchi, Y.; Naka-Kaneda, H. Growth differentiation factor 6 derived from mesenchymal stem/stromal cells reduces age-related functional deterioration in multiple tissues. Aging 2016, 8, 1259–1275. [Google Scholar] [CrossRef]
- Vassilieva, I.O.; Reshetnikova, G.F.; Shatrova, A.N.; Tsupkina, N.V.; Kharchenko, M.V.; Alekseenko, L.L.; Nikolsky, N.N.; Burova, E.B. Senescence-messaging secretome factors trigger premature senescence in human endometrium-derived stem cells. Biochem. Biophys. Res. Commun. 2018, 496, 1162–1168. [Google Scholar] [CrossRef]
- Fulzele, S.; Mendhe, B.; Khayrullin, A.; Johnson, M.; Kaiser, H.; Liu, Y.; Isales, C.M.; Hamrick, M.W. Muscle-derived mir-34a increases with age in circulating extracellular vesicles and induces senescence of bone marrow stem cells. Aging 2019, 11, 1791–1803. [Google Scholar] [CrossRef]
- Whitehead, J.; Zhang, J.; Harvestine, J.N.; Kothambawala, A.; Liu, G.Y.; Leach, J.K. Tunneling nanotubes mediate the expression of senescence markers in mesenchymal stem/stromal cell spheroids. Stem Cells 2019. [Google Scholar] [CrossRef]
- Ozcan, S.; Alessio, N.; Acar, M.B.; Toprak, G.; Gonen, Z.B.; Peluso, G.; Galderisi, U. Myeloma cells can corrupt senescent mesenchymal stromal cells and impair their anti-tumor activity. Oncotarget. 2015, 6, 39482–39492. [Google Scholar]
- Alessio, N.; Aprile, D.; Squillaro, T.; Di Bernardo, G.; Finicelli, M.; Melone, M.A.; Peluso, G.; Galderisi, U. The senescence-associated secretory phenotype (sasp) from mesenchymal stromal cells impairs growth of immortalized prostate cells but has no effect on metastatic prostatic cancer cells. Aging 2019, 11, 5817–5828. [Google Scholar] [CrossRef]
- Rhee, K.J.; Lee, J.I.; Eom, Y.W. Mesenchymal stem cell-mediated effects of tumor support or suppression. Int. J. Mol. Sci. 2015, 16, 30015–30033. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, X.; Wang, L.; Liu, G.; Wu, X.; Jing, Y.; Li, H.; Wang, G. Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. 2015, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Di, G.H.; Liu, Y.; Lu, Y.; Liu, J.; Wu, C.; Duan, H.F. Il-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS ONE 2014, 9, e113572. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Yong, D.; El-Jawhari, J.J.; Cuthbert, R.; McGonagle, D.; Win Naing, M.; Jones, E. Identification of senescent cells in multipotent mesenchymal stromal cell cultures: Current methods and future directions. Cytotherapy 2019, 21, 803–819. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Guidotti, S.; Minguzzi, M.; Platano, D.; Cattini, L.; Trisolino, G.; Mariani, E.; Borzi, R.M. Lithium chloride dependent glycogen synthase kinase 3 inactivation links oxidative DNA damage, hypertrophy and senescence in human articular chondrocytes and reproduces chondrocyte phenotype of obese osteoarthritis patients. PLoS ONE 2015, 10, e0143865. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970, 3, 393–403. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Noppe, G.; Dekker, P.; de Koning-Treurniet, C.; Blom, J.; van Heemst, D.; Dirks, R.W.; Tanke, H.J.; Westendorp, R.G.; Maier, A.B. Rapid flow cytometric method for measuring senescence associated beta-galactosidase activity in human fibroblasts. Cytometry A. 2009, 75, 910–916. [Google Scholar] [CrossRef]
- Shimada, H.; Sakakima, H.; Tsuchimochi, K.; Matsuda, F.; Komiya, S.; Goldring, M.B.; Ijiri, K. Senescence of chondrocytes in aging articular cartilage: Gadd45beta mediates p21 expression in association with c/ebpbeta in senescence-accelerated mice. Pathol. Res. Pract. 2011, 207, 225–231. [Google Scholar] [CrossRef]
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res. 2000, 257, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Chiou, C.C.; Chang, P.Y.; Wu, J.T. Urinary 8-ohdg: A marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta 2004, 339, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Fasth, A.; Ek, T.; Hammarsten, O. Validation of a flow cytometry-based detection of gamma-h2ax, to measure DNA damage for clinical applications. Cytometry B. Clin. Cytom. 2017, 92, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.S.V.; Kirschner, M.; Halfmeyer, I.; Estrada, N.; Xicoy, B.; Isfort, S.; Vieri, M.; Zamora, L.; Abels, A.; Bouillon, A.S.; et al. Comparison of flow-fish and mm-qpcr telomere length assessment techniques for the screening of telomeropathies. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]
