Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation



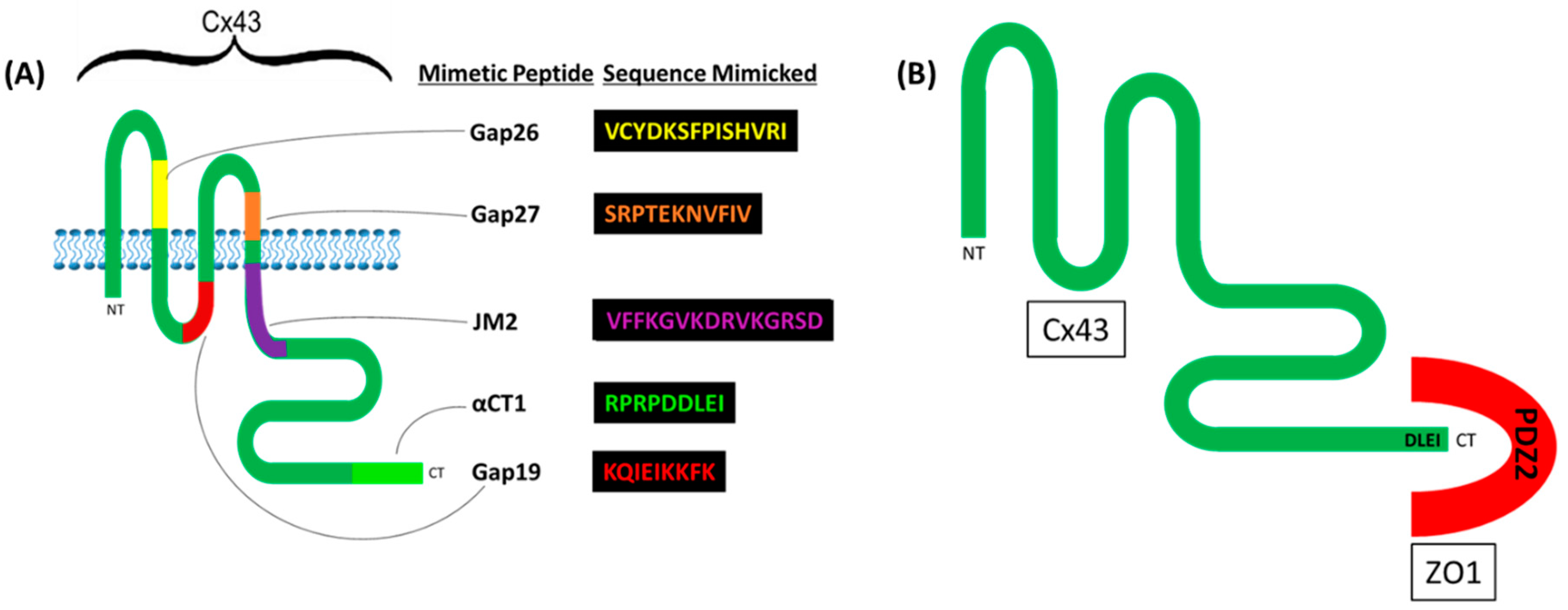{kind=link}
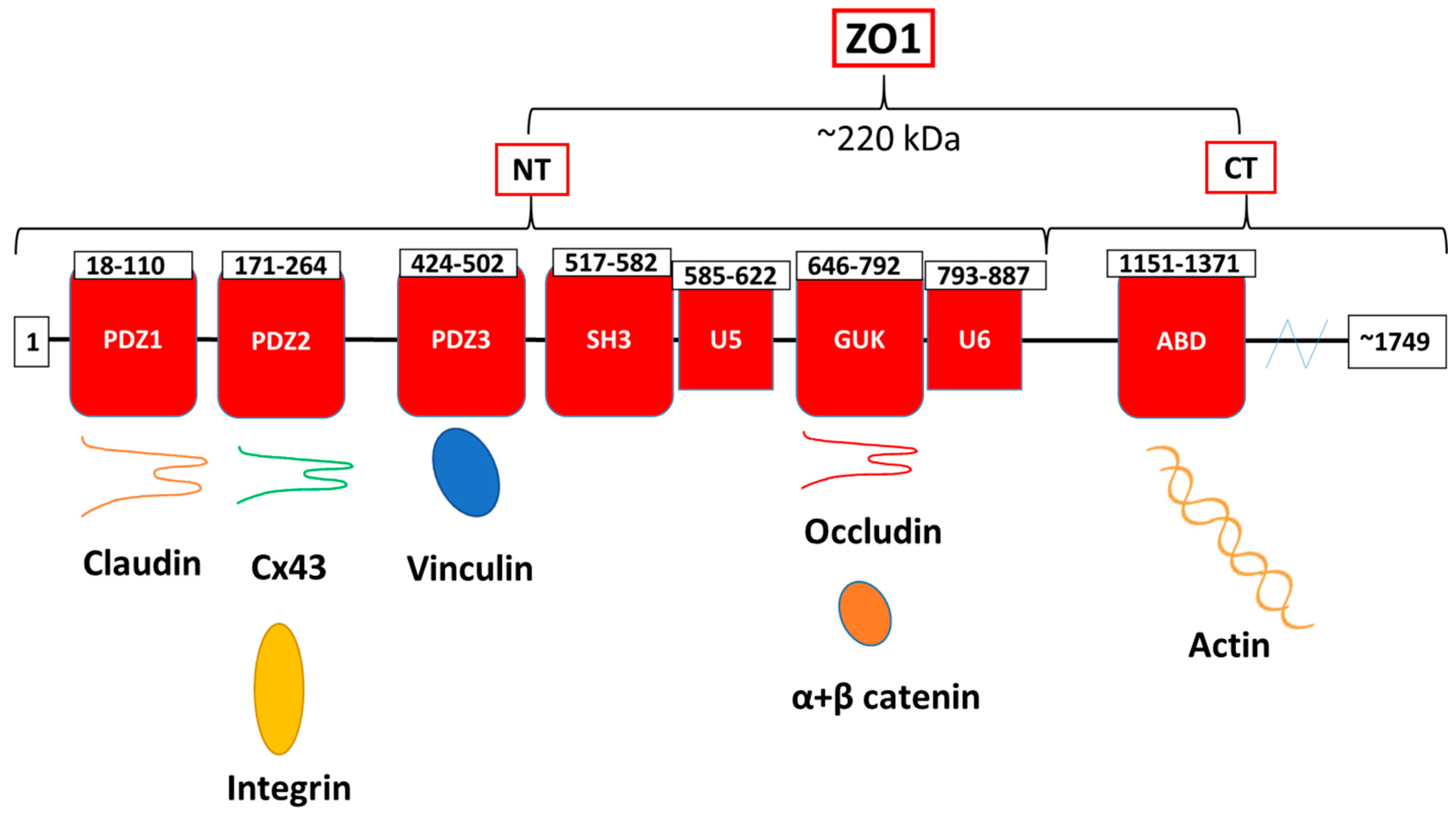{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Connexin Structure and Function
2.1. Cx43
2.2. Canonical Channel-Dependent Functions of Cx43
2.3. GJs and Cell–Cell Adhesion
2.4. Connexins and Barrier Function
3. Barrier-Modulating Structures
3.1. Tight Junction Structure and Function
3.2. Occludin
3.3. Claudin
3.4. Adherens Junctions
3.5. Actin
3.6. Vinculin-Based Focal Adhesions
4. Channel-Dependent Roles of Connexins in Barrier Function
4.1. The Role of GJIC in Barrier Function Regulation
4.2. The Role of HCs in Barrier Function Regulation
5. Channel-Independent Roles of Cx43 in Barrier Function
5.1. Key Studies
5.2. Connexins, Actin, and Potential Implications for Barrier Function Regulation
5.3. Cx43, Focal Adhesions, PI3K/PKCα Signaling, the Actin Cytoskeleton, and Barrier Function
5.4. Cx43, Cell Chirality, and Actin-Mediated Barrier Function Regulation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Förster, C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008, 130, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.G.; Palade, G.E. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Miller, A.L. Tricellular junctions: How to build junctions at the TRICkiest points of epithelial cells. Mol. Biol. Cell 2017, 28, 2023–2034. [Google Scholar] [CrossRef]
- Escribano, J.; Chen, M.B.; Moeendarbary, E.; Cao, X.; Shenoy, V.; Garcia-Aznar, J.M.; Kamm, R.D.; Spill, F. Balance of mechanical forces drives endothelial gap formation and may facilitate cancer and immune-cell extravasation. PLoS Comput. Biol. 2019, 15, e1006395. [Google Scholar] [CrossRef]
- Herrero, R.; Sanchez, G.; Lorente, J.A. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 2017, 6, 11. [Google Scholar] [CrossRef]
- Heusch, G. The coronary circulation as a target of cardioprotection. Circ. Res. 2016, 118, 1643–1658. [Google Scholar] [CrossRef]
- Heusch, G. Protection of the human coronary circulation by remote ischemic conditioning. Int. J. Cardiol. 2018, 252, 35–36. [Google Scholar] [CrossRef]
- Simmons, S.; Erfinanda, L.; Bartz, C.; Kuebler, W. Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 2018, 597, 997–1021. [Google Scholar] [CrossRef]
- Soon, A.S.; Chua, J.W.; Becker, D.L. Connexins in endothelial barrier function—Novel therapeutic targets countering vascular hyperpermeability. Thromb. Haemost. 2016, 116, 852–867. [Google Scholar] [CrossRef]
- Aghajanian, A.; Wittchen, E.S.; Allingham, M.J.; Garrett, T.A.; Burridge, K. Endothelial cell junctions and the regulation of vascular permeability and leukocyte transmigration. J. Thromb. Haemost. 2008, 6, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Derangeon, M.; Spray, D.C.; Bourmeyster, N.; Sarrouilhe, D.; Hervé, J.-C. Reciprocal influence of connexins and apical junction proteins on their expressions and functions. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Hervé, J.-C.; Bourmeyster, N.; Sarrouilhe, D.; Duffy, H.S. Gap junctional complexes: From partners to functions. Prog. Biophys. Mol. Biol. 2007, 94, 29–65. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G. Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc. Res. 2009, 77, 1–3. [Google Scholar] [CrossRef]
- Francis, R.; Xu, X.; Park, H.; Wei, C.-J.; Chang, S.; Chatterjee, B.; Lo, C. Connexin43 Modulates Cell Polarity and Directional Cell Migration by Regulating Microtubule Dynamics. PLoS ONE 2011, 6, e26379. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Kameritsch, P.; Pogoda, K.; Pohl, U. Channel-independent influence of connexin 43 on cell migration. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 1993–2001. [Google Scholar] [CrossRef]
- Olk, S.; Zoidl, G.; Dermietzel, R. Connexins, cell motility, and the cytoskeleton. Cell Motil. Cytoskelet. 2009, 66, 1000–1016. [Google Scholar] [CrossRef]
- Matsuuchi, L.; Naus, C.C. Gap junction proteins on the move: Connexins, the cytoskeleton and migration. Biochim. Biophys. Acta (BBA) Biomembr. 2013, 1828, 94–108. [Google Scholar] [CrossRef]
- Brisset, A.C.; Isakson, B.E.; Kwak, B.R. Connexins in vascular physiology and pathology. Antioxid. Redox Signal. 2009, 11, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Takebe, K.; Endo, A.; Hara, S.; Oyamada, Y. Connexin expression and gap-junctional intercellular communication in ES cells and iPS cells. Front. Pharmacol. 2013, 4, 85. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nat. Cell Biol. 2009, 458, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A.; Tani, K.; Hiroaki, Y.; Fujiyoshi, Y.; Sosinsky, G.E. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc. Natl. Acad. Sci. USA 2007, 104, 10034–10039. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.; Harris, A.L. Gap junction channel structure in the early 21st century: Facts and fantasies. Curr. Opin. Cell Biol. 2007, 19, 521–528. [Google Scholar] [CrossRef]
- Giepmans, B.N.G.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef]
- Chen, J.; Pan, L.; Wei, Z.; Zhao, Y.; Zhang, M. Domain-swapped dimerization of ZO-1 PDZ2 generates specific and regulatory connexin43-binding sites. EMBO J. 2008, 27, 2113–2123. [Google Scholar] [CrossRef][Green Version]
- Hervé, J.-C.; Derangeon, M.; Sarrouilhe, D.; Bourmeyster, N. Influence of the scaffolding protein Zonula Occludens (ZOs) on membrane channels. Biochim. et Biophys. Acta (BBA) Biomembr. 2014, 1838, 595–604. [Google Scholar] [CrossRef]
