Reduced Sulfation Enhanced Oxytosis and Ferroptosis in Mouse Hippocampal HT22 Cells


Abstract
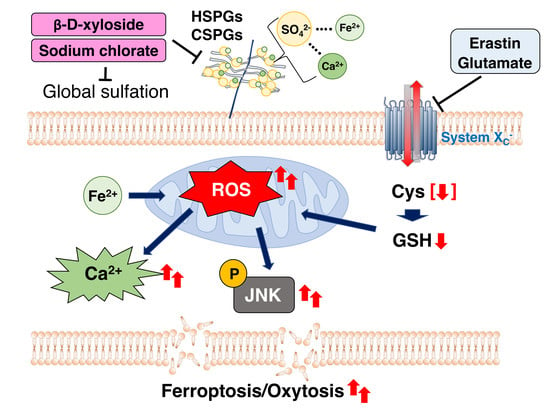
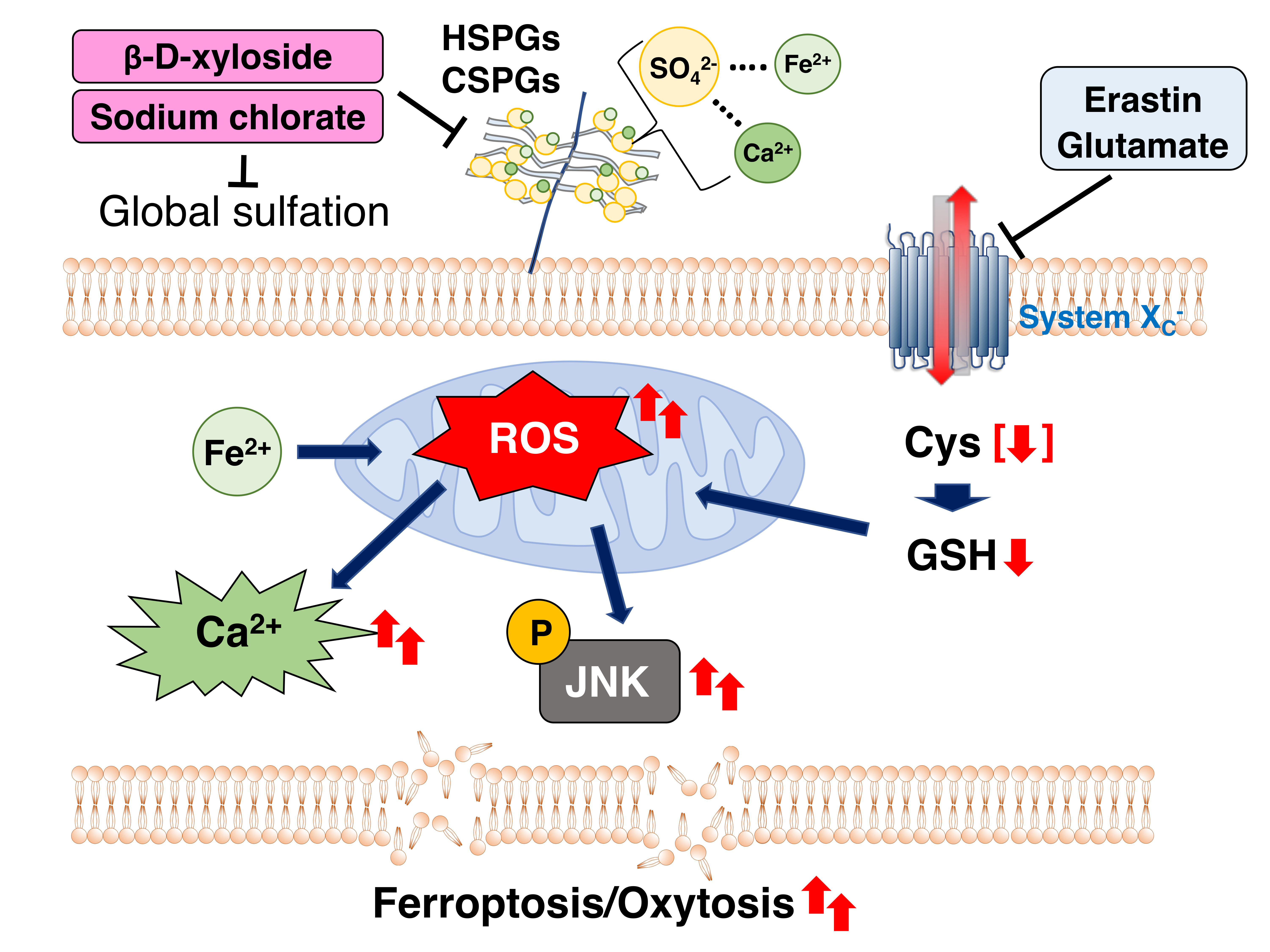{