Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy


 ,
,  and
and
Abstract
1. Introduction
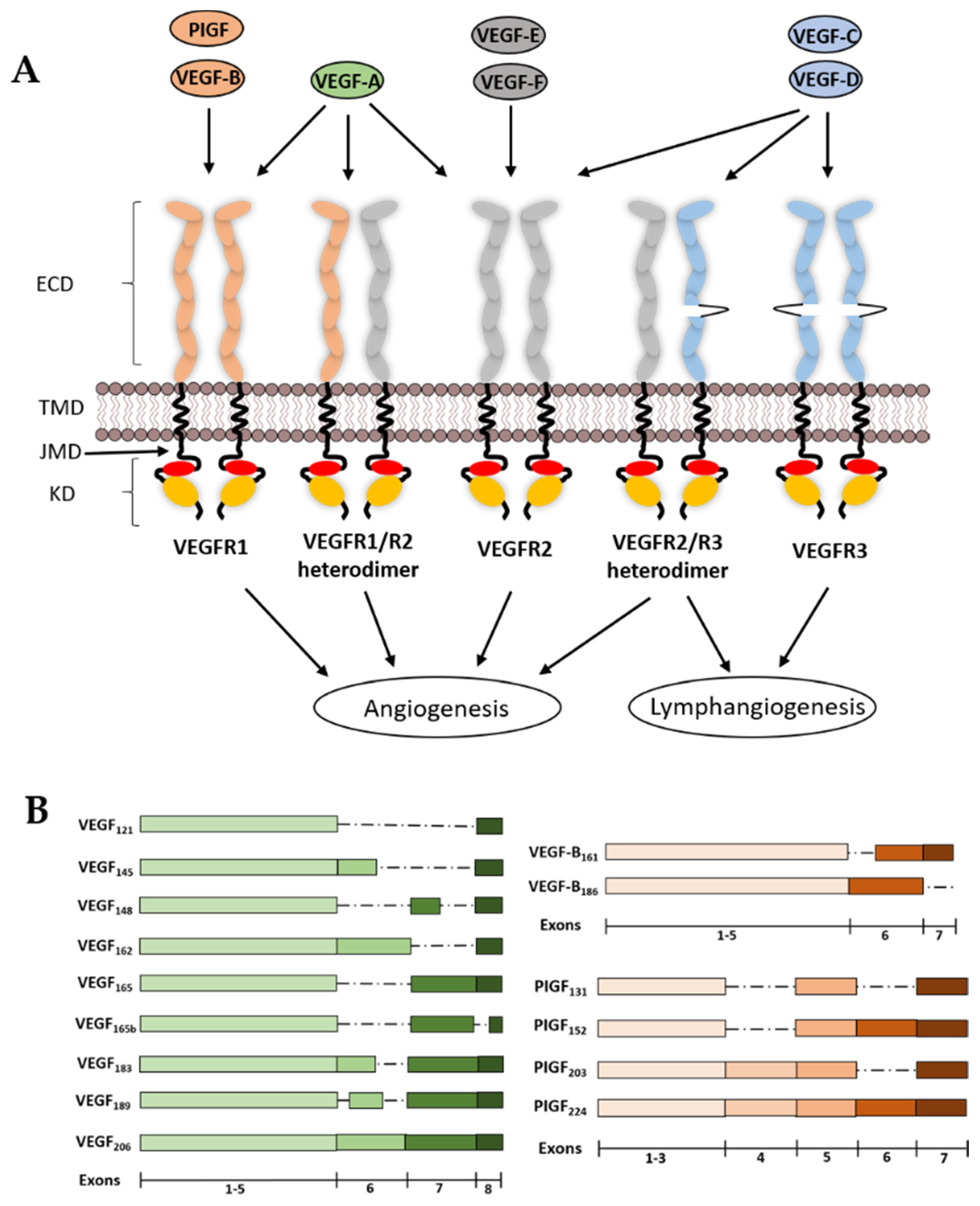
2. VEGF Ligands and Receptor Diversity
2.1. VEGF-A
2.2. VEGF-B
2.3. VEGF-C and VEGF-D
2.4. PIGF
2.5. VEGF-E
2.6. VEGF-F
2.7. VEGFR1
2.8. VEGFR2
2.9. VEGFR3
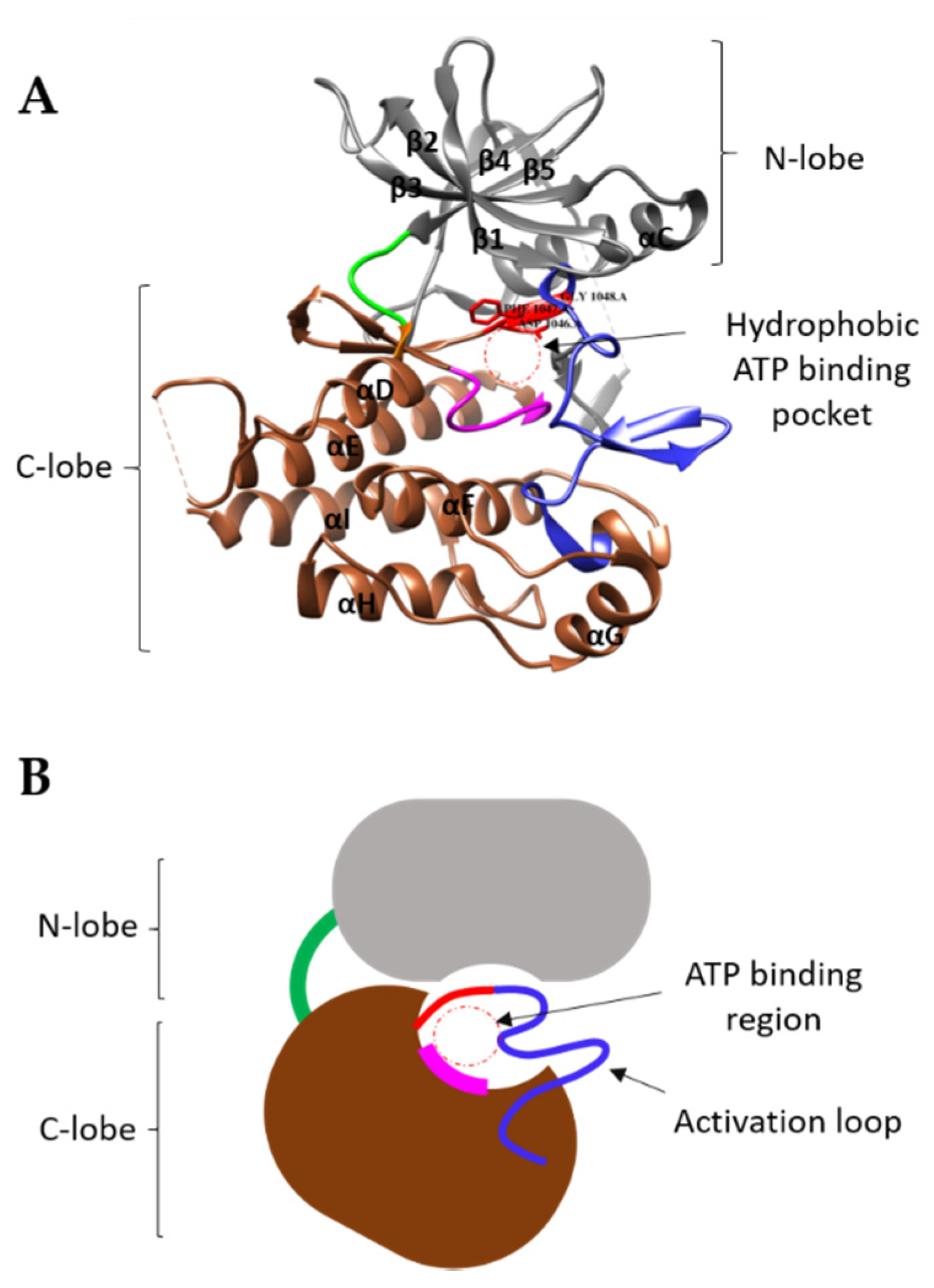
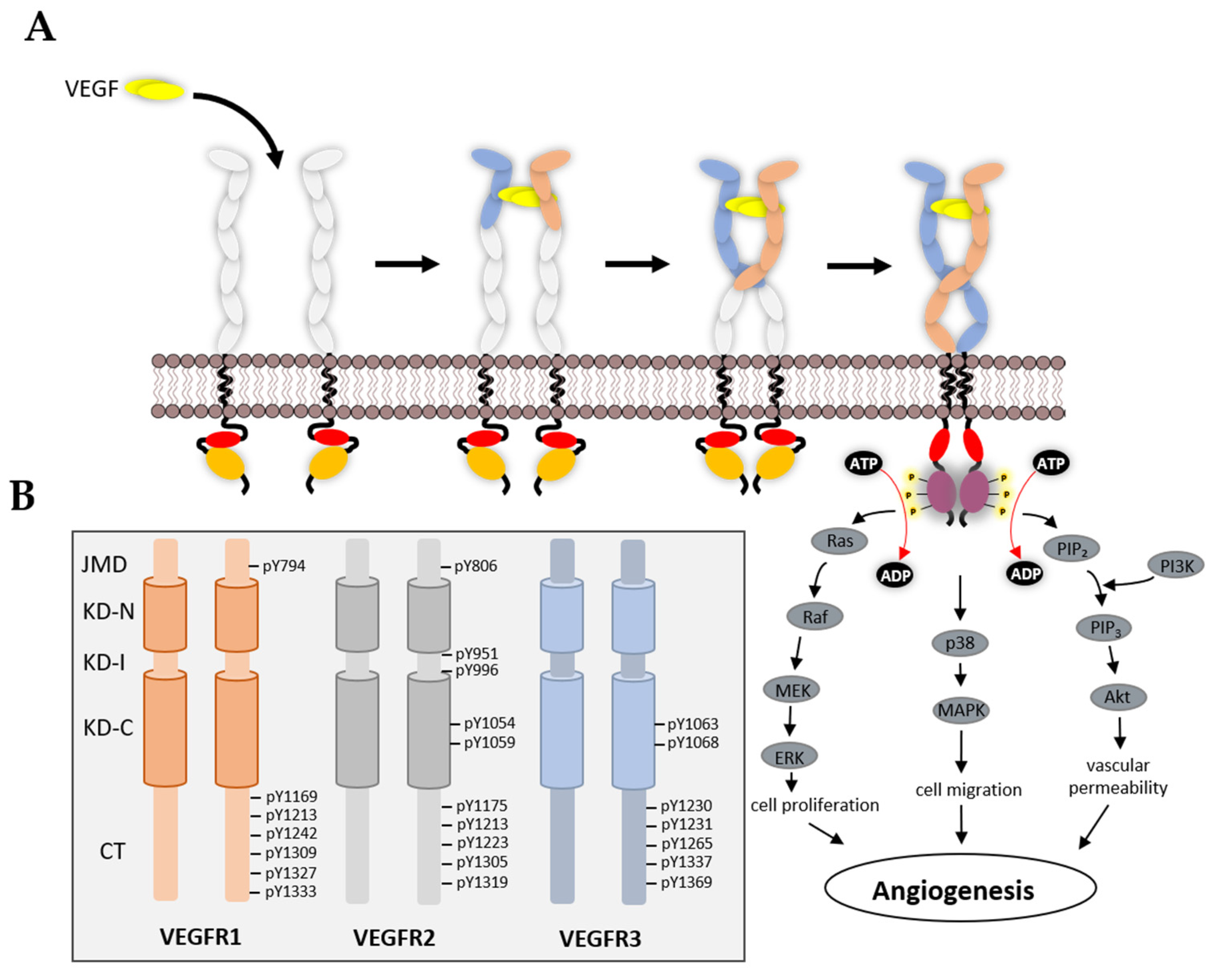
3. Structural Features of VEGFRs and Their Functions
3.1. VEGFR Extracellular Domain
3.2. VEGFR Transmembrane Domain
3.3. VEGFR Cytoplasmic Domain
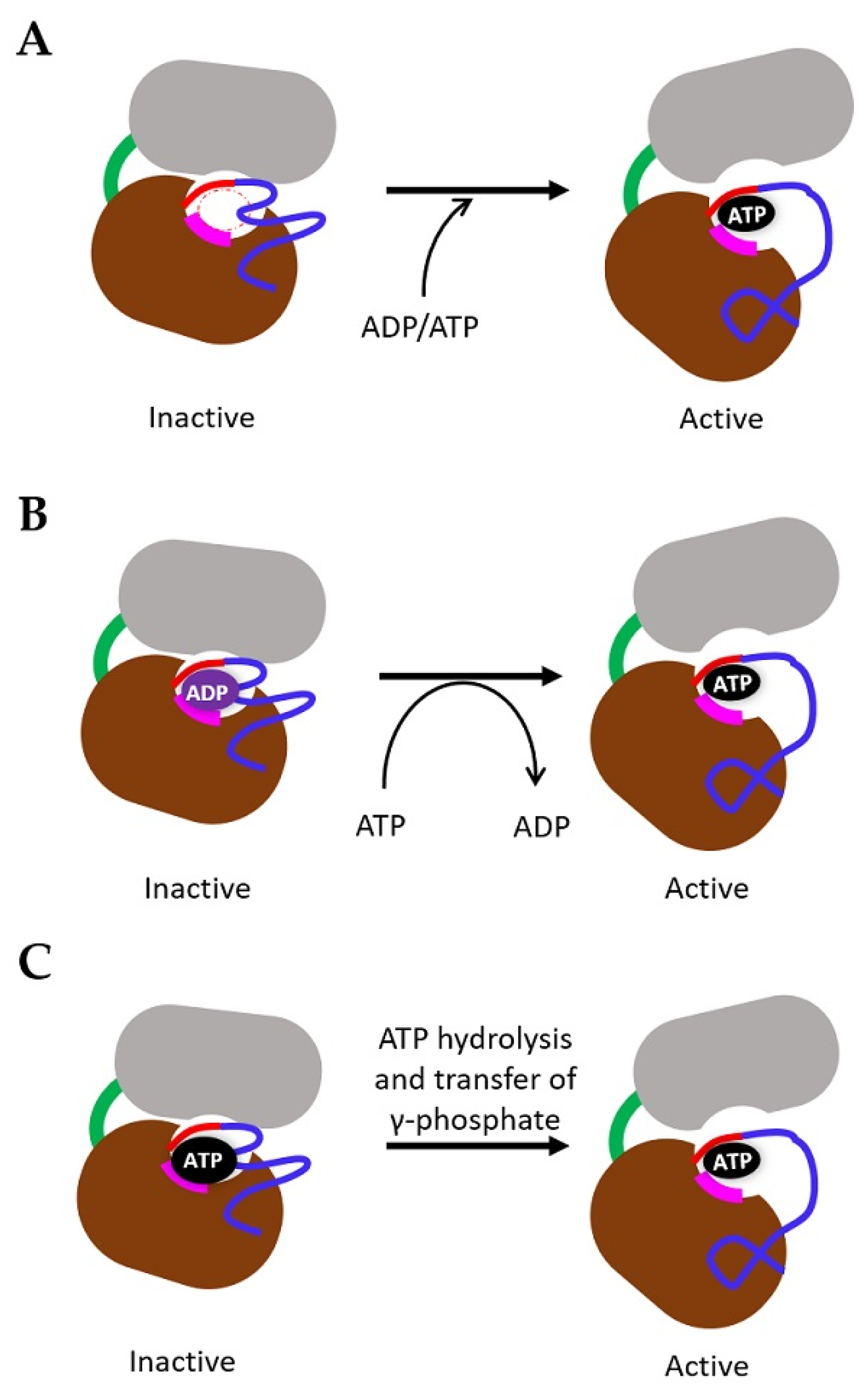
4. Mechanism of TK Activation
5. VEGFR Signal Transduction
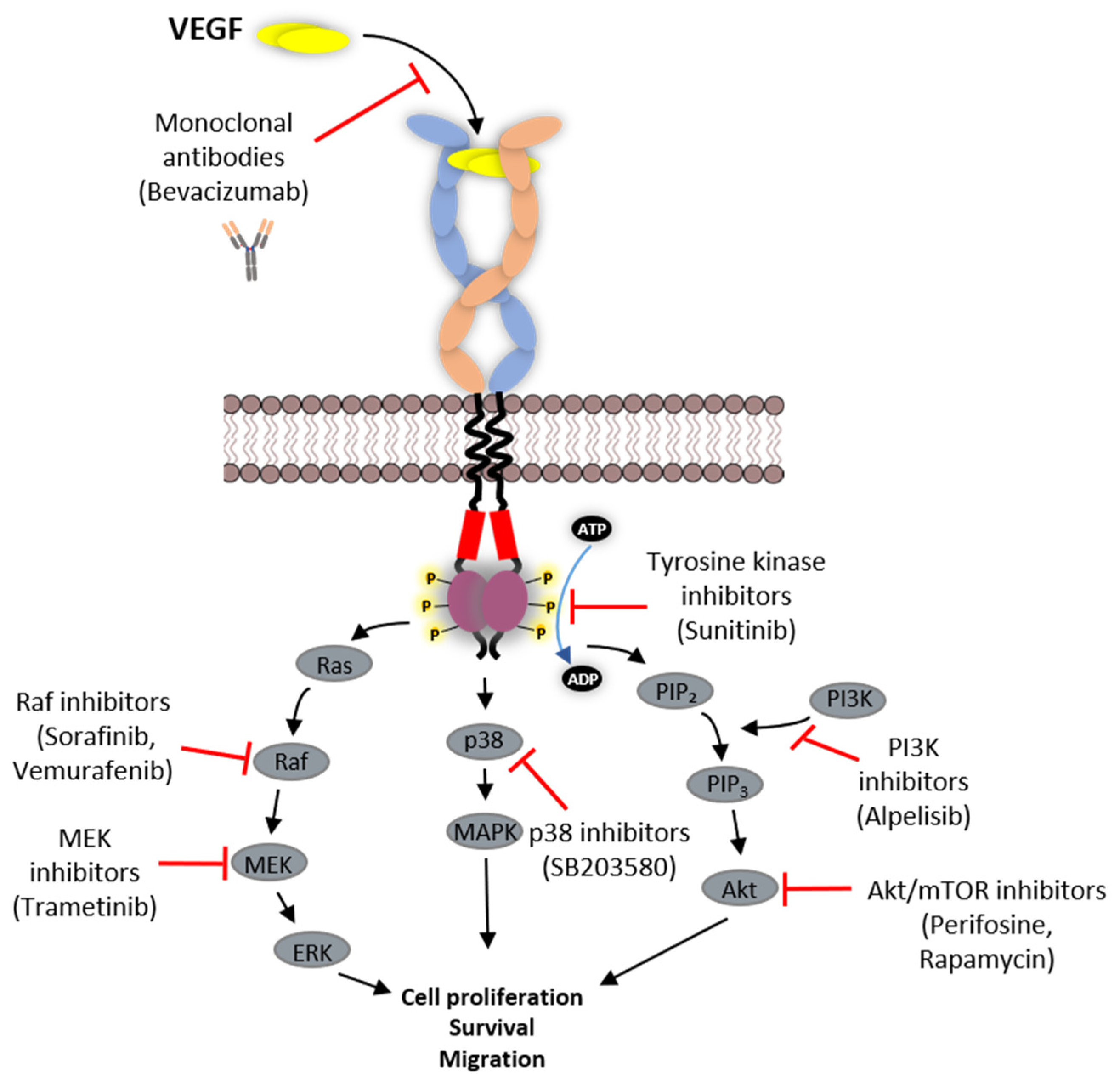
6. Strategies Employed in Inhibition of VEGFR Function
6.1. Protein-Based Therapies
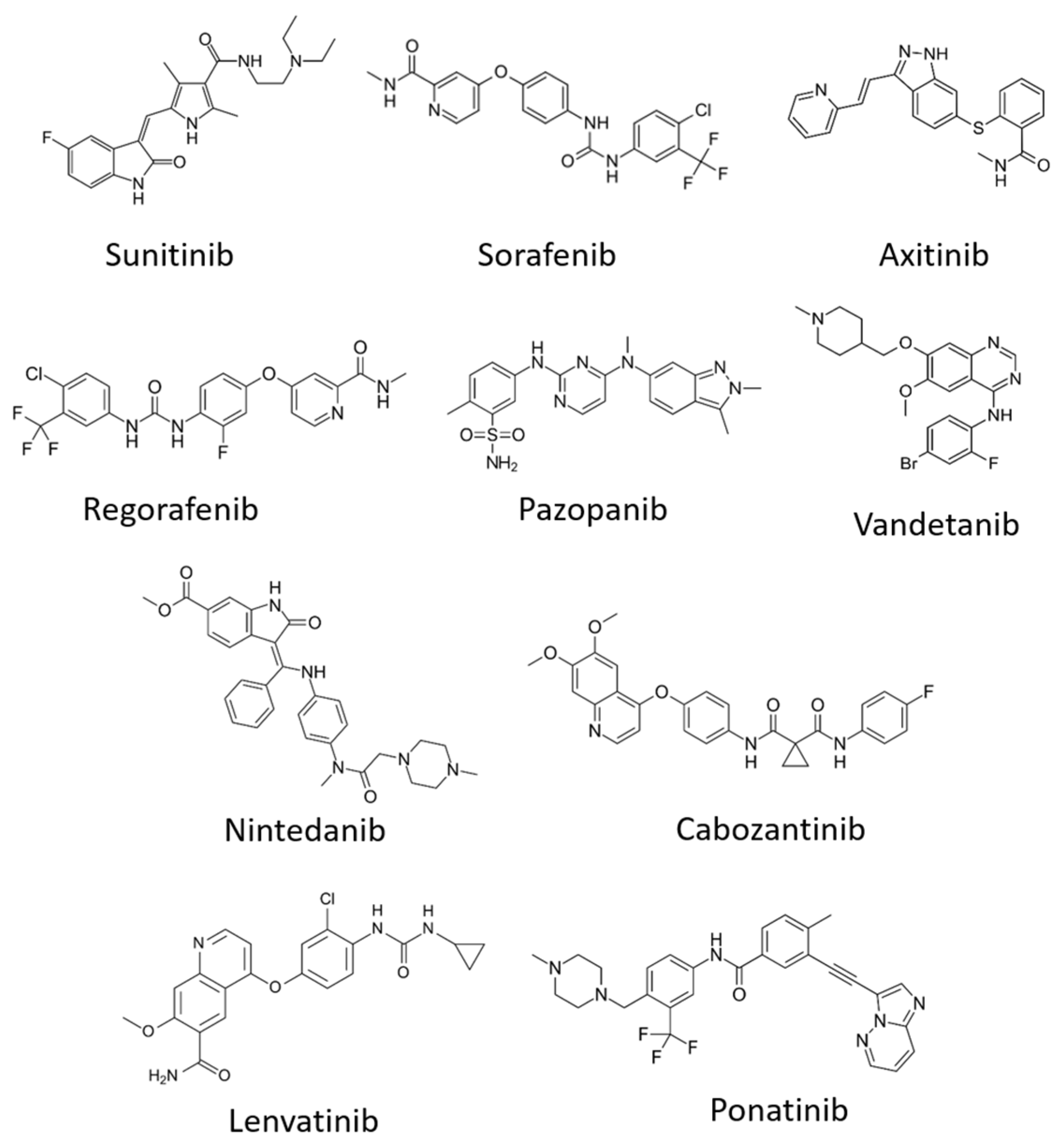
6.2. Tyrosine Kinase Inhibitors (TKIs)
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Holmes, D.I.R.; Zachary, I. The vascular endothelial growth factor (VEGF) family: Angiogenic factors in health and disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kipryushina, Y.O.; Yakovlev, K.V.; Odintsova, N.A. Vascular endothelial growth factors: A comparison between invertebrates and vexrtebrates. Cytokine Growth Factor Rev. 2015, 26, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; de la Cruz, O.N.H.; Lopez-Gonzalez, J.S. Contribution of angiogenesis to inflammation and cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Beazley-Long, N.; Benest, A.V.; Ye, X.; Ved, N.; Hulse, R.P.; Barratt, S.; Machado, M.J.; Donaldson, L.F.; Harper, S.J.; et al. Physiological role of vascular endothelial growth factors as homeostatic regulators. Compr. Physiol. 2018, 8, 955–979. [Google Scholar] [CrossRef]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Hsu, M.-C.; Pan, M.-R.; Hung, W.-C. Two birds, one stone: Double hits on tumor growth and lymphangiogenesis by targeting vascular endothelial growth factor receptor 3. Cells 2019, 8, 270. [Google Scholar] [CrossRef]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and its Receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Guyot, M.; Pagès, G. VEGF splicing and the role of VEGF splice variants: From physiological-pathological conditions to specific pre-mrna splicing. Methods Mol. Biol. 2015, 1332, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Makarem, J.A.; Shamseddine, A.I. Vascular endothelial growth factor family of ligands and receptors: Review. Blood Cells Mol. Dis. 2007, 38, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, Y.; Yamazaki, Y.; Morita, T. Specific distribution of VEGF-F in Viperinae snake venoms: Isolation and characterization of a VGEF-F from the venom of Daboia russelli siamensis. Arch Biochem. Biophys. 2005, 439, 241–247. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor receptor-2: Its unique signaling and specific ligand, VEGF-E. Cancer Sci. 2003, 94, 751–756. [Google Scholar] [CrossRef]
- Beckouche, N.; Bignon, M.; Lelarge, V.; Mathivet, T.; Pichol-Thievend, C.; Berndt, S.; Hardouin, J.; Garand, M.; Ardidie-Robouant, C.; Barret, A.; et al. The interaction of heparan sulfate proteoglycans with endothelial transglutaminase-2 limits VEGF165-induced angiogenesis. Sci. Signal 2015, 8, ra70. [Google Scholar] [CrossRef]
- Stringer, S.E. The role of heparan sulphate proteoglycans in angiogenesis. Biochem. Soc. Trans. 2006, 34, 451–453. [Google Scholar] [CrossRef]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J. Biol. Chem. 2012, 287, 11082–11089. [Google Scholar] [CrossRef]
- Karpanen, T.; Heckman, C.A.; Keskitalo, S.; Jeltsch, M.; Ollila, H.; Neufeld, G.; Tamagnone, L.; Alitalo, K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006, 20, 1462–1472. [Google Scholar] [CrossRef]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling–In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; van Oosterom, A.T.; de Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Staels, W.; Heremans, Y.; Heimberg, H.; de Leu, N. VEGF-A and blood vessels: A beta cell perspective. Diabetologia 2019, 62, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. (Lond.) 2005, 109, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef]
- Grunewald, F.S.; Prota, A.E.; Giese, A.; Ballmer-Hofer, K. Structure-function analysis of VEGF receptor activation and the role of coreceptors in angiogenic signaling. Biochim. Biophys. Acta 2010, 1804, 567–580. [Google Scholar] [CrossRef]
- Abraham, D.; Hofbauer, R.; Schafer, R.; Blumer, R.; Paulus, P.; Miksovsky, A.; Traxler, H.; Kocher, A.; Aharinejad, S. Selective downregulation of VEGF-A(165), VEGF-R(1), and decreased capillary density in patients with dilative but not ischemic cardiomyopathy. Circ. Res. 2000, 87, 644–647. [Google Scholar] [CrossRef]
- Li, X.; Lee, C.; Tang, Z.; Zhang, F.; Arjunan, P.; Li, Y.; Hou, X.; Kumar, A.; Dong, L. VEGF-B: A survival, or an angiogenic factor? Cell Adh. Migr. 2009, 3, 322–327. [Google Scholar] [CrossRef]