- Jiang, J.; Hoagland, D.; Palatinus, J.A.; He, H.; Iyyathurai, J.; Jourdan, L.J.; Bultynck, G.; Wang, Z.; Zhang, Z.; Schey, K.; et al. Interaction of α Carboxyl Terminus 1 Peptide With the Connexin 43 Carboxyl Terminus Preserves Left Ventricular Function After Ischemia-Reperfusion Injury. J. Am. Hear. Assoc. 2019, 8, e012385. [Google Scholar] [CrossRef]
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 40–47. [Google Scholar] [CrossRef]
- Loewenstein, W.R. Junctional intercellular communication: The cell-to-cell membrane channel. Physiol. Rev. 1981, 61, 829–913. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Cell-to-cell passage of large molecules. Nature 1966, 212, 629–630. [Google Scholar]
- Meşe, G.; Richard, G.; White, T.W. Gap junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2516–2524. [Google Scholar] [CrossRef]
- Revel, J.P.; Karnovsky, M.J. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J. Cell Biol. 1967, 33, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Friend, D.S. Vitamin-A-induced mucous metaplasia. An. in vitro system for modulating tight and gap junction differentiation. J. Cell Biol. 1976, 68, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef]
- Dunn, C.A.; Lampe, P.D. Injury-triggered Akt phosphorylation of Cx43: A ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2014, 127 Pt 2, 455–464. [Google Scholar] [CrossRef]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef]
- Rhett, J.M.; Gourdie, R.G. The perinexus: A new feature of Cx43 gap junction organization. Heart Rhythm 2012, 9, 619–623. [Google Scholar] [CrossRef]
- Thévenin, A.F.; Margraf, R.A.; Fisher, C.G.; Kells-Andrews, R.M.; Falk, M.M. Phosphorylation regulates connexin43/ZO-1 binding and release, an important step in gap junction turnover. Mol. Biol. Cell 2017, 28, 3595–3608. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.J.; Hackam, D.J. The role of gap junctions in health and disease. Crit. Care Med. 2005, 33, S535–S538. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Mese, G.; Valiuniene, L.; White, T.W.; Brink, P.R.; Valiunas, V. Gap Junction Channels Exhibit Connexin-specific Permeability to Cyclic Nucleotides. J. Gen. Physiol. 2008, 131, 293–305. [Google Scholar] [CrossRef]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17, 11. [Google Scholar] [CrossRef]
- Peracchia, C. Chemical gating of gap junction channels: Roles of calcium, pH and calmodulin. Biochim. Biophys. Acta (BBA) Biomembr. 2004, 1662, 61–80. [Google Scholar] [CrossRef]
- Moyer, K.; Davis, A.; Saggers, G.; Mackay, D.R.; Ehrlich, H.P. Wound Healing: The Role of Gap Junctional Communication in Rat Granulation Tissue Maturation. Exp. Mol. Pathol. 2002, 72, 10–16. [Google Scholar] [CrossRef]
- Jongsma, H.J.; Wilders, R. Gap junctions in cardiovascular disease. Circ. Res. 2000, 86, 1193–1197. [Google Scholar] [CrossRef]
- Rohr, S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 2004, 62, 309–322. [Google Scholar] [CrossRef]
- Hoagland, D.T.; Santos, W.L.; Poelzing, S.; Gourdie, R.G. The role of the gap junction perinexus in cardiac conduction: Potential as a novel anti-arrhythmic drug target. Prog. Biophys. Mol. Biol. 2019, 144, 41–50. [Google Scholar] [CrossRef]
- Gourdie, R.G. The cardiac gap junction has discrete functions in electrotonic and ephaptic coupling. Anat. Rec. 2019, 302, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Hoeker, G.S.; Alvarez-Laviada, A.; Hoagland, D.T.; Wan, X.; King, R.; Sanchez-Alonso, J.; Chen, C.; Jourdan, J.; Isom, L.L.; et al. The adhesion function of the sodium channel beta subunit (β1) contributes to cardiac action potential propagation. eLife 2018, 7, 37610. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Reyes, E.P.; García, I.E.; Pinto, B.; Martínez, A.D.; González, C. Diseases associated with leaky hemichannels. Front. Cell. Neurosci. 2015, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- D’Hondt, C.; Iyyathurai, J.; Himpens, B.; Leybaert, L.; Bultynck, G. Cx43-hemichannel function and regulation in physiology and pathophysiology: Insights from the bovine corneal endothelial cell system and beyond. Front. Physiol. 2014, 5, 348. [Google Scholar] [CrossRef] [PubMed]
- Iyyathurai, J.; Wang, N.; D’Hondt, C.; Jiang, J.X.; Leybaert, L.; Bultynck, G. The SH3-binding domain of Cx43 participates in loop/tail interactions critical for Cx43-hemichannel activity. Cell. Mol. Life Sci. 2018, 75, 2059–2073. [Google Scholar] [CrossRef]
- Iyyathurai, J.; D’Hondt, C.; Wang, N.; De Bock, M.; Himpens, B.; Retamal, M.A.; Stehberg, J.; Leybaert, L.; Bultynck, G. Peptides and peptide-derived molecules targeting the intracellular domains of Cx43: Gap junctions versus hemichannels. Neuropharmacol. 2013, 75, 491–505. [Google Scholar] [CrossRef]
- De Bock, M.; Wang, N.; Decrock, E.; Bultynck, G.; Leybaert, L. Intracellular cleavage of the Cx43 C-terminal domain by matrix-metalloproteases: A novel contributor to inflammation? Mediat. Inflamm. 2015, 2015, 257471. [Google Scholar] [CrossRef]
- Lopez, W.; Ramachandran, J.; Alsamarah, A.; Luo, Y.; Harris, A.L.; Contreras, J.E. Mechanism of gating by calcium in connexin hemichannels. Proc. Natl. Acad. Sci. USA 2016, 113, E7986–E7995. [Google Scholar] [CrossRef]
- Cotrina, M.L.; Lin, J.H.-C.; Nedergaard, M. Adhesive properties of connexin hemichannels. Glia 2008, 56, 1791–1798. [Google Scholar] [CrossRef]
- Lin, J.H.-C.; Takano, T.; Cotrina, M.L.; Arcuino, G.; Kang, J.; Liu, S.; Gao, Q.; Jiang, L.; Li, F.; Lichtenberg-Frate, H.; et al. Connexin 43 Enhances the Adhesivity and Mediates the Invasion of Malignant Glioma Cells. J. Neurosci. 2002, 22, 4302–4311. [Google Scholar] [CrossRef]
