Dissecting the Cytochrome P450 OleP Substrate Specificity: Evidence for a Preferential Substrate

 , , , ,
, , , ,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. OleP Expression and Purification
2.3. Equilibrium Binding Analysis
2.4. Crystallization, X-Ray Data Collection, and Analysis
2.5. Structure Determination and Refinement
2.6. Molecular Docking
3. Results and Discussion
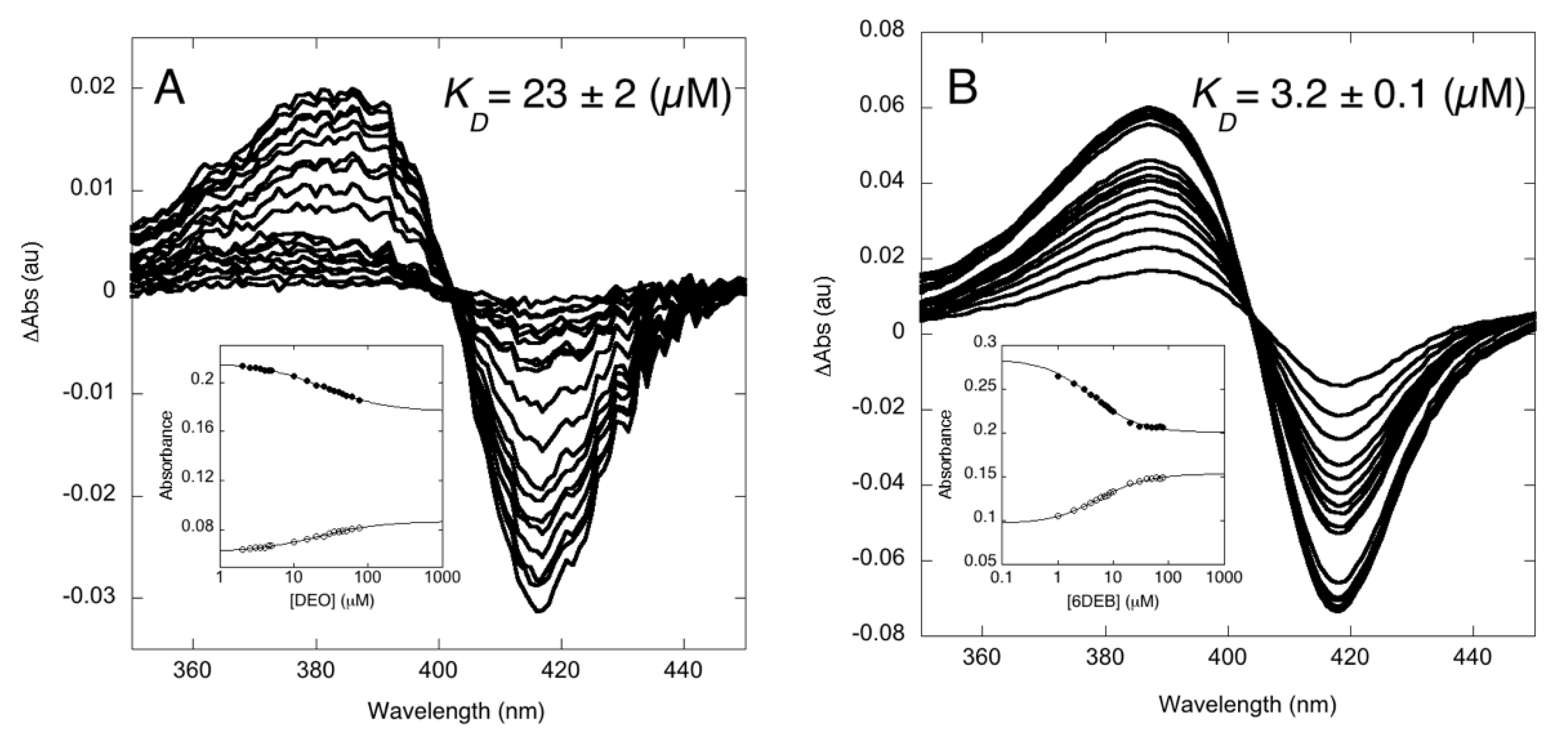
3.1. Binding Properties of OleP to the Aglycone Substrate
3.2. The Structure of OleP Bound to the Oleandolide, DEO
3.3. Docking of L-O-DEO and Crystallographic Structure of OleP Bound to DEO and L-Rhamnose
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McLean, K.J.; Sabri, M.; Marshall, K.R.; Lawson, R.J.; Lewis, D.G.; Clift, D.; Balding, P.R.; Warman, A.J.; McVey, J.P.; Quinn, A.-M.; et al. Biodiversity of cytochrome P450 redox systems. Biochem. Soc. Trans. 2005, 33, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.L.; Lamb, D.C.; Kelly, D.E. Cytochrome P450 biodiversity and biotechnology. Biochem. Soc. Trans. 2006, 34, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Nagini, S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure Mechanism and Biochemistry, 4th ed.; Kluwer Academic/Plenum Publisher: New York, NY, USA, 2015. [Google Scholar]
- Coon, M.C. CYTOCHROME P450: Nature's most versatile biological catalyst. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 1–25. [Google Scholar] [CrossRef]
- Grogan, G. Cytochomes P450: Exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, Y.; Guengerich, F.P.; Ma, L.; Li, S.; Zhang, W. Engineering cytochrome P450 enzyme systems for biomedical and biotechnological applications. J. Biol. Chem. 2020, 295, 833–849. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schilichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Olano, C.; Mendez, C.; Hutchinson, C.R.; Salas, J.A. A cytochrome P450-like gene possibly involved in oleandomycin biosynthesis in Streptomyces antibioticus. FEMS Microbiol. Lett. 1995, 127, 117–120. [Google Scholar] [CrossRef]
- Grobe, S.; Wszołek, A.; Brundiek, H.; Fekete, M.; Bornscheuer, U.T. Highly selective bile acid hydroxylation by the multifunctional bacterial P450 monooxygenase CYP107D1 (OleP). Biotechnol. Lett. 2020, 42, 819–824. [Google Scholar] [CrossRef]
- Agematu, H.; Matsumoto, N.; Fujii, Y.; Kabumoto, H.; Doi, S.; Machida, K.; Ishikawa, J.; Arisawa, A. Hydroxylation of testosterone by bacterial cytochromes P450 using the Escherichia coli expression system. Biosci. Biotechnol. Biochem. 2006, 70, 307–311. [Google Scholar] [CrossRef]