- Stacker, S.A.; Stenvers, K.; Caesar, C.; Vitali, A.; Domagala, T.; Nice, E.; Roufail, S.; Simpson, R.J.; Moritz, R.; Karpanen, T.; et al. Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J. Biol. Chem. 1999, 274, 32127–32136. [Google Scholar] [CrossRef]
- Bui, H.M.; Enis, D.; Robciuc, M.R.; Nurmi, H.J.; Cohen, J.; Chen, M.; Yang, Y.; Dhillon, V.; Johnson, K.; Zhang, H.; et al. Proteolytic activation defines distinct lymphangiogenic mechanisms for VEGFC and VEGFD. J. Clin. Investig. 2016, 126, 2167–2180. [Google Scholar] [CrossRef]
- Stuttfeld, E.; Ballmer-Hofer, K. Structure and function of VEGF receptors. IUBMB Life 2009, 61, 915–922. [Google Scholar] [CrossRef]
- Ikeda, K.; Oki, E.; Saeki, H.; Ando, K.; Morita, M.; Oda, Y.; Imamura, M.; Kakeji, Y.; Maehara, Y. Intratumoral lymphangiogenesis and prognostic significance of VEGFC expression in gastric cancer. Anticancer Res. 2014, 34, 3911–3915. [Google Scholar] [PubMed]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Caesar, C.; Baldwin, M.E.; Thornton, G.E.; Williams, R.A.; Prevo, R.; Jackson, D.G.; Nishikawa, S.; Kubo, H.; Achen, M.G. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 2001, 7, 186–191. [Google Scholar] [CrossRef]
- Baldwin, M.E.; Halford, M.M.; Roufail, S.; Williams, R.A.; Hibbs, M.L.; Grail, D.; Kubo, H.; Stacker, S.A.; Achen, M.G. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol. Cell. Biol. 2005, 25, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Autiero, M.; Luttun, A.; Tjwa, M.; Carmeliet, P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: Novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J. Thromb. Haemost. 2003, 1, 1356–1370. [Google Scholar] [CrossRef]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; de Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef]
- Odorisio, T.; Schietroma, C.; Zaccaria, M.L.; Cianfarani, F.; Tiveron, C.; Tatangelo, L.; Failla, C.M.; Zambruno, G. Mice overexpressing placenta growth factor exhibit increased vascularization and vessel permeability. J. Cell Sci. 2002, 115, 2559–2567. [Google Scholar]
- Kiba, A.; Yabana, N.; Shibuya, M. A set of loop-1 and -3 structures in the novel vascular endothelial growth factor (VEGF) family member, VEGF-ENZ-7, is essential for the activation of VEGFR-2 signaling. J. Biol. Chem. 2003, 278, 13453–13461. [Google Scholar] [CrossRef]
- Cebe-Suarez, S.; Grunewald, F.S.; Jaussi, R.; Li, X.; Claesson-Welsh, L.; Spillmann, D.; Mercer, A.A.; Prota, A.E.; Ballmer-Hofer, K. Orf virus VEGF-E NZ2 promotes paracellular NRP-1/VEGFR-2 coreceptor assembly via the peptide RPPR. FASEB J. 2008, 22, 3078–3086. [Google Scholar] [CrossRef]
- Takahashi, H.; Hattori, S.; Iwamatsu, A.; Takizawa, H.; Shibuya, M. A novel snake venom vascular endothelial growth factor (VEGF) predominantly induces vascular permeability through preferential signaling via VEGF receptor-1. J. Biol. Chem. 2004, 279, 46304–46314. [Google Scholar] [CrossRef]
- Sawano, A.; Takahashi, T.; Yamaguchi, S.; Aonuma, M.; Shibuya, M. Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related to vascular endothelial growth factor. Cell Growth Differ 1996, 7, 213–221. [Google Scholar] [PubMed]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharm. 2014, 9, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Olenyuk, B.Z.; Zhang, G.-J.; Klco, J.M.; Nickols, N.G.; Kaelin, W.G., Jr.; Dervan, P.B. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc. Natl. Acad. Sci. USA 2004, 101, 16768–16773. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.E.; Liang, X.H.; Ruchatz, A.; Busch, J.; Gu, Z.; Roose-Girma, M.; Olsson, C.; Erickson, S.; Ferrara, N.; Gerber, H.-P. Generation of mice carrying floxed VEGFR-1 and VEGFR-2 alleles to study the effects of postnatal gene ablation on angiogenesis and hematopoiesis. Cancer Res. 2004, 64, 596. [Google Scholar]
- Hiratsuka, S.; Minowa, O.; Kuno, J.; Noda, T.; Shibuya, M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Neagoe, P.E.; Lemieux, C.; Sirois, M.G. Vascular endothelial growth factor (VEGF)-A165-induced prostacyclin synthesis requires the activation of VEGF receptor-1 and -2 heterodimer. J. Biol. Chem. 2005, 280, 9904–9912. [Google Scholar] [CrossRef]
- Cudmore, M.J.; Hewett, P.W.; Ahmad, S.; Wang, K.Q.; Cai, M.; Al-Ani, B.; Fujisawa, T.; Ma, B.; Sissaoui, S.; Ramma, W.; et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat. Commun. 2012, 3, 972. [Google Scholar] [CrossRef]
- Mac Gabhann, F.; Popel, A.S. Dimerization of VEGF receptors and implications for signal transduction: A computational study. Biophys. Chem. 2007, 128, 125–139. [Google Scholar] [CrossRef]
- Lopez-Garcia, M.; Nowicka, M.; Bendtsen, C.; Lythe, G.; Ponnambalam, S.; Molina-Paris, C. Quantifying the phosphorylation timescales of receptor-ligand complexes: A Markovian matrix-analytic approach. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef]