- Elias, L.A.; Wang, D.D.; Kriegstein, A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nature 2007, 448, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Ludidi, S.; Jonkers, D.; Elamin, E.; Pieters, H.-J.; Schaepkens, E.; Bours, P.; Kruimel, J.; Conchillo, J.; Masclee, A. The Intestinal Barrier in Irritable Bowel Syndrome: Subtype-Specific Effects of the Systemic Compartment in an In Vitro Model. PLOS ONE 2015, 10, e0123498. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Lee, A.; Hendargo, K.J.; Reddy, V.S.; Shlykov, M.A.; Kuppusamykrishnan, H.; Medrano-Soto, A.; Saier, M.H., Jr. Characterization of the Tetraspan Junctional Complex (4JC) superfamily. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- Sawada, N.; Murata, M.; Kikuchi, K.; Osanai, M.; Tobioka, H.; Kojima, T.; Chiba, H. Tight junctions and human diseases. Med Mol. Morphol. 2003, 36, 147–156. [Google Scholar] [CrossRef]
- Mandel, L.J.; Bacallao, R.; Zampighi, G. Uncoupling of the molecular ‘fence’ and paracellular ‘gate’ functions in epithelial tight junctions. Nature 1993, 361, 552–555. [Google Scholar] [CrossRef]
- Wade, J.B.; Karnovsky, M.J. The structure of the zonula occludens. A single fibril model based on freeze-fracture. J. Cell Biol. 1974, 60, 168–180. [Google Scholar] [CrossRef]
- Belardi, B.; Hamkins-Indik, T.; Harris, A.R.; Kim, J.; Xu, K.; Fletcher, D.A. A Weak Link with Actin Organizes Tight Junctions to Control Epithelial Permeability. Dev. Cell 2020, 54, 792–804. [Google Scholar] [CrossRef]
- Fanning, A.S.; Anderson, J.M. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct Binding of Three Tight Junction-Associated Maguks, Zo-1, Zo-2, and Zo-3, with the Cooh Termini of Claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef]
- Zemljic-Harpf, A.E.; Godoy, J.C.; Platoshyn, O.; Asfaw, E.K.; Busija, A.R.; Domenighetti, A.A.; Ross, R.S. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J. Cell Sci. 2014, 127, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Lye, M.F.; Fanning, A.S.; Su, Y.; Van Itallie, C.M.; Lavie, A. Insights into Regulated Ligand Binding Sites from the Structure of ZO-1 Src Homology 3-Guanylate Kinase Module. J. Biol. Chem. 2010, 285, 13907–13917. [Google Scholar] [CrossRef]
- Itoh, M.; Nagafuchi, A.; Moroi, S.; Tsukita, S. Involvement of ZO-1 in Cadherin-based Cell Adhesion through Its Direct Binding to α Catenin and Actin Filaments. J. Cell Biol. 1997, 138, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.C.; Zweimueller-Mayer, J.; Steinbacher, P.; Lametschwandtner, A. The Dual Role of Zonula Occludens (ZO) Proteins. J. Biomed. Biotechnol. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, D.; Le, S.; Laroche, T.; Mean, I.; Jond, L.; Yan, J.; Citi, S. Tension-Dependent Stretching Activates ZO-1 to Control the Junctional Localization of Its Interactors. Curr. Biol. 2017, 27, 3783–3795. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.J.; Zihni, C.; Ruppel, A.; Hartmann, C.; Ebnet, K.; Tada, M.; Balda, M.S.; Matter, K. Interplay between Extracellular Matrix Stiffness and JAM-A Regulates Mechanical Load on ZO-1 and Tight Junction Assembly. Cell Rep. 2020, 32, 107924. [Google Scholar] [CrossRef]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Buckley, A.; Turner, J.R. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Itoh, M.; Bissell, M.J. The organization of tight junctions in epithelia: Implications for mammary gland biology and breast tumorigenesis. J. Mammary Gland Biol. Neoplasia 2003, 8, 449–462. [Google Scholar] [CrossRef]
- Feldman, G.J.; Mullin, J.M.; Ryan, M.P. Occludin: Structure, function and regulation. Adv. Drug Deliv. Rev. 2005, 57, 883–917. [Google Scholar] [CrossRef]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.-D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex Phenotype of Mice Lacking Occludin, a Component of Tight Junction Strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Mariano, C.; Sasaki, H.; Brites, D.; Brito, M.A. A look at tricellulin and its role in tight junction formation and maintenance. Eur. J. Cell Biol. 2011, 90, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Staehelin, L.A. Further observations on the fine structure of freeze-cleaved tight junctions. J. Cell Sci. 1973, 13, 763–786. [Google Scholar] [PubMed]
- Walker, D.C.; MacKenzie, A.; Hosford, S. The structure of the tricellular region of endothelial tight junctions of pulmonary capillaries analyzed by freeze-fracture. Microvasc. Res. 1994, 48, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Sasaki, H.; Tsukita, S.; Furuse, M.; Tsukita, S. Loss of Occludin Affects Tricellular Localization of Tricellulin. Mol. Biol. Cell 2008, 19, 4687–4693. [Google Scholar] [CrossRef] [PubMed]
- Cording, J.; Berg, J.; Käding, N.; Bellmann, C.; Tscheik, C.; Westphal, J.K.; Milatz, S.; Günzel, D.; Wolburg, H.; Piontek, J.; et al. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J. Cell Sci. 2012, 126, 554–564. [Google Scholar] [CrossRef]
- Krug, S.M.; Amasheh, S.; Richter, J.F.; Milatz, S.; Günzel, D.; Westphal, J.K.; Huber, O.; Schulzke, J.D.; Fromm, M. Tricellulin Forms a Barrier to Macromolecules in Tricellular Tight Junctions without Affecting Ion Permeability. Mol. Biol. Cell 2009, 20, 3713–3724. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Anderson, J.M. Occludin confers adhesiveness when expressed in fibroblasts. J. Cell Sci. 1997, 110 Pt 9, 1113–1121. [Google Scholar]