- Gaisser, S.; Lill, R.; Staunton, J.; Mendez, C.; Salas, J.A.; Leadlay, P.F. Parallel pathways for oxydation of 14-membered polyketide macrolactones in Saccharopolyspora erythraea. Mol. Microbiol. 2002, 44, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Xue, Q.; Tang, L.; Carney, J.R.; Betlach, M.; McDaniel, R. Cloning, characterization and heterologous expression of a polyketide synthase and a P-450 oxidase involved in the biosynthesis of the antibiotic oleandomycin. J. Antibiot. 2000, 53, 502–508. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tatsuta, K.; Gunji, H.; Tajima, S.; Ishiyama, T.; Imai, S.; Okuyama, S.; Fukatsu, S. Biosynthetic studies on oleandomycin by incorporation of the chemically synthesized aglycones. J. Antibiot. 1990, 43, 909–911. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Rodriguez, D.; Olano, C.; Brana, A.F.; Mendez, C.; Salas, J.A. Functional analysis of OleY 1-oleandrosyl 3-O-methyltransferase of the oleandomycin biosynthetic pathway in Streptomyces antibioticus. J. Bacteriol. 2001, 183, 5358–5363. [Google Scholar] [CrossRef] [PubMed]
- Lambalot, R.H.; Cane, D.E.; Aparicio, J.J.; Katz, L. Overproduction and characterization of the erythromycin C-12 hydroxylase, EryK. Biochemistry 1995, 34, 1858–1866. [Google Scholar] [CrossRef]
- Savino, C.; Montemiglio, L.C.; Sciara, G.; Miele, A.E.; Kendrew, S.G.; Jemth, P.; Gianni, S.; Vallone, B. Investigating the structural plasticity of a cytochrome P450: Three-dimensional structures of P450 EryK and binding to its physiological substrate. J. Biol. Chem. 2009, 284, 29170–29179. [Google Scholar] [CrossRef]
- DeMars, M.D.n.; Samora, N.L.; Yang, S.; Garcia-Borràs, M.; Sanders, J.N.; Houk, K.N.; Podust, L.M.; Sherman, D.H. Exploring the molecular basis for substrate specificity in homologous macrolide biosynthetic cytochromes P450. J. Biol. Chem. 2019, 294, 15947–15961. [Google Scholar] [CrossRef]
- Montemiglio, L.C.; Parisi, G.; Scaglione, A.; Sciara, G.; Savino, C.; Vallone, B. Functional analysis and crystallographic structure of clotrimazol bound OleP, a cytochrome P450 epoxidase from Streptomyces antibioticus involved in oleandomycin biosynthesis. Biochim. Biophys. Acta 2016, 1860, 465–475. [Google Scholar] [CrossRef]