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Ebos, J.M.; Bocci, G.; Man, S.; Thorpe, P.E.; Hicklin, D.J.; Zhou, D.; Jia, X.; Kerbel, R.S. A naturally occurring soluble form of vascular endothelial growth factor receptor 2 detected in mouse and human plasma. Mol. Cancer Res. 2004, 2, 315–326. [Google Scholar] [PubMed]
- Harris, H.; Wolk, A.; Larsson, A.; Vasson, M.-P.; Basu, S. Soluble vascular endothelial growth factor receptors 2 (sVEGFR-2) and 3 (sVEGFR-3) and breast cancer risk in the Swedish Mammography Cohort. Int. J. Mol. Epidemiol. Genet. 2016, 7, 81–86. [Google Scholar] [PubMed]
- Cross, M.J.; Dixelius, J.; Matsumoto, T.; Claesson-Welsh, L. VEGF-receptor signal transduction. Trends Biochem. Sci. 2003, 28, 488–494. [Google Scholar] [CrossRef]
- Nilsson, I.; Bahram, F.; Li, X.; Gualandi, L.; Koch, S.; Jarvius, M.; Soderberg, O.; Anisimov, A.; Kholova, I.; Pytowski, B.; et al. VEGF receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. Embo J. 2010, 29, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect Med. 2012, 2, a006502. [Google Scholar] [CrossRef]
- Dumont, D.J.; Jussila, L.; Taipale, J.; Lymboussaki, A.; Mustonen, T.; Pajusola, K.; Breitman, M.; Alitalo, K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 1998, 282, 946–949. [Google Scholar] [CrossRef]
- Singh, N.; Tiem, M.; Watkins, R.; Cho, Y.K.; Wang, Y.; Olsen, T.; Uehara, H.; Mamalis, C.; Luo, L.; Oakey, Z.; et al. Soluble vascular endothelial growth factor receptor 3 is essential for corneal alymphaticity. Blood 2013, 121, 4242–4249. [Google Scholar] [CrossRef]
- Emami-Naeini, P.; Dohlman, T.H.; Omoto, M.; Hattori, T.; Chen, Y.; Lee, H.S.; Chauhan, S.K.; Dana, R. Soluble vascular endothelial growth factor receptor-3 suppresses allosensitization and promotes corneal allograft survival. Graefes Arch Clin. Exp. Ophthalmol. 2014, 252, 1755–1762. [Google Scholar] [CrossRef]
- Park, S.A.; Jeong, M.S.; Ha, K.-T.; Jang, S.B. Structure and function of vascular endothelial growth factor and its receptor system. BMB Rep. 2018, 51, 73–78. [Google Scholar] [CrossRef]
- King, C.; Hristova, K. Direct measurements of VEGF-VEGFR2 binding affinities reveal the coupling between ligand binding and receptor dimerization. J. Biol. Chem. 2019, 294, 9064–9075. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. VEGF receptor protein-tyrosine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2008, 375, 287–291. [Google Scholar] [CrossRef]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the full-length vegfr-1 extracellular domain in complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Ruch, C.; Skiniotis, G.; Steinmetz, M.O.; Walz, T.; Ballmer-Hofer, K. Structure of a VEGF–VEGF receptor complex determined by electron microscopy. Nat. Struct. Amp Mol. Biol. 2007, 14, 249. [Google Scholar] [CrossRef] [PubMed]
- Christinger, H.W.; Fuh, G.; de Vos, A.M.; Wiesmann, C. The crystal structure of placental growth factor in complex with domain 2 of vascular endothelial growth factor receptor-1. J. Biol. Chem. 2004, 279, 10382–10388. [Google Scholar] [CrossRef]
- Leppanen, V.M.; Tvorogov, D.; Kisko, K.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Markovic-Mueller, S.; Stuttfeld, E.; Goldie, K.N.; Ballmer-Hofer, K.; et al. Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc. Natl. Acad. Sci. USA 2013, 110, 12960–12965. [Google Scholar] [CrossRef]
- Rahimi, N.; Dayanir, V.; Lashkari, K. Receptor chimeras indicate that the vascular endothelial growth factor receptor-1 (VEGFR-1) modulates mitogenic activity of VEGFR-2 in endothelial cells. J. Biol. Chem. 2000, 275, 16986–16992. [Google Scholar] [CrossRef]
- Wiesmann, C.; Fuh, G.; Christinger, H.W.; Eigenbrot, C.; Wells, J.A.; de Vos, A.M. Crystal Structure at 1.7 Å resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 1997, 91, 695–704. [Google Scholar] [CrossRef]
- Starovasnik, M.A.; Christinger, H.W.; Wiesmann, C.; Champe, M.A.; de Vos, A.M.; Skelton, N.J. Solution structure of the VEGF-binding domain of Flt-1: Comparison of its free and bound states11Edited by P. E. Wright. J. Mol. Biol. 1999, 293, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Darley, P.I.; Acharya, K.R. Structural insights into the binding of vascular endothelial growth factor-B by VEGFR-1(D2): Recognition and specificity. J. Biol. Chem. 2010, 285, 23779–23789. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xie, P.; Opatowsky, Y.; Schlessinger, J. Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 1906–1911. [Google Scholar] [CrossRef] [PubMed]
- Brozzo, M.S.; Bjelic, S.; Kisko, K.; Schleier, T.; Leppanen, V.M.; Alitalo, K.; Winkler, F.K.; Ballmer-Hofer, K. Thermodynamic and structural description of allosterically regulated VEGFR-2 dimerization. Blood 2012, 119, 1781–1788. [Google Scholar] [CrossRef]