- Buschmann, M.M.; Shen, L.; Rajapakse, H.; Raleigh, D.R.; Wang, Y.; Wang, Y.; Lingaraju, A.; Zha, J.; Abbott, E.; McAuley, E.M.; et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol. Biol. Cell 2013, 24, 3056–3068. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5–deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Günzel, D.; Krug, S.M.; Schulzke, J.-D.; Fromm, M.; Yu, A.S.L. Claudin-2-mediated cation and water transport share a common pore. Acta Physiol. 2016, 219, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Milatz, S.; Krug, S.M.; Oelrich, B.B.; Schulzke, J.-D.; Amasheh, S.; Günzel, D.; Fromm, M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Kluger, M.S.; Clark, P.R.; Tellides, G.; Gerke, V.; Pober, J.S. Claudin-5 Controls Intercellular Barriers of Human Dermal Microvascular but Not Human Umbilical Vein Endothelial Cells. Arter. Thromb. Vasc. Biol. 2013, 33, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Tai, C.-Y.; Mok, L.-P.; Mosser, E.A.; Schuman, E.M. Calcium-dependent dynamics of cadherin interactions at cell-cell junctions. Proc. Natl. Acad. Sci. USA 2011, 108, 9857–9862. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Nose, A.; Hatta, K.; Kawakami, A.; Takeichi, M. Calcium-dependent cell-cell adhesion molecules (cadherins): Subclass specificities and possible involvement of actin bundles. J. Cell Biol. 1987, 105, 2501–2510. [Google Scholar] [CrossRef]
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- Watabe-Uchida, M.; Uchida, N.; Imamura, Y.; Nagafuchi, A.; Fujimoto, K.; Uemura, T.; Vermeulen, S.; Van Roy, F.; Adamson, E.D.; Takeichi, M. α-Catenin-Vinculin Interaction Functions to Organize the Apical Junctional Complex in Epithelial Cells. J. Cell Biol. 1998, 142, 847–857. [Google Scholar] [CrossRef]
- Panorchan, P.; Thompson, M.S.; Davis, K.J.; Tseng, Y.; Konstantopoulos, K.; Wirtz, D. Single-molecule analysis of cadherin-mediated cell-cell adhesion. J. Cell Sci. 2006, 119, 66–74. [Google Scholar] [CrossRef]
- Liu, Z.; Tan, J.L.; Cohen, D.M.; Yang, M.T.; Sniadecki, N.J.; Ruiz, S.A.; Nelson, C.M.; Chen, C.S. Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA 2010, 107, 9944–9949. [Google Scholar] [CrossRef]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef] [PubMed]
- Merkel, C.D.; Li, Y.; Raza, Q.; Stolz, D.B.; Kwiatkowski, A.V. Vinculin anchors contractile actin to the cardiomyocyte adherens junction. Mol. Biol. Cell 2019, 30, 2639–2650. [Google Scholar] [CrossRef] [PubMed]
- Bertocchi, C.; Ravasio, A.; Ong, H.T.; Toyama, Y.; Kanchanawong, P. Mechanical roles of vinculin/β-catenin interaction in adherens junction. bioRxiv 2019, 770735. [Google Scholar] [CrossRef]
- Higashi, T.; Arnold, T.R.; Stephenson, R.E.; Dinshaw, K.M.; Miller, A.L. Maintenance of the Epithelial Barrier and Remodeling of Cell-Cell Junctions during Cytokinesis. Curr. Biol. 2016, 26, 1829–1842. [Google Scholar] [CrossRef]
- Chugh, P.; Paluch, E.K. The actin cortex at a glance. J. Cell Sci. 2018, 131, jcs186254. [Google Scholar] [CrossRef]
- Belvitch, P.; Htwe, Y.M.; Brown, M.E.; Dudek, S. Cortical actin dynamics in endothelial permeability. Curr. Top. Membr. 2018, 82, 141–195. [Google Scholar]
- Burridge, K.; Wittchen, E.S. The tension mounts: SFs as force-generating mechanotransducers. J. Cell Biol. 2013, 200, 9–19. [Google Scholar] [CrossRef]
- Rodgers, L.S.; Beam, M.T.; Anderson, J.M.; Fanning, A.S. Epithelial barrier assembly requires coordinated activity of multiple domains of the tight junction protein ZO-1. J. Cell Sci. 2013, 126, 1565–1575. [Google Scholar] [CrossRef]
- Rodgers, L.S.; Fanning, A.S. Regulation of epithelial permeability by the actin cytoskeleton. Cytoskeleton 2011, 68, 653–660. [Google Scholar] [CrossRef]
- Belvitch, P.; Brown, M.E.; Brinley, B.N.; Letsiou, E.; Rizzo, A.N.; Garcia, J.G.N.; Dudek, S.M. The ARP 2/3 complex mediates endothelial barrier function and recovery. Pulm. Circ. 2017, 7, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Dominguez, R. Regulation of actin cytoskeleton dynamics in cells. Mol. Cells 2010, 29, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Tanislav, C.; Troidl, C.; Schulz, R.; Hamm, C.; Gündüz, D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol. Rep. 2014, 2, e12175. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Springer, T.A. Trans.-cellular migration: Cell-cell contacts get intimate. Curr. Opin. Cell Biol. 2008, 20, 533–540. [Google Scholar] [CrossRef]
- Yu, D.; Marchiando, A.M.; Weber, C.R.; Raleigh, D.R.; Wang, Y.; Shen, L.; Turner, J.R. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc. Natl. Acad. Sci. USA 2010, 107, 8237–8241. [Google Scholar] [CrossRef]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2019, 9, 1942. [Google Scholar] [CrossRef]
- Kugelmann, D.; Rotkopf, L.T.; Radeva, M.Y.; Garcia-Ponce, A.; Walter, E.; Waschke, J. Histamine causes endothelial barrier disruption via Ca2+-mediated RhoA activation and tension at adherens junctions. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Mehta, D.; Tiruppathi, C.; Sandoval, R.; Minshall, R.D.; Holinstat, M.; Malik, A.B. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J. Physiol. 2002, 539, 779–789. [Google Scholar] [CrossRef]
- Bogatcheva, N.V.; Zemskova, M.A.; Kovalenkov, Y.; Poirier, C.; Verin, A.D. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J. Cell. Physiol. 2009, 221, 750–759. [Google Scholar] [CrossRef]