- Parisi, G.; Montemiglio, L.C.; Giuffrè, A.; Macone, A.; Scaglione, A.; Cerutti, G.; Exertier, C.; Savino, C.; Vallone, B. Substrate-induced conformational change in cytochrome P450 OleP. FASEB J. 2019, 33, 1787–1800. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, P. Substrate binding to cytochromes P450. Anal. Bioanal. Chem. 2008, 392, 1019–1030. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Computational Project. The CCP4 suite: Programs for protein crystallography. Acta Cryst. 1994, D50, 7690–7763. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are mydata and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. Molecular replacementwith MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. 2004, D60, 2126–2132. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Cryst. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement ofmacromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Brunger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Cryst. 2019, D75, 861–877. [Google Scholar] [CrossRef]
- Williams, C.J.; Hintze, B.J.; Headd, J.J.; Moriarty, N.W.; Chen, V.B.; Jain, S.; Prisant MG Lewis, S.M.; Videau, L.L.; Keedy, D.A.; Deis, L.N.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG—A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Section 1.3: Chemical Bonds in Biochemistry. In Biochemistry, 5th ed.; W.H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Conner, K.P.; Woods, C.; Atkins, W.M. Interactions of cytochrome P450s with their ligands. Arch. Biochem. Biophys. 2011, 507, 56–65. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | OleP–DEOLS | OleP–DEOHS | OleP–DEO-Rhamnose | OleP–6DEB–Rhamnose |
|---|---|---|---|---|
| PDB ID | 6ZI2 | 6ZHZ | 6ZI7 | 6ZI3 |
| Crystallization Conditions | 0.2 M NaCl 0.1 M Tris·HCl pH 7.4 25% PEG 3350 (seeding) | 4 M HCOONa (10% glycerol) | 4.4 M HCOONa | 4.2 M HCOONa (20% glycerol) |
| Space group | P1 | C2 | C2 | C2 |
| Unit cell (Å, °) | a = 112.07, b = 116.64, c = 125.17, α = 104.43, β = 104.25, γ = 113.91 | a = 247.47, b = 111.22, c = 159.20, β = 129.4 | a = 247.53, b = 110.68, c = 159.28, β = 129.5 | a = 247.38, b = 111.15, c = 159.14, β = 129.4 |
| Resolution (å) | 39.89–2.87 (3.05–2.87) | 37.7–2.20 (2.33–2.20) | 50.0–2.11 (2.24–2.11) | 50.0–1.96 (2.08–1.96) |
| Total measurements | 411,472 | 574,285 | 1,262,482 | 1,251,578 |
| Unique reflections | 115,590 | 168,218 | 373,419 | 462,663 |
| Completeness (%) | 98.2 (95.9) | 98.9 (97.3) | 99.1 (99.0) | 97.7 (96.7) |
| Redundancy | 3.56 (3.64) | 3.41 (3.37) | 3.38 (3.30) | 2.70 (2.54) |
| Rmerge a (%) | 16.5 (92.4) | 5.6 (112.7) | 7.2 (147.1) | 6.4 (163.6) |
| cc/2 (%) | 99.0 (68.8) | 99.8 (51.4) | 99.8 (48.4) | 99.7 (36.2) |
| I/σ (I) | 7.29 (1.36) | 12.10 (1.05) | 10.52 (0.72) | 7.80 (0.52) |
| Wilson B-value (Å2) | 44.1 | 44.9 | 56.7 | 53.5 |
| Refinement | ||||