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Immunofluorescent Staining and Imaging
2.4. Western Blotting
2.5. Cell Viability Assay
2.6. Cell Death Assay
2.7. GSH Determination
2.8. ROS Production
2.9. Ca2+ Influx
2.10. Statistical Analyses
3. Results
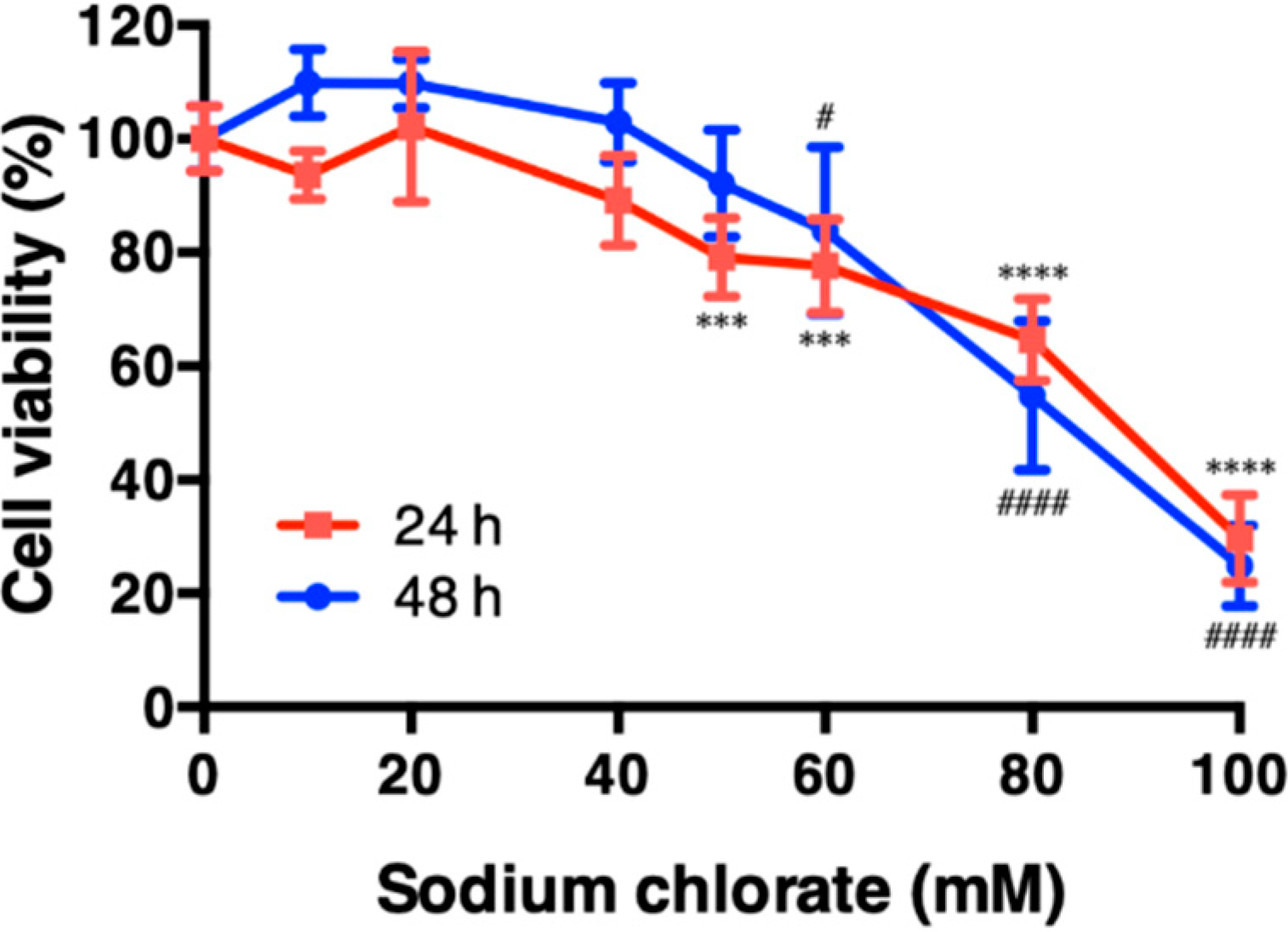
3.1. Effect of Sodium Chlorate on Cell Viability in HT22 Cells
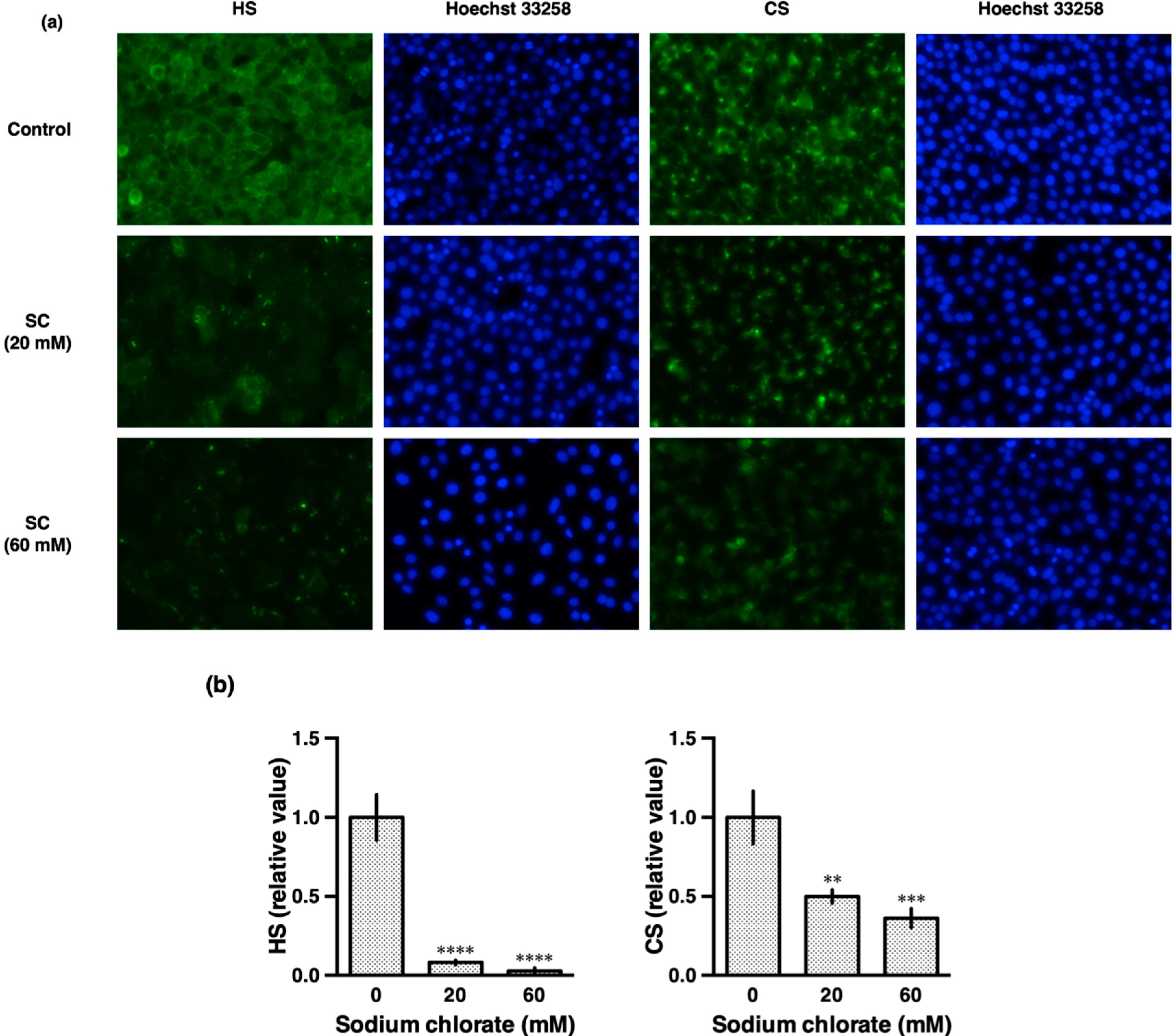