- Thompson, P.M.; Tolbert, C.E.; Shen, K.; Kota, P.; Palmer, S.M.; Plevock, K.M.; Orlova, A.; Galkin, V.E.; Burridge, K.; Egelman, E.H.; et al. Identification of an Actin Binding Surface on Vinculin that Mediates Mechanical Cell and Focal Adhesion Properties. Structure 2014, 22, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Chinthalapudi, K.; Rangarajan, E.S.; Izard, T. The interaction of talin with the cell membrane is essential for integrin activation and focal adhesion formation. Proc. Natl. Acad. Sci. USA 2018, 115, 10339–10344. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Guilluy, C. Focal adhesions, SFs and mechanical tension. Exp. Cell Res. 2016, 343, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Hotulainen, P.; Lappalainen, P. SFs are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 2006, 173, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kassianidou, E.; Kumar, S. Actomyosin SF subtypes have unique viscoelastic properties and roles in tension generation. Mol. Biol. Cell 2018, 29, 1992–2004. [Google Scholar] [CrossRef]
- Parsons, J.T.; Martin, K.H.; Slack, J.K.; Taylor, J.M.; A Weed, S. Focal Adhesion Kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 2000, 19, 5606–5613. [Google Scholar] [CrossRef]
- Birukova, A.A.; Shah, A.S.; Tian, Y.; Gawlak, G.; Sarich, N.; Birukov, K.G. Selective Role of Vinculin in Contractile Mechanisms of Endothelial Permeability. Am. J. Respir. Cell Mol. Biol. 2016, 55, 476–486. [Google Scholar] [CrossRef]
- Huveneers, S.; Oldenburg, J.; Spanjaard, E.; Van Der Krogt, G.; Grigoriev, I.; Akhmanova, A.; Rehmann, H.; De Rooij, J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 2012, 196, 641–652. [Google Scholar] [CrossRef]
- Xie, X.; Chen, C.; Huang, K.; Wang, S.; Hao, J.; Huang, J.; Huang, H. RhoA/rho kinase signaling reduces connexin43 expression in high glucose-treated glomerular mesangial cells with zonula occludens-1 involvement. Exp. Cell Res. 2014, 327, 276–286. [Google Scholar] [CrossRef]
- De Bock, M.; Culot, M.; Wang, N.; Da Costa, A.; Decrock, E.; Bol, M.; Bultynck, G.; Cecchelli, R.; Leybaert, L. Low extracellular Ca2+ conditions induce an increase in brain endothelial permeability that involves intercellular Ca2+ waves. Brain Res. 2012, 1487, 78–87. [Google Scholar] [CrossRef]
- De Bock, M.; Vandenbroucke, R.E.; Decrock, E.; Culot, M.; Cecchelli, R.; Leybaert, L. A new angle on blood-CNS interfaces: A role for connexins? FEBS Lett. 2014, 588, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Decrock, E.; Bol, M.; Gadicherla, A.K.; Culot, M.; Cecchelli, R.; Bultynck, G.; Leybaert, L. Endothelial calcium dynamics, connexin channels and blood–brain barrier function. Prog. Neurobiol. 2013, 108, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Goeckeler, Z.; Wysolmerski, R. Myosin light chain kinase-regulated endothelial cell contraction: The relationship between isometric tension, actin polymerization, and myosin phosphorylation. J. Cell Biol. 1995, 130, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Garcia, J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.J., 3rd; Birukova, A.A.; Beyer, E.C.; Birukov, K.G. Gap junction protein connexin43 exacerbates lung vascular permeability. PLoS ONE 2014, 9, e100931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Han, Y.; Wang, B.; Li, S.; Gan, W. Beyond gap junction channel function: The expression of Cx43 contributes to aldosterone-induced mesangial cell proliferation via the ERK1/2 and PKC pathways. Cell Physiol. Biochem. 2015, 36, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Polontchouk, L.; Ebelt, B.; Jackels, M.; Dhein, S. Chronic effects of endothelin-1 and angiotensin-II on gap junctions and intercellular communication in cardiac cells. FASEB J. 2001, 16, 1–15. [Google Scholar] [CrossRef]
- Koyama, Y.; Baba, A. Endothelin-induced cytoskeletal actin re-organization in cultured astrocytes: Inhibition by C3 ADP-ribosyltransferase. Glia 1996, 16, 342–350. [Google Scholar] [CrossRef]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef]
- Nagasawa, K.; Chiba, H.; Fujita, H.; Kojima, T.; Saito, T.; Endo, T.; Sawada, N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell. Physiol. 2006, 208, 123–132. [Google Scholar] [CrossRef]
- Simon, A.M.; McWhorter, A.R. Vascular abnormalities in mice lacking the endothelial gap junction proteins connexin37 and connexin40. Dev. Biol. 2002, 251, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Salmon, M.; Regnault, B.; Cayet, N.; Caille, R.; DeMuth, K.; Hardelin, J.-P.; Janel, N.; Meda, P.; Petit, C. Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc. Natl. Acad. Sci. 2007, 104, 6229–6234. [Google Scholar] [CrossRef] [PubMed]
- Ezan, P.; André, P.; Cisternino, S.; Saubaméa, B.; Boulay, A.-C.; Doutremer, S.; Thomas, M.-A.; Quenech’du, N.; Giaume, C.; Cohen-Salmon, M. Deletion of astroglial connexins weakens the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Katsuno, T.; Hoshimoto, A.; Hirano, N.; Saito, Y.; Suzuki, Y. Connexin 26-mediated gap junctional intercellular communication suppresses paracellular permeability of human intestinal epithelial cell monolayers. Exp. Cell Res. 2004, 298, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Sanderson, M.J. Intercellular Ca(2+) waves: Mechanisms and function. Physiol. Rev. 2012, 92, 1359–1392. [Google Scholar] [CrossRef]
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium oscillations. Biochem. Soc. Symp. 2007, 74, 1–7. [Google Scholar] [CrossRef]
- Pearson, R.A.; Dale, N.; Llaudet, E.; Mobbs, P. ATP Released via Gap Junction Hemichannels from the Pigment Epithelium Regulates Neural Retinal Progenitor Proliferation. Neuron 2005, 46, 731–744. [Google Scholar] [CrossRef]
- Arcuino, G.; Lin, J.H.-C.; Takano, T.; Liu, C.; Jiang, L.; Gao, Q.; Kang, J.; Nedergaard, M. Intercellular calcium signaling mediated by point-source burst release of ATP. Proc. Natl. Acad. Sci. USA 2002, 99, 9840–9845. [Google Scholar] [CrossRef]