- Serakinci, N.; Cagsin, H.; Mavis, M. Use of u-stela for accurate measurement of extremely short telomeres. Methods Mol. Biol. 2019, 2045, 217–224. [Google Scholar]
- Neri, S.; Mariani, E.; Cattini, L.; Facchini, A. Long-term in vitro expansion of osteoarthritic human articular chondrocytes do not alter genetic stability: A microsatellite instability analysis. J. Cell Physiol. 2011, 226, 2579–2585. [Google Scholar] [CrossRef]
- Lamichane, S.; Baek, S.H.; Kim, Y.J.; Park, J.H.; Dahal Lamichane, B.; Jang, W.B.; Ji, S.; Lee, N.K.; Dehua, L.; Kim, D.Y.; et al. Mhy2233 attenuates replicative cellular senescence in human endothelial progenitor cells via sirt1 signaling. Oxid. Med. Cell Longev. 2019, 2019, 6492029. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, Y.C.; Xiao, B.J.; Guo, X.D.; Zheng, Q.X.; Wu, B. Age-related changes in the global DNA methylation profile of oligodendrocyte progenitor cells derived from rat spinal cords. Curr. Med. Sci. 2019, 39, 67–74. [Google Scholar] [CrossRef]
- Madsen, S.D.; Russell, K.C.; Tucker, H.A.; Glowacki, J.; Bunnell, B.A.; O’Connor, K.C. Decoy trail receptor cd264: A cell surface marker of cellular aging for human bone marrow-derived mesenchymal stem cells. Stem Cell Res. Ther. 2017, 8, 201. [Google Scholar] [CrossRef]
- Amati, E.; Perbellini, O.; Rotta, G.; Bernardi, M.; Chieregato, K.; Sella, S.; Rodeghiero, F.; Ruggeri, M.; Astori, G. High-throughput immunophenotypic characterization of bone marrow- and cord blood-derived mesenchymal stromal cells reveals common and differentially expressed markers: Identification of angiotensin-converting enzyme (cd143) as a marker differentially expressed between adult and perinatal tissue sources. Stem Cell Res. Ther. 2018, 9, 10. [Google Scholar]
- Bertolo, A.; Baur, M.; Guerrero, J.; Potzel, T.; Stoyanov, J. Autofluorescence is a reliable in vitro marker of cellular senescence in human mesenchymal stromal cells. Sci. Rep. 2019, 9, 2074. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Martin, D.C.; Androulakis, I.P.; Moghe, P.V. Fluorescence imaging of actin turnover parses early stem cell lineage divergence and senescence. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ang, J.; Lee, Y.A.; Raghothaman, D.; Jayaraman, P.; Teo, K.L.; Khan, F.J.; Reuveny, S.; Chang, Y.T.; Kang, N.Y.; Oh, S. Rapid detection of senescent mesenchymal stromal cells by a fluorescent probe. Biotechnol. J. 2019, 14, e1800691. [Google Scholar] [CrossRef] [PubMed]
- Khong, D.; Li, M.; Singleton, A.; Chin, L.Y.; Mukundan, S.; Parekkadan, B. Orthogonal potency analysis of mesenchymal stromal cell function during ex vivo expansion. Exp. Cell Res. 2018, 362, 102–110. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking aging to chronic disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Scudellari, M. To stay young, kill zombie cells. Nature 2017, 550, 448–450. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147.e116. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of hsp90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D. Hsp90 inhibitors as senolytic drugs to extend healthy aging. Cell Cycle 2018, 17, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Gurau, F.; Baldoni, S.; Prattichizzo, F.; Espinosa, E.; Amenta, F.; Procopio, A.D.; Albertini, M.C.; Bonafe, M.; Olivieri, F. Anti-senescence compounds: A potential nutraceutical approach to healthy aging. Ageing Res. Rev. 2018, 46, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cui, J.; Liu, X.; Lv, B.; Xie, Z.; Yu, B. Roles of microrna-34a targeting sirt1 in mesenchymal stem cells. Stem Cell Res. Ther. 2015, 6, 195. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.J.; Yang, Y.; Sui, B.D.; Hu, C.H.; Zhao, P.; Liao, L.; Chen, J.; Zhang, L.Q.; Yang, T.T.; Zhang, S.F.; et al. Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics 2018, 8, 2387–2406. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hong, Y.; He, H.; Jiang, G.; You, W.; Liang, X.; Fu, Q.; Han, S.; Lian, Q.; Zhang, Y. Fgf21 mediates mesenchymal stem cell senescence via regulation of mitochondrial dynamics. Oxid. Med. Cell Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Antonioli, E.; Torres, N.; Ferretti, M.; Piccinato, C.A.; Sertie, A.L. Individual response to mtor inhibition in delaying replicative senescence of mesenchymal stromal cells. PLoS ONE 2019, 14, e0204784. [Google Scholar] [CrossRef]
- He, Y.; Li, W.; Hu, G.; Sun, H.; Kong, Q. Bioactivities of ef24, a novel curcumin analog: A review. Front. Oncol. 2018, 8, 614. [Google Scholar] [CrossRef]
- Oh, Y.S.; Jeong, S.G.; Cho, G.W. Anti-senescence effects of DNA methyltransferase inhibitor rg108 in human bone marrow mesenchymal stromal cells. Biotechnol. Appl. Biochem. 2015, 62, 583–590. [Google Scholar] [CrossRef]
- Assis, R.I.F.; Wiench, M.; Silverio, K.G.; da Silva, R.A.; Feltran, G.D.S.; Sallum, E.A.; Casati, M.Z.; Nociti, F.H., Jr.; Andia, D.C. Rg108 increases nanog and oct4 in bone marrow-derived mesenchymal cells through global changes in DNA modifications and epigenetic activation. PLoS ONE 2018, 13, e0207873. [Google Scholar] [CrossRef] [PubMed]
- Grezella, C.; Fernandez-Rebollo, E.; Franzen, J.; Ventura Ferreira, M.S.; Beier, F.; Wagner, W. Effects of senolytic drugs on human mesenchymal stromal cells. Stem Cell Res. Ther. 2018, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by abt263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Kruger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 2019, 29, 1233. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. eBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Frobel, J.; Hemeda, H.; Lenz, M.; Abagnale, G.; Joussen, S.; Denecke, B.; Saric, T.; Zenke, M.; Wagner, W. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Reports 2014, 3, 414–422. [Google Scholar] [CrossRef]
- Spitzhorn, L.S.; Megges, M.; Wruck, W.; Rahman, M.S.; Otte, J.; Degistirici, O.; Meisel, R.; Sorg, R.V.; Oreffo, R.O.C.; Adjaye, J. Human ipsc-derived mscs (imscs) from aged individuals acquire a rejuvenation signature. Stem Cell Res. Ther. 2019, 10, 100. [Google Scholar] [CrossRef]
- Kimbrel, E.A.; Kouris, N.A.; Yavanian, G.J.; Chu, J.; Qin, Y.; Chan, A.; Singh, R.P.; McCurdy, D.; Gordon, L.; Levinson, R.D.; et al. Mesenchymal stem cell population derived from human pluripotent stem cells displays potent immunomodulatory and therapeutic properties. Stem Cells Dev. 2014, 23, 1611–1624. [Google Scholar] [CrossRef]
- Gobel, C.; Goetzke, R.; Eggermann, T.; Wagner, W. Interrupted reprogramming into induced pluripotent stem cells does not rejuvenate human mesenchymal stromal cells. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bigot, N.; Mouche, A.; Preti, M.; Loisel, S.; Renoud, M.L.; Le Guevel, R.; Sensebe, L.; Tarte, K.; Pedeux, R. Hypoxia differentially modulates the genomic stability of clinical-grade adscs and bm-mscs in long-term culture. Stem Cells 2015, 33, 3608–3620. [Google Scholar] [CrossRef]
- Jin, Y.; Kato, T.; Furu, M.; Nasu, A.; Kajita, Y.; Mitsui, H.; Ueda, M.; Aoyama, T.; Nakayama, T.; Nakamura, T.; et al. Mesenchymal stem cells cultured under hypoxia escape from senescence via down-regulation of p16 and extracellular signal regulated kinase. Biochem. Biophys. Res. Commun. 2010, 391, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Chen, Y.J.; Yew, T.L.; Chen, L.L.; Wang, J.Y.; Chiu, C.H.; Hung, S.C. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of e2a-p21 by hif-twist. Blood 2011, 117, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Park, J.M.; Song, Y.; Kim, S.; Moon, J. Hif1alpha-mediated aimp3 suppression delays stem cell aging via the induction of autophagy. Aging Cell 2019, 18, e12909. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.Y.; Chun, S.Y.; Ha, Y.S.; Kim, D.H.; Kim, J.; Song, P.H.; Kim, H.T.; Yoo, E.S.; Kim, B.S.; Kwon, T.G. Hypoxia enhances cell properties of human mesenchymal stem cells. Tissue Eng. Regen. Med. 2017, 14, 595–604. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, A.J.; Kim, G.Y.; Jo, A.; Lee, J.E.; Leem, S.H.; Yoon, J.H.; Ye, S.K.; Chung, J.W. Lactoferrin protects human mesenchymal stem cells from oxidative stress-induced senescence and apoptosis. J. Microbiol. Biotechnol. 2017, 27, 1877–1884. [Google Scholar] [CrossRef]
- Chang, T.C.; Hsu, M.F.; Shih, C.Y.; Wu, K.K. 5-methoxytryptophan protects mscs from stress induced premature senescence by upregulating foxo3a and mtor. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Choi, Y.; Yoon, D.S.; Lee, K.M.; Choi, S.M.; Lee, M.H.; Park, K.H.; Han, S.H.; Lee, J.W. Enhancement of mesenchymal stem cell-driven bone regeneration by resveratrol-mediated sox2 regulation. Aging Dis. 2019, 10, 818–833. [Google Scholar] [CrossRef]
- Wong, T.Y.; Chang, C.H.; Yu, C.H.; Huang, L.L.H. Hyaluronan keeps mesenchymal stem cells quiescent and maintains the differentiation potential over time. Aging Cell 2017, 16, 451–460. [Google Scholar] [CrossRef]
- Block, T.J.; Marinkovic, M.; Tran, O.N.; Gonzalez, A.O.; Marshall, A.; Dean, D.D.; Chen, X.D. Restoring the quantity and quality of elderly human mesenchymal stem cells for autologous cell-based therapies. Stem Cell Res. Ther. 2017, 8, 239. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Senescence Marker | Techniques for Detection | Senescent Features, Pros and Cons | ||
|---|---|---|---|---|
| Cell morphology |  |  | Microscopy [185] Flow cytometry [186] (FSC for size, SSC for granularity) | Senescent MSCs show enlarged and granular cell morphology. Microscopic assessment is easy but only qualitative. Flow cytometric assessment is quantitative |
| CFU |  | Colony formation in agar culture [187] | The CFU number is a measure of cell clonogeneity and decreases with MSC age. CFU assessment requires careful plating at low density. | |
| Sa-β-gal |  | Microscopy (colorimetric activity assay with X-gal) [188] Flow cytometry (fluorimetric activity assay with C12FDG) [189] IHC, IF or WB with specific Abs (protein expression) [190] | Senescent cells at low density express a lysosomal β-galactosidase active at pH 6.0, detectable either with activity assays or with a specific antibody. The activity assays can give altered results on high density cultures [191] | |
| 8-oxo-dG |  | IHC, IF, ELISA [192], HPLC [192]-MS/MS | 8-oxodG is a DNA base derivative, robust marker of oxidative DNA and RNA damage | |
| γH2AX |    | IF Flow cytometry [193] WB | Histone H2AX phosphorylation is an indirect measure of DNA double strand breaks due to physical, chemical, oxidative stress. It indicates that cells organize a DNA damage response, but its persistence sustains senescence of the cells. | |
| Telomeres |   | Southern blotting [170] Flow FISH [194] Real-time PCR [194] STELA [195] | Telomere attrition is directly correlated to replicative senescence, but it also occurs after exposure to oxidative damage. The subpopulation heterogeneity must be taken into account and may be addressed with emerging techniques such as STELA, detecting individual telomere length. | |
| MSI |  | PCR followed by gel or capillary electrophoresis [196] | Repeated sequences variations are an indirect indication of genomic instability and deficient DNA repair due to replicative or oxidative stress. They increase with cell aging. | |
| Gene expression of senescent markers at mRNA level |  | Real-time RT-PCR [33] Microarray RNAseq | Expression of genes related to senescence. Several pathways can be analyzed, but gene expression analysis prevalently focuses on p53 and cyclin dependent kinase inhibitors (p16 and p21) | |
| Expression of senescent markers at protein level |   | WB [197] IHC IF Flow cytometry | Evaluation of the expression levels of proteins related to senescence (p16, p21, p53, etc.) | |
| Global methylation |  | NGS after bisulfite treatment [198] | Genome wide analysis of methylated cytosines. | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neri, S.; Borzì, R.M. Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging. Biomolecules 2020, 10, 340. https://doi.org/10.3390/biom10020340
Neri S, Borzì RM. Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging. Biomolecules. 2020; 10(2):340. https://doi.org/10.3390/biom10020340
Chicago/Turabian StyleNeri, Simona, and Rosa Maria Borzì. 2020. "Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging" Biomolecules 10, no. 2: 340. https://doi.org/10.3390/biom10020340
APA StyleNeri, S., & Borzì, R. M. (2020). Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging. Biomolecules, 10(2), 340. https://doi.org/10.3390/biom10020340