| Molecules per asymmetric unit | 9 | 6 | 6 | 6 |
| Resolution Range (Å) | 39.89–2.93 | 37.73–2.2 | 47.92–2.28 | 48.09–2.08 |
| Rwork/Rfree b (%) | 24.0/29.1 | 18.7/24.3 | 18.4/24.7 | 17.2/22.5 |
| Deviations from ideal geometry | ||||
| Bond (Å) | 0.011 | 0.012 | 0.013 | 0.011 |
| Angles (°) | 1.78 | 2.16 | 2.16 | 1.75 |
| Ramachandran (%) Favored/allowed/outliers * | 96.6/3.4/0.0 | 96.1/3.9/0.0 | 96.6/3.4/0.0 | 97.0/3.0/0.0 |
| Mean B-factors (Å2) | ||||
| Protein | 56.9 | 45.2 | 59.5 | 59.6 |
| HEM/DEO/6DEB/RAM | 51.3/53.5/-/- | 31.7/44.7/-/- | 43.1/46.8/-/54.6 | 42.8/-/47.2/75.7 |
| H2O/Na/FMT/GOL/TRS | 13.5/-/-/-/- | 62.2/58.5/87.5/81.6/64.4 | 59.6/56.1/76.4/- | 63.9/86.7/55.9/83.3/87.6 |
| RSCC (per monomer in the asymmetric unit) | ||||
| DEO | 0.88/0.96/0.95/0.91/0.88/0.88/0.88/0.88/0.80 | 0.97/0.96/0.97/0.95/0.95/0.95 | 0.97/0.97/0.97/0.97/0.96/0.96 | |
| 6DEB | 0.98/0.93/0.96/0.93/0.97/0.97 | |||
| RAM (bound to the solvent cavity) | 0.91;0.86/0.81/-/0.85/-/- | 0.87/0.80/0.86/0.87/0.85/- | ||
| No of Atoms | ||||
| Protein | 26350 | 18824 | 19903 | 20575 |
| HEM/DEO/6DEB/RAM | 387/234/-/- | 258/156/-/- | 258/156/-/33 | 258/-/162/77+ |
| H2O/Na+/FMT/GOL/TRS | 96/-/-/-/- | 763/5/249/90/24 | 1369/6/642/-/- | 1145/7/609/36/24 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parisi, G.; Freda, I.; Exertier, C.; Cecchetti, C.; Gugole, E.; Cerutti, G.; D’Auria, L.; Macone, A.; Vallone, B.; Savino, C.; et al. Dissecting the Cytochrome P450 OleP Substrate Specificity: Evidence for a Preferential Substrate. Biomolecules 2020, 10, 1411. https://doi.org/10.3390/biom10101411
Parisi G, Freda I, Exertier C, Cecchetti C, Gugole E, Cerutti G, D’Auria L, Macone A, Vallone B, Savino C, et al. Dissecting the Cytochrome P450 OleP Substrate Specificity: Evidence for a Preferential Substrate. Biomolecules. 2020; 10(10):1411. https://doi.org/10.3390/biom10101411
Chicago/Turabian StyleParisi, Giacomo, Ida Freda, Cécile Exertier, Cristina Cecchetti, Elena Gugole, Gabriele Cerutti, Lucia D’Auria, Alberto Macone, Beatrice Vallone, Carmelinda Savino, and et al. 2020. "Dissecting the Cytochrome P450 OleP Substrate Specificity: Evidence for a Preferential Substrate" Biomolecules 10, no. 10: 1411. https://doi.org/10.3390/biom10101411
APA StyleParisi, G., Freda, I., Exertier, C., Cecchetti, C., Gugole, E., Cerutti, G., D’Auria, L., Macone, A., Vallone, B., Savino, C., & Montemiglio, L. C. (2020). Dissecting the Cytochrome P450 OleP Substrate Specificity: Evidence for a Preferential Substrate. Biomolecules, 10(10), 1411. https://doi.org/10.3390/biom10101411