- Cotrina, M.L.; Lin, J.H.-C.; Alves-Rodrigues, A.; Liu, S.; Li, J.; Azmi-Ghadimi, H.; Kang, J.; Naus, C.C.G.; Nedergaard, M. Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. USA 1998, 95, 15735–15740. [Google Scholar] [CrossRef]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; Da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin Channels Provide a Target to Manipulate Brain Endothelial Calcium Dynamics and Blood—Brain Barrier Permeability. Br. J. Pharmacol. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [PubMed]
- Delvaeye, T.; De Smet, M.A.J.; Verwaerde, S.; Decrock, E.; Czekaj, A.; Vandenbroucke, R.E.; Lemeire, K.; Gonçalves, A.; Declercq, W.; Vandenabeele, P.; et al. Blocking connexin43 hemichannels protects mice against tumour necrosis factor-induced inflammatory shock. Sci. Rep. 2019, 9, 16623. [Google Scholar] [CrossRef]
- Palatinus, J.A.; Rhett, J.M.; Gourdie, R.G. Enhanced PKCε mediated phosphorylation of connexin43 at serine 368 by a carboxyl-terminal mimetic peptide is dependent on injury. Channels 2011, 5, 236–240. [Google Scholar] [CrossRef]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A Peptide Mimetic of the Connexin43 Carboxyl-Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.S.; Dunn, C.A.; Carter, W.G.; Usui, M.L.; Olerud, J.E.; Lampe, P.D. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J. Cell Biol. 2004, 167, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Manjarrez-Marmolejo, J.; Franco-Pérez, J. Gap junction blockers: An. overview of their effects on induced seizures in animal models. Curr. Neuropharmacol. 2016, 14, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Maass, K.; Ghanem, A.; Kim, J.-S.; Saathoff, M.; Urschel, S.; Kirfel, G.; Grümmer, R.; Kretz, M.; Lewalter, T.; Tiemann, K.; et al. Defective Epidermal Barrier in Neonatal Mice Lacking the C-Terminal Region of Connexin43. Mol. Biol. Cell 2004, 15, 4597–4608. [Google Scholar] [CrossRef]
- Obert, E.; Strauss, R.; Brandon, C.; Grek, C.; Ghatnekar, G.; Gourdie, R.; Rohrer, B. Targeting the tight junction protein, zonula occludens-1, with the connexin43 mimetic peptide, αCT1, reduces VEGF-dependent RPE pathophysiology. J. Mol. Med. 2017, 95, 535–552. [Google Scholar] [CrossRef]
- Butkevich, E.; Hülsmann, S.; Wenzel, D.; Shirao, T.; Duden, R.; Majoul, I. Drebrin Is a Novel Connexin-43 Binding Partner that Links Gap Junctions to the Submembrane Cytoskeleton. Curr. Biol. 2004, 14, 650–658. [Google Scholar] [CrossRef]
- Ambrosi, C.; Ren, C.; Spagnol, G.; Cavin, G.; Cone, A.; Grintsevich, E.E.; Sosinsky, G.E.; Sorgen, P.L. Connexin43 Forms Supramolecular Complexes through Non-Overlapping Binding Sites for Drebrin, Tubulin, and ZO-1. PLOS ONE 2016, 11, e0157073. [Google Scholar] [CrossRef]
- Sorgen, P.L.; Trease, A.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein⁻Protein Interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Lampe, P.D. Identification of connexin-43 interacting proteins. Cell Commun. Adhes. 2003, 10, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Francis, R.; Wei, C.J.; Linask, K.L.; Lo, C.W. Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development 2006, 133, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.G.; Zhu, D.; Todd, S.D.L.; Kidder, G.M. Characteristics of C6 glioma cells overexpressing a gap junction protein. Cell. Mol. Neurobiol. 1992, 12, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Ionta, M.; Ferreira, R.A.S.; Pfister, S.C.; Machado-Santelli, G.M. Exogenous Cx43 expression decrease cell proliferation rate in rat hepatocarcinoma cells independently of functional gap junction. Cancer Cell Int. 2009, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, M.L.; Lin, J.H.-C.; López-García, J.C.; Naus, C.C.G.; Nedergaard, M. ATP-Mediated Glia Signaling. J. Neurosci. 2000, 20, 2835–2844. [Google Scholar] [CrossRef]
- Wall, M.E.; Otey, C.; Qi, J.; Banes, A.J. Connexin 43 is localized with actin in tenocytes. Cell Motil. Cytoskelet. 2007, 64, 121–130. [Google Scholar] [CrossRef]
- Basheer, W.; Shaw, R. The “tail” of Connexin43: An. unexpected journey from alternative translation to trafficking. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1848–1856. [Google Scholar] [CrossRef]
- Basheer, W.A.; Xiao, S.; Epifantseva, I.; Fu, Y.; Kleber, A.G.; Hong, T.; Shaw, R.M. GJA1-20k Arranges Actin to Guide Cx43 Delivery to Cardiac Intercalated Discs. Circ. Res. 2017, 121, 1069–1080. [Google Scholar] [CrossRef]
- Ponsaerts, R.; De Vuyst, E.; Retamal, M.; D’Hondt, C.; Vermeire, D.; Wang, N.; De Smedt, H.; Zimmermann, P.; Himpens, B.; Vereecke, J.; et al. Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 2010, 24, 4378–4395. [Google Scholar] [CrossRef] [PubMed]
- Kameritsch, P.; Kiemer, F.; Beck, H.; Pohl, U.; Pogoda, K. Cx43 increases serum induced filopodia formation via activation of p21-activated protein kinase 1. Biochim. et Biophys. Acta (BBA) Bioenerg. 2015, 1853, 2907–2917. [Google Scholar] [CrossRef] [PubMed]
- Ponce, A.G.; Madrid, A.F.C.; Robles, H.V.; Paredes, S.C.; Nava, P.; Betanzos, A.; Zarbock, A.; Rottner, K.; Vestweber, D.; Schnoor, M. Loss of cortactin causes endothelial barrier dysfunction via disturbed adrenomedullin secretion and actomyosin contractility. Sci. Rep. 2016, 6, 29003. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D.; Zeuschner, D.; Rottner, K.; Schnoor, M. Cortactin regulates the activity of small GTPases and ICAM-1 clustering in endothelium: Implications for the formation of docking structures. Tissue Barriers 2013, 1, e23862. [Google Scholar] [CrossRef][Green Version]