- Leppanen, V.M.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Kalkkinen, N.; Strandin, T.; Lankinen, H.; Goldman, A.; Ballmer-Hofer, K.; Alitalo, K. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc. Natl. Acad. Sci. USA 2010, 107, 2425–2430. [Google Scholar] [CrossRef]
- McTigue, M.A.; Wickersham, J.A.; Pinko, C.; Showalter, R.E.; Parast, C.V.; Tempczyk-Russell, A.; Gehring, M.R.; Mroczkowski, B.; Kan, C.C.; Villafranca, J.E.; et al. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: A key enzyme in angiogenesis. Structure 1999, 7, 319–330. [Google Scholar] [CrossRef]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef]
- Thieltges, K.M.; Avramovic, D.; Piscitelli, C.L.; Markovic-Mueller, S.; Binz, H.K.; Ballmer-Hofer, K. Characterization of a drug-targetable allosteric site regulating vascular endothelial growth factor signaling. Angiogenesis 2018, 21, 533–543. [Google Scholar] [CrossRef]
- Manni, S.; Mineev, K.S.; Usmanova, D.; Lyukmanova, E.N.; Shulepko, M.A.; Kirpichnikov, M.P.; Winter, J.; Matkovic, M.; Deupi, X.; Arseniev, A.S.; et al. Structural and functional characterization of alternative transmembrane domain conformations in VEGF receptor 2 activation. Structure 2014, 22, 1077–1089. [Google Scholar] [CrossRef]
- Dosch, D.D.E.; Ballmer-Hofer, K. Transmembrane domain-mediated orientation of receptor monomers in active VEGFR-2 dimers. FASEB J. 2010, 24, 32–38. [Google Scholar] [CrossRef]
- Gille, H.; Kowalski, J.; Yu, L.; Chen, H.; Pisabarro, M.T.; Davis-Smyth, T.; Ferrara, N. A repressor sequence in the juxtamembrane domain of Flt-1 (VEGFR-1) constitutively inhibits vascular endothelial growth factor-dependent phosphatidylinositol 3’-kinase activation and endothelial cell migration. EMBO J. 2000, 19, 4064–4073. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, C.; Wang, G.; Xu, Z.; Luo, Y.; Wang, J.; Zhu, W. Exploring binding mechanisms of VEGFR2 with three drugs lenvatinib, sorafenib, and sunitinib by molecular dynamics simulation and free energy calculation. Chem. Biol. Drug Des. 2019, 93, 934–948. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Meharena, H.S.; Kornev, A.P. Evolution of a dynamic molecular switch. IUBMB Life 2019, 71, 672–684. [Google Scholar] [CrossRef]
- Bessman, N.J.; Freed, D.M.; Lemmon, M.A. Putting together structures of epidermal growth factor receptors. Curr. Opin. Struct. Biol. 2014, 29, 95–101. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Freed, D.M.; Schlessinger, J.; Kiyatkin, A. The dark side of cell signaling: Positive roles for negative regulators. Cell 2016, 164, 1172–1184. [Google Scholar] [CrossRef]
- Zhao, W.; Jamshidiha, M.; Lanyon-Hogg, T.; Recchi, C.; Cota, E.; Tate, E.W. Direct targeting of the ras GTPase superfamily through structure- based design. Curr. Top. Med. Chem. 2017, 17, 16–29. [Google Scholar] [CrossRef]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Zhang, F.; Prahst, C.; Mathivet, T.; Pibouin-Fragner, L.; Zhang, J.; Genet, G.; Tong, R.; Dubrac, A.; Eichmann, A. The Robo4 cytoplasmic domain is dispensable for vascular permeability and neovascularization. Nat. Commun. 2016, 7, 13517. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.A.; Giese, A.; Stuttfeld, E.; Abram Saliba, J.; Villemagne, D.; Schleier, T.; Binz, H.K.; Ballmer-Hofer, K. Targeting extracellular domains D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor regulatory sites. Mol. Cell. Biol. 2012, 32, 3802–3813. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Eyck, L.F.T. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Chen, T.T.; Luque, A.; Lee, S.; Anderson, S.M.; Segura, T.; Iruela-Arispe, M.L. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J. Cell Biol. 2010, 188, 595–609. [Google Scholar] [CrossRef]
- Mokhdomi, T.A.; Bukhari, S.; Chikan, N.A.; Amin, A.; Wafai, A.H.; Wani, S.H.; Chowdri, N.A.; Qadri, R.A. A novel kinase mutation in VEGFR-1 predisposes its αC-helix/activation loop towards allosteric activation: Atomic insights from protein simulation. Eur. J. Hum. Genet. 2016, 24, 1287–1293. [Google Scholar] [CrossRef]
- Meyer, R.D.; Singh, A.; Majnoun, F.; Latz, C.; Lashkari, K.; Rahimi, N. Substitution of C-terminus of VEGFR-2 with VEGFR-1 promotes VEGFR-1 activation and endothelial cell proliferation. Oncogene 2004, 23, 5523–5531. [Google Scholar] [CrossRef]
- Meyer, R.D.; Singh, A.J.; Rahimi, N. The carboxyl terminus controls ligand-dependent activation of VEGFR-2 and its signaling. J. Biol. Chem. 2004, 279, 735–742. [Google Scholar] [CrossRef]
- Clayton, A.H.; Walker, F.; Orchard, S.G.; Henderson, C.; Fuchs, D.; Rothacker, J.; Nice, E.C.; Burgess, A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J. Biol. Chem. 2005, 280, 30392–30399. [Google Scholar] [CrossRef]