- Li, B.; He, J.; Lv, H.; Liu, Y.; Lv, X.; Zhang, C.; Zhu, Y.; Ai, D. c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J. Clin. Investig. 2019, 129, 1167–1179. [Google Scholar] [CrossRef]
- Batra, N.; Burra, S.; Siller-Jackson, A.J.; Gu, S.; Xia, X.; Weber, G.F.; DeSimone, D.; Bonewald, L.F.; Lafer, E.M.; Sprague, E.; et al. Mechanical stress-activated integrin α5β1 induces opening of connexin 43 hemichannels. Proc. Natl. Acad. Sci. USA 2012, 109, 3359–3364. [Google Scholar] [CrossRef]
- Shi, Y.; Li, R.; Yang, J.; Li, X. No tight junctions in tight junction protein-1 expressing HeLa and fibroblast cells. Int. J. Physiol. Pathophysiol. Pharmacol. 2020, 12, 70–78. [Google Scholar]
- Fischer, R.S.; Lam, P.-Y.; Huttenlocher, A.; Waterman, C.M. Filopodia and focal adhesions: An integrated system driving branching morphogenesis in neuronal pathfinding and angiogenesis. Dev. Biol. 2019, 451, 86–95. [Google Scholar] [CrossRef]
- Massoumi, R.; Sjolander, A. Leukotriene D(4) affects localisation of vinculin in intestinal epithelial cells via distinct tyrosine kinase and protein kinase C controlled events. J. Cell Sci. 2001, 114, 1925–1934. [Google Scholar]
- Fogh, B.S.; Multhaupt, H.A.B.; Couchman, J.R. Protein kinase C, focal adhesions and the regulation of cell migration. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2014, 62, 172–184. [Google Scholar] [CrossRef]
- Husøy, T.; Cruciani, V.; Sanner, T.; Mikalsen, S.-O. Phosphorylation of connexin43 and inhibition of gap junctional communication in 12-O-tetradecanoylphorbol-13-acetate-exposed R6 fibroblasts: Minor role of protein kinase CβI and μ. Carcinogenesis 2001, 22, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J.; Mayr-Wohlfart, U.; Kessler, S.; Breitig, D.; Günther, K.-P. Adsorption and release properties of growth factors from biodegradable implants. J. Biomed. Mater. Res. 2001, 59, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Long, B.; Liu, X.; Li, Y.; Han, N.; Zhao, P.; Chen, W. Effects of sevoflurane on tight junction protein expression and PKC-α translocation after pulmonary ischemia–reperfusion injury. Exp. Mol. Med. 2015, 47, e167. [Google Scholar] [CrossRef] [PubMed]
- Rosson, D.; O’Brien, T.G.; Kampherstein, J.A.; Szallasi, Z.; Bogi, K.; Blumberg, P.M.; Mullin, J.M. Protein Kinase C-α Activity Modulates Transepithelial Permeability and Cell Junctions in the LLC-PK1Epithelial Cell Line. J. Biol. Chem. 1997, 272, 14950–14953. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.; Soler, A.P.; Mullin, J.M. Protein kinase C activation leads to dephosphorylation of occludin and tight junction permeability increase in LLC-PK1 epithelial cell sheets. J. Cell Sci. 2000, 113 Pt 18, 3187–3196. [Google Scholar]
- Letiges, M. Knockout of PKC Enhances Insulin Signaling Through PI3K. Mol. Endocrinol. 2002, 16, 847–858. [Google Scholar] [CrossRef]
- Hsu, A.H.; Lum, M.A.; Shim, K.-S.; Frederick, P.J.; Morrison, C.D.; Chen, B.; Lele, S.M.; Sheinin, Y.M.; Daikoku, T.; Dey, S.K.; et al. Crosstalk between PKCα and PI3K/AKT Signaling Is Tumor Suppressive in the Endometrium. Cell Rep. 2018, 24, 655–669. [Google Scholar] [CrossRef]
- Bruewer, M.; Hopkins, A.M.; Hobert, M.E.; Nusrat, A.; Madara, J.L. RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am. J. Physiol. Physiol. 2004, 287, C327–C335. [Google Scholar] [CrossRef]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef]
- Khan, N.; Binder, L.; Pantakani, D.V.K.; Asif, A.R. MPA Modulates Tight Junctions’ Permeability via Midkine/PI3K Pathway in Caco-2 Cells: A Possible Mechanism of Leak-Flux Diarrhea in Organ Transplanted Patients. Front. Physiol. 2017, 8, 438. [Google Scholar] [CrossRef]
- Beier, F.; Loeser, R.F. Biology and pathology of Rho GTPase, PI-3 kinase-Akt, and MAP kinase signaling pathways in chondrocytes. J. Cell. Biochem. 2010, 110, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.N.; Berthiaume, L.G.; Kikuchi, Y.; Begg, D.; Bourgoin, S.; Brindley, D.N. Tumor necrosis factor-alpha induces SF formation through ceramide production: Role of sphingosine kinase. Mol. Biol. Cell 2001, 12, 3618–3630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Fang, Z.; Wang, G.; Wu, S. Gap junctional intercellular communication dysfunction mediates the cognitive impairment induced by cerebral ischemia-reperfusion injury: PI3K/Akt pathway involved. Am. J. Transl. Res. 2017, 9, 5442–5451. [Google Scholar]
- Bhattacharjee, R.; Kaneda, M.; Nakahama, K.-I.; Morita, I. The steady-state expression of connexin43 is maintained by the PI3K/Akt in osteoblasts. Biochem. Biophys. Res. Commun. 2009, 382, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Qin, G.; Luo, M.; Chen, J.; Zhang, Q.; Li, L.; Pan, L.; Qin, S. Reciprocal positive regulation between Cx26 and PI3K/Akt pathway confers acquired gefitinib resistance in NSCLC cells via GJIC-independent induction of EMT. Cell Death Dis. 2015, 6, e1829. [Google Scholar] [CrossRef]
- Ishikawa, S.; Kuno, A.; Tanno, M.; Miki, T.; Kouzu, H.; Itoh, T.; Sato, T.; Sunaga, D.; Murase, H.; Miura, T. Role of connexin-43 in protective PI3K-Akt-GSK-3β signaling in cardiomyocytes. Am. J. Physiol. Circ. Physiol. 2012, 302, H2536–H2544. [Google Scholar] [CrossRef]
- Murphy, S.F.; Varghese, R.T.; Lamouille, S.; Guo, S.; Pridham, K.J.; Kanabur, P.; Osimani, A.; Sharma, S.; Jourdan, J.; Rodgers, C.M.; et al. Connexin 43 inhibition sensitizes chemoresistant glioblastoma cells to temozolomide. Cancer Res. 2016, 76, 139–149. [Google Scholar] [CrossRef]
- Janoštiak, R.; Pataki, A.C.; Brábek, J.; Rösel, D. Mechanosensors in integrin signaling: The emerging role of p130Cas. Eur. J. Cell Biol. 2014, 93, 445–454. [Google Scholar] [CrossRef]
- Riquelme, M.A.; Cardenas, E.R.; Xu, H.; Jiang, J.X. The role of connexin channels in the response of mechanical loading and unloading of bone. Int. J. Mol. Sci. 2020, 21, 1146. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Speacht, T.L.; Donahue, H.J. Cx43 and mechanotransduction in bone. Curr. Osteoporos. Rep. 2015, 13, 67–72. [Google Scholar] [CrossRef]
- Gerilechaogetu, F.; Feng, H.; Golden, H.; Nizamutdinov, D.; Dostal, J.; Jacob, J.; Afroze, S.; Foster, D.; Bowman, J.; Ochoa, B. Current concepts in the role of mechanosensing in the regulation of cardiac contractile function. Austin. J. Clin. Med. 2014, 1, 11015. [Google Scholar]
- Geng, W.; Bošković, G.; Fultz, M.; Li, C.; Niles, R.; Ohno, S.; Wright, G.L. Regulation of expression and activity of four PKC isozymes in confluent and mechanically stimulated UMR-108 osteoblastic cells. J. Cell. Physiol. 2001, 189, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.; Richardson, W.J.; Rhett, J.M.; Bustos, F.; Degen, K.; Ghatnekar, G.S.; Grek, C.L.; Marsh, S.; Jourdan, L.J.; Holmes, J.W.; et al. The connexin 43 carboxyl terminal mimetic peptide αCT1 prompts differentiation of a collagen scar matrix resembling unwounded skin. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sugawara, M.; Miyoshi, H.; Miura, T.; Tanaka, H.; Tsubota, K.-I.; Liu, H. Dynamics of actin SFs and focal adhesions during slow migration in swiss 3T3 fibroblasts: Intracellular mechanism of cell turning. BioMed Res. Int. 2016, 2016, 5749749. [Google Scholar] [CrossRef]
- Pellegrin, S.; Mellor, H. Actin stress fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef]
- Inaki, M.; Liu, J.; Matsuno, K. Cell chirality: Its origin and roles in left-right asymmetric development. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2016, 371, 20150403. [Google Scholar] [CrossRef]
- Inaki, M.; Sasamura, T.; Matsuno, K. Cell chirality drives left-right asymmetric morphogenesis. Front. Cell Dev. Biol. 2018, 6, 34. [Google Scholar] [CrossRef]
- Wan, L.Q.; Chin, A.S.; Worley, K.E.; Ray, P. Cell chirality: Emergence of asymmetry from cell culture. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150413. [Google Scholar] [CrossRef]
- Wan, L.Q.; Ronaldson, K.; Park, M.; Taylor, G.; Zhang, Y.; Gimble, J.M.; Vunjak-Novakovic, G. Micropatterned mammalian cells exhibit phenotype-specific left-right asymmetry. Proc. Natl. Acad. Sci. USA 2011, 108, 12295–12300. [Google Scholar] [CrossRef]
- Tee, Y.H.; Shemesh, T.; Thiagarajan, V.; Hariadi, R.F.; Anderson, K.L.; Page, C.; Volkmann, N.; Hanein, D.; Sivaramakrishnan, S.; Kozlov, M.M.; et al. Cellular chirality arising from the self-organization of the actin cytoskeleton. Nat. Cell Biol. 2015, 17, 445–457. [Google Scholar] [CrossRef]
- Naganathan, S.R.; Fürthauer, S.; Nishikawa, M.; Jülicher, F.; Grill, S.W. Active torque generation by the actomyosin cell cortex drives left–right symmetry breaking. eLife 2014, 3, e04165. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Uemura, S.; Adachi, K.; Itoh, H.; Kinosita, K.; Ishiwata, S. Myosin V is a left-handed spiral motor on the right-handed actin helix. Nat. Genet. 2002, 9, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ray, P.; Lu, Y.W.; Kaur, G.; Schwarz, J.J.; Wan, L.Q. Cell chirality regulates intercellular junctions and endothelial permeability. Sci. Adv. 2018, 4, eaat2111. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D.Y.; Zhao, X.-Q.; Francis, R.; Huang, G.Y.; Mably, J.D.; Lo, C.W. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Dev. 2009, 136, 3185–3193. [Google Scholar] [CrossRef]
- Xu, X.; Li, W.; Huang, G.; Meyer, R.; Chen, T.; Luo, Y.; Thomas, M.; Radice, G.; Lo, C. Modulation of mouse neural crest cell motility by N-cadherin and connexin 43 gap junctions. J. Cell Biol. 2001, 154, 217–230. [Google Scholar] [CrossRef]
- Ewart, J.L.; Cohen, M.F.; A Meyer, R.; Huang, G.Y.; Wessels, A.; Gourdie, R.G.; Chin, A.J.; Park, S.M.; O Lazatin, B.; Villabon, S.; et al. Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development 1997, 124, 1281–1292. [Google Scholar]
- Levin, M.; Nascone, N. Two molecular models of initial left-right asymmetry generation. Med. Hypotheses 1997, 49, 429–435. [Google Scholar] [CrossRef]
- Britz-Cunningham, S.H.; Shah, M.M.; Zuppan, C.W.; Fletcher, W.H. Mutations of theConnexin43Gap-Junction Gene in Patients with Heart Malformations and Defects of Laterality. N. Engl. J. Med. 1995, 332, 1323–1330. [Google Scholar] [CrossRef]
- TenBroek, E.M.; Lampe, P.D.; Solan, J.L.; Reynhout, J.K.; Johnson, R.G. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J. Cell Biol. 2001, 155, 1307–1318. [Google Scholar] [CrossRef]
- Chen, W.C.; Tsai, F.J.; Wu, J.Y.; Wu, H.C.; Li, C.W. Does Ser364Pro mutation of connexin 43 exist in Taiwanese patients with Ivemark syndrome? Zhonghua Yi Xue Za Zhi 2000, 63, 691–695. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strauss, R.E.; Gourdie, R.G. Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. Biomolecules 2020, 10, 1656. https://doi.org/10.3390/biom10121656
Strauss RE, Gourdie RG. Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. Biomolecules. 2020; 10(12):1656. https://doi.org/10.3390/biom10121656
Chicago/Turabian StyleStrauss, Randy E., and Robert G. Gourdie. 2020. "Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation" Biomolecules 10, no. 12: 1656. https://doi.org/10.3390/biom10121656
APA StyleStrauss, R. E., & Gourdie, R. G. (2020). Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. Biomolecules, 10(12), 1656. https://doi.org/10.3390/biom10121656