- Kozer, N.; Barua, D.; Orchard, S.; Nice, E.C.; Burgess, A.W.; Hlavacek, W.S.; Clayton, A.H. Exploring higher-order EGFR oligomerisation and phosphorylation—A combined experimental and theoretical approach. Mol. Biosyst. 2013, 9, 1849–1863. [Google Scholar] [CrossRef]
- Kozer, N.; Barua, D.; Henderson, C.; Nice, E.C.; Burgess, A.W.; Hlavacek, W.S.; Clayton, A.H. Recruitment of the adaptor protein Grb2 to EGFR tetramers. Biochemistry 2014, 53, 2594–2604. [Google Scholar] [CrossRef] [PubMed]
- Sawano, A.; Takahashi, T.; Yamaguchi, S.; Shibuya, M. The phosphorylated 1169-tyrosine containing region of flt-1 kinase (VEGFR-1) is a major binding site for PLCgamma. Biochem. Biophys. Res. Commun. 1997, 238, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Isohara, T.; Kato, T.; Shigeta, K.; Yamano, T.; Uno, I. Tyrosine 1213 of Flt-1 is a major binding site of Nck and SHP-2. Biochem. Biophys. Res. Commun. 1998, 246, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Shigeta, K.; Isohara, T.; Yamano, T.; Uno, I. Sck interacts with KDR and Flt-1 via its SH2 domain. Biochem. Biophys. Res. Commun. 1998, 251, 77–82. [Google Scholar] [CrossRef]
- Vogel, C.; Bauer, A.; Wiesnet, M.; Preissner, K.T.; Schaper, W.; Marti, H.H.; Fischer, S. Flt-1, but not Flk-1 mediates hyperpermeability through activation of the PI3-K/Akt pathway. J. Cell. Physiol. 2007, 212, 236–243. [Google Scholar] [CrossRef]
- Solowiej, J.; Chen, J.H.; Zou, H.Y.; Grant, S.K.; Murray, B.W. Substrate-specific conformational regulation of the receptor tyrosine kinase VEGFR2 catalytic domain. ACS Chem. Biol. 2013, 8, 978–986. [Google Scholar] [CrossRef]
- Blanes, M.G.; Oubaha, M.; Rautureau, Y.; Gratton, J.P. Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. J. Biol. Chem. 2007, 282, 10660–10669. [Google Scholar] [CrossRef]
- Zeng, H.; Sanyal, S.; Mukhopadhyay, D. Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 2001, 276, 32714–32719. [Google Scholar] [CrossRef]
- Sase, H.; Watabe, T.; Kawasaki, K.; Miyazono, K.; Miyazawa, K. VEGFR2-PLCgamma1 axis is essential for endothelial specification of VEGFR2+ vascular progenitor cells. J. Cell Sci. 2009, 122, 3303–3311. [Google Scholar] [CrossRef]
- Karpov, O.A.; Fearnley, G.W.; Smith, G.A.; Kankanala, J.; McPherson, M.J.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. Receptor tyrosine kinase structure and function in health and disease. AIMS Biophys. 2015, 2, 476–502. [Google Scholar] [CrossRef]
- Kong, D.-H.; Kim, M.R.; Jang, J.H.; Na, H.-J.; Lee, S. A Review of Anti-angiogenic targets for monoclonal antibody cancer therapy. Int. J. Mol. Sci. 2017, 18, 1786. [Google Scholar] [CrossRef] [PubMed]
- Sumner, G.; Georgaros, C.; Rafique, A.; DiCioccio, T.; Martin, J.; Papadopoulos, N.; Daly, T.; Torri, A. Anti-VEGF drug interference with VEGF quantitation in the R&D systems human quantikine VEGF ELISA kit. Bioanalysis 2019, 11, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J. Analysis of the binding affinity of vascular endothelial growth factor A (VEGF) to ranibizumab, aflibercept and bevacizumab. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1961. [Google Scholar]
- Mukherji, S.K. Bevacizumab (Avastin). Am. J. Neuroradiol. 2010, 31, 235–236. [Google Scholar] [CrossRef]
- Kazazi-Hyseni, F.; Beijnen, J.H.; Schellens, J.H.M. Bevacizumab. Oncologist 2010, 15, 819–825. [Google Scholar] [CrossRef]
- Ferrara, N.; Damico, L.; Shams, N.; Lowman, H.; Kim, R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006, 26, 859–870. [Google Scholar] [CrossRef]
- Shahsuvaryan, M.L. Therapeutic potential of ranibizumab in corneal neovascularization. Trends Pharm. Sci. 2017, 38, 667–668. [Google Scholar] [CrossRef]
- Singh, A.D.; Parmar, S. Ramucirumab (Cyramza): A Breakthrough Treatment for Gastric Cancer. P T Peer Rev. J. Formul. Manag. 2015, 40, 430–468. [Google Scholar]
- Arrieta, O.; Zatarain-Barron, Z.L.; Cardona, A.F.; Carmona, A.; Lopez-Mejia, M. Ramucirumab in the treatment of non-small cell lung cancer. Expert Opin. Drug Saf. 2017, 16, 637–644. [Google Scholar] [CrossRef]
- Al-Halafi, A.M. Vascular endothelial growth factor trap-eye and trap technology: Aflibercept from bench to bedside. Oman J. Ophthalmol. 2014, 7, 112–115. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Freund, K.B.; Mrejen, S.; Gallego-Pinazo, R. An update on the pharmacotherapy of neovascular age-related macular degeneration. Expert Opin. Pharmacother. 2013, 14, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Wylegala, E.; Teper, S.J. [VEGF in age-related macular degeneration. Part II. VEGF inhibitors use in age-related macular degeneration treatment]. Klin Ocz. 2007, 109, 97–100. [Google Scholar]
- Vinores, S.A. Pegaptanib in the treatment of wet, age-related macular degeneration. Int. J. Nanomed. 2006, 1, 263–268. [Google Scholar]
- Spratlin, J.L.; Cohen, R.B.; Eadens, M.; Gore, L.; Camidge, D.R.; Diab, S.; Leong, S.; O’Bryant, C.; Chow, L.Q.M.; Serkova, N.J.; et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J. Clin. Oncol. 2010, 28, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Spratlin, J. Ramucirumab (IMC-1121B): Monoclonal antibody inhibition of vascular endothelial growth factor receptor-2. Curr. Oncol. Rep. 2011, 13, 97–102. [Google Scholar] [CrossRef]
- Lee, W.S.; Pyun, B.-J.; Kim, S.-W.; Shim, S.R.; Nam, J.R.; Yoo, J.Y.; Jin, Y.; Jin, J.; Kwon, Y.-G.; Yun, C.-O.; et al. TTAC-0001, a human monoclonal antibody targeting VEGFR-2/KDR, blocks tumor angiogenesis. mAbs 2015, 7, 957–968. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, S.Y.; Lee, W.S.; Yoo, J.S.; Sun, J.-M.; Lee, J.; Park, S.H.; Park, J.O.; Ahn, M.-J.; Lim, H.Y.; et al. Phase I trial and pharmacokinetic study of tanibirumab, a fully human monoclonal antibody to vascular endothelial growth factor receptor 2, in patients with refractory solid tumors. Investig. New Drugs 2017, 35, 782–790. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Krishnamurthi, S.; Youssoufian, H.; Hall, N.; Fox, F.; Dontabhaktuni, A.; Grebennik, D.; Remick, S. Icrucumab, a fully human monoclonal antibody against the vascular endothelial growth factor receptor-1, in the treatment of patients with advanced solid malignancies: A Phase 1 study. Investig. New Drugs 2014, 32, 303–311. [Google Scholar] [CrossRef]
- Vahdat, L.T.; Layman, R.; Yardley, D.A.; Gradishar, W.; Salkeni, M.A.; Joy, A.A.; Garcia, A.A.; Ward, P.; Khatcheressian, J.; Sparano, J.; et al. Randomized phase II study of ramucirumab or icrucumab in combination with capecitabine in patients with previously treated locally advanced or metastatic breast cancer. Oncologist 2017, 22, 245–254. [Google Scholar] [CrossRef][Green Version]
- Graziani, G.; Ruffini, F.; Tentori, L.; Scimeca, M.; Dorio, A.S.; Atzori, M.G.; Failla, C.M.; Morea, V.; Bonanno, E.; D’Atri, S.; et al. Antitumor activity of a novel anti-vascular endothelial growth factor receptor-1 monoclonal antibody that does not interfere with ligand binding. Oncotarget 2016, 7, 72868–72885. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.G.; Tentori, L.; Ruffini, F.; Ceci, C.; Lisi, L.; Bonanno, E.; Scimeca, M.; Eskilsson, E.; Daubon, T.; Miletic, H.; et al. The anti-vascular endothelial growth factor receptor-1 monoclonal antibody D16F7 inhibits invasiveness of human glioblastoma and glioblastoma stem cells. J. Exp. Clin. Cancer Res. 2017, 36, 106. [Google Scholar] [CrossRef] [PubMed]
- Persaud, K.; Tille, J.C.; Liu, M.; Zhu, Z.; Jimenez, X.; Pereira, D.S.; Miao, H.Q.; Brennan, L.A.; Witte, L.; Pepper, M.S.; et al. Involvement of the VEGF receptor 3 in tubular morphogenesis demonstrated with a human anti-human VEGFR-3 monoclonal antibody that antagonizes receptor activation by VEGF-C. J. Cell Sci. 2004, 117, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, X.; Lu, D.; Brennan, L.; Persaud, K.; Liu, M.; Miao, H.; Witte, L.; Zhu, Z. A recombinant, fully human, bispecific antibody neutralizes the biological activities mediated by both vascular endothelial growth factor receptors 2 and 3. Mol. Cancer Ther. 2005, 4, 427–434. [Google Scholar] [PubMed]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N. Structural basis for substrate recognition and control in protein kinases. Ernst Scher. Res. Found Workshop 2001. [Google Scholar] [CrossRef]
- Adams, V.R.; Leggas, M. Sunitinib malate for the treatment of metastatic renal cell carcinoma and gastrointestinal stromal tumors. Clin. Ther. 2007, 29, 1338–1353. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Sunitinib: A VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 2007, 356, 323–328. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Berretta, M.; Rinaldi, L.; Di Benedetto, F.; Lleshi, A.; de Re, V.; Facchini, G.; de Paoli, P.; di Francia, R. Angiogenesis inhibitors for the treatment of hepatocellular carcinoma. Front. Pharmacol. 2016, 7, 428. [Google Scholar] [CrossRef]
- Escudier, B.; Lougheed, J.C.; Albiges, L. Cabozantinib for the treatment of renal cell carcinoma. Expert Opin. Pharm. 2016, 17, 2499–2504. [Google Scholar] [CrossRef] [PubMed]
- Grullich, C. Cabozantinib: A MET, RET, and VEGFR2 tyrosine kinase inhibitor. Recent Results Cancer Res. 2014, 201, 207–214. [Google Scholar] [PubMed]
- Sonpavde, G.; Hutson, T.E. Pazopanib: A novel multitargeted tyrosine kinase inhibitor. Curr. Oncol. Rep. 2007, 9, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Kantarjian, H.; Jabbour, E.; Gonzalez, G.N.; Borthakur, G.; Pemmaraju, N.; Daver, N.; Gachimova, E.; Ferrajoli, A.; Kornblau, S.; et al. Ponatinib as first-line treatment for patients with chronic myeloid leukaemia in chronic phase: A phase 2 study. Lancet Haematol. 2015, 2, e376–e383. [Google Scholar] [CrossRef]
- Rey, J.-B.; Launay-Vacher, V.; Tournigand, C. Regorafenib as a single-agent in the treatment of patients with gastrointestinal tumors: An overview for pharmacists. Target. Oncol. 2015, 10, 199–213. [Google Scholar] [CrossRef]
- Omata, M.; Cheng, A.-L.; Kokudo, N.; Kudo, M.; Lee, J.M.; Jia, J.; Tateishi, R.; Han, K.-H.; Chawla, Y.K.; Shiina, S.; et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: A 2017 update. Hepatol. Int. 2017, 11, 317–370. [Google Scholar] [CrossRef]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef]
- Hewett, Y.; Ghimire, S.; Farooqi, B.; Shah, B.K. Lenvatinib—A multikinase inhibitor for radioiodine-refractory differentiated thyroid cancer. J. Oncol. Pharm. Pract. 2018, 24, 28–32. [Google Scholar] [CrossRef]
- Kawalec, P.; Malinowska-Lipien, I.; Brzostek, T.; Kozka, M. Lenvatinib for the treatment of radioiodine-refractory differentiated thyroid carcinoma: A systematic review and indirect comparison with sorafenib. Expert Rev. Anticancer Ther. 2016, 16, 1303–1309. [Google Scholar] [CrossRef]
- Ton, G.N.; Banaszynski, M.E.; Kolesar, J.M. Vandetanib: A novel targeted therapy for the treatment of metastatic or locally advanced medullary thyroid cancer. Am. J. Health Syst. Pharm. 2013, 70, 849–855. [Google Scholar] [CrossRef]
- Fala, L. Ofev (Nintedanib): First tyrosine kinase inhibitor approved for the treatment of patients with idiopathic pulmonary fibrosis. Am. Health Drug Benefits 2015, 8, 101–104. [Google Scholar] [PubMed]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kwong-Kwok, W. Recent developments in anti-cancer agents targeting the Ras/Raf/ MEK/ERK Pathway. Recent Patents Anti-Cancer Drug Discov. 2009, 4, 28–35. [Google Scholar] [CrossRef]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Go, Z.; Iyer, R.; Kolis, S.; Zhao, S.; Lee, R.; Grippo, J.F.; et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010, 70, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; McKee, A.E.; Ning, Y.M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X.; et al. FDA approval summary: Vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef]
- Cuenda, A.; Rouse, J.; Doza, Y.N.; Meier, R.; Cohen, P.; Gallagher, T.F.; Young, P.R.; Lee, J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995, 364, 229–233. [Google Scholar]
- Lali, F.V.; Hunt, A.E.; Turner, S.J.; Foxwell, B.M. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J. Biol. Chem. 2000, 275, 7395–7402. [Google Scholar]
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef]
- Wander, S.A.; Hennessy, B.T.; Slingerland, J.M. Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J. Clin. Investig. 2011, 121, 1231–1241. [Google Scholar] [CrossRef]
- Petiot, A.; Faure, J.; Stenmark, H.; Gruenberg, J. PI3P signaling regulates receptor sorting but not transport in the endosomal pathway. J. Cell Biol. 2003, 162, 971–979. [Google Scholar] [CrossRef]
- Studentova, H.; Vitaskova, D.; Melichar, B. Lenvatinib for the treatment of kidney cancer. Expert Rev. Anticancer Ther. 2018, 18, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Glen, H. Lenvatinib therapy for the treatment of patients with advanced renal cell carcinoma. Future Oncol. 2016, 12, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. Alpelisib effective in advanced-stage disease. Nat. Rev. Clin. Oncol. 2019, 24, 019–0234. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Mesange, P.; Poindessous, V.; Sabbah, M.; Escargueil, A.E.; de Gramont, A.; Larsen, A.K. Intrinsic bevacizumab resistance is associated with prolonged activation of autocrine VEGF signaling and hypoxia tolerance in colorectal cancer cells and can be overcome by nintedanib, a small molecule angiokinase inhibitor. Oncotarget 2014, 5, 4709–4721. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Details | VEGFR1 | VEGFR2 | VEGFR3 |
|---|---|---|---|
| Swiss UniProt ID | P17948 | P35968 | P35916 |
| Full length | 1338 | 1356 | 1363 |
| Signal peptide | 26 (1–26) | 19 (1–19) | 24 (1–24) |
| Receptor chain | 1312 (27–1338) | 1337 (20–1356) | 1339 (25–1363) |
| Extracellular domain | 732 (27–758) | 745 (20–764) | 751 (25–775) |
| Immunoglobulin (Ig)-like 1 | 92 (32–123) | 65 (46–110) | 98 (30–127) |
| Immunoglobulin (Ig)-like 2 | 64 (151–214) | 67 (141–207) | 63 (151–213) |
| Immunoglobulin (Ig)-like 3 | 98 (230–327) | 97 (224–320) | 108 (219–326) |
| Immunoglobulin (Ig)-like 4 | 87 (335–421) | 87 (328–414) | 85 (331–415) |
| Immunoglobulin (Ig)-like 5 | 126 (428–553) | 128 (421–548) | 131 (422–552) |
| Immunoglobulin (Ig)-like 6 | 99 (556–654) | 110 (551–660) | 117 (555–671) |
| Immunoglobulin (Ig)-like 7 | 87 (661–747) | 87 (667–753) | 87 (678–764) |
| Transmembrane domain | 22 (759–780) | 21 (765–785) | 21 (776–796) |
| Cytoplasmic domain | 558 (781–1338) | 571 (786–1356) | 567 (797–1363) |
| Complex | Protein Data Bank (PDB) Code | Domains | Method | Reference |
|---|---|---|---|---|
| VEGFR1/VEGF-A | 1FLT | Domain 2 | X-ray diffraction | [70] |
| VEGFR1/VEGF-A | 1QTY | Domain 2 | X-ray diffraction | [71] |
| VEGFR1/PIGF | 1RV6 | Domain 2 | X-ray diffraction | [67] |
| VEGFR1/VEGF-B | 2XAC | Domain 2 | X-ray diffraction | [72] |
| VEGFR1/VEGF-A | 5T89 | Domains 1–6 | X-ray diffraction and negative stain EM | [65] |
| VEGFR2 | 3KVQ | Domain 7 | X-ray diffraction | [73] |
| VEGFR2/VEGF-A | 3V2A | Domains 2 and 3 | X-ray diffraction | [74] |
| VEGFR2/VEGF-C | 2X1W | Domains 2 and 3 | X-ray diffraction | [75] |
| VEGFR2/VEGF-C | 2X1X | Domains 2 and 3 in tetragonal crystal form | X-ray diffraction | [75] |
| VEGFR2 kinase domain | 1VR2 | Kinase domain | X-ray diffraction | [76] |
| VEGFR2 kinase domain with sunitinib | 4AGD | Juxta membrane and kinase domains | X-ray diffraction | [77] |
| VEGFR2 kinase domain with sorafenib | 4ASD | Juxta membrane and kinase domains | X-ray diffraction | [77] |
| VEGFR2 kinase domain with axitinib | 4AGC | Juxta membrane and Kinase domains | X-ray diffraction | [77] |
| VEGFR2 kinase domain with tivozanib | 4ASE | Juxta membrane and kinase domains | X-ray diffraction | [77] |
| VEGFR2/VEGF-E | 3V6B | Domains 2 and 3 | X-ray diffraction | [74] |
| VEGFR2/DARPin Db4 | 5OYJ | Domains 4 and 5 | X-ray diffraction | [78] |
| VEGFR2 trimeric mutant transmembrane domain | 2MET | Transmembrane domain | NMR | [79] |
| VEGFR2 mutant dimeric transmembrane domain | 2MEU | Transmembrane domain | NMR | [79] |
| VEGFR2 dimeric membrane domain in DPC micelles | 2M59 | Transmembrane domain | NMR | [79] |
| VEGFR3/VEGF-C | 4BSK | Domains 1 and 2 | X-ray diffraction | [68] |
| VEGFR3 ECD | 4BSJ | Domains 4 and 5 | X-ray diffraction | [68] |
| Name | Target | Type | Treatment | Reference |
|---|---|---|---|---|
| Bevacizumab (Avastin) | VEGF-A | Human monoclonal IgG1 antibody | Non-small cell lung cancer (NSCLC), colorectal cancer, metastatic breast cancer, renal cell cancer and advanced glioblastoma multiforme | [115,116] |
| Ranibizumab (Lucentis) | VEGF-A | Antibody Fab fragment | Corneal neovascularisation | [117,118] |
| Ramucirumab (Cyramza) | VEGFR2 | Human monoclonal IgG1 antibody | Advanced gastric cancer and non-small cell lung cancer (NSCLC) | [119,120] |
| Aflibercept (Zaltrap) | VEGF-A, VEGF-B and PIGF | Soluble decoy receptor | Neovascular age-related macular degeneration (AMD), Diabetic macular edema (DME) | [121,122,123] |
| Pegaptanib (Macugen) | VEGF 165 | Polynucleotide aptamer | Neovascular age-related macular degeneration (AMD) | [124,125] |
| Inhibitors | VEGFR1 | VEGFR2 | VEGFR3 | Treatment | Other Targets | References |
|---|---|---|---|---|---|---|
| Sunitinib (Sutent) | + | + | + | Metastatic renal cell carcinoma (mRCC) and gastrointestinal stromal tumour (GIST) | PDGFRβ, FLT3 | [138,139] |
| Sorafenib (Nexavar) | + | Advanced renal cell carcinoma, advanced hepatocellular carcinoma | PDGFR, c-kit, Raf-1, B-Raf | [140,141] | ||
| Cabozantinib (Cabometyx) | + | Advanced renal cell carcinoma | MET, RET | [142,143] | ||
| Pazopanib (GW78603) | + | + | + | Renal cell carcinoma | PDGFR-α, PDGFR-β and c-kit | [144] |
| Ponatinib (AP24534) | + | Chronic myeloid leukaemia | FGFR-1, FGFR-2, FGFR-3, PDGFR-α | [145] | ||
| Regorafenib (BAY 73-4506) | + | + | + | Metastatic colorectal cancer (mCRC), gastrointestinal stromal tumours (GIST) and hepatocellular carcinoma | FGFR-1, FGFR-2, PDGFR-α, PDGFR-β, KIT, TIE2, TrkA | [146,147] |
| Axitinib | + | + | + | Renal cell carcinoma | PDGFRβ, c-Kit | [148] |
| Lenvatinib (E7080) | + | + | + | Radioactive iodine (RAI)-refractory thyroid cancer | PDGFR-α, FGFR-1, FGFR-2, FGFR-3, FGFR-4, KIT and RET | [149,150] |
| Vandetanib (ZD6474) | + | + | Locally advanced and metastatic medullary thyroid cancer | EGFR, RET | [151] | |
| Nintedanib (BIBF 1120) | + | + | + | Idiopathic pulmonary fibrosis (IPF) | FGFR-1, FGFR2, FGFR3, PDGFR-α, PDGFR-β and FLT3 | [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaik, F.; Cuthbert, G.A.; Homer-Vanniasinkam, S.; Muench, S.P.; Ponnambalam, S.; Harrison, M.A. Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy. Biomolecules 2020, 10, 1673. https://doi.org/10.3390/biom10121673
Shaik F, Cuthbert GA, Homer-Vanniasinkam S, Muench SP, Ponnambalam S, Harrison MA. Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy. Biomolecules. 2020; 10(12):1673. https://doi.org/10.3390/biom10121673
Chicago/Turabian StyleShaik, Faheem, Gary A. Cuthbert, Shervanthi Homer-Vanniasinkam, Stephen P. Muench, Sreenivasan Ponnambalam, and Michael A. Harrison. 2020. "Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy" Biomolecules 10, no. 12: 1673. https://doi.org/10.3390/biom10121673
APA StyleShaik, F., Cuthbert, G. A., Homer-Vanniasinkam, S., Muench, S. P., Ponnambalam, S., & Harrison, M. A. (2020). Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy. Biomolecules, 10(12), 1673. https://doi.org/10.3390/biom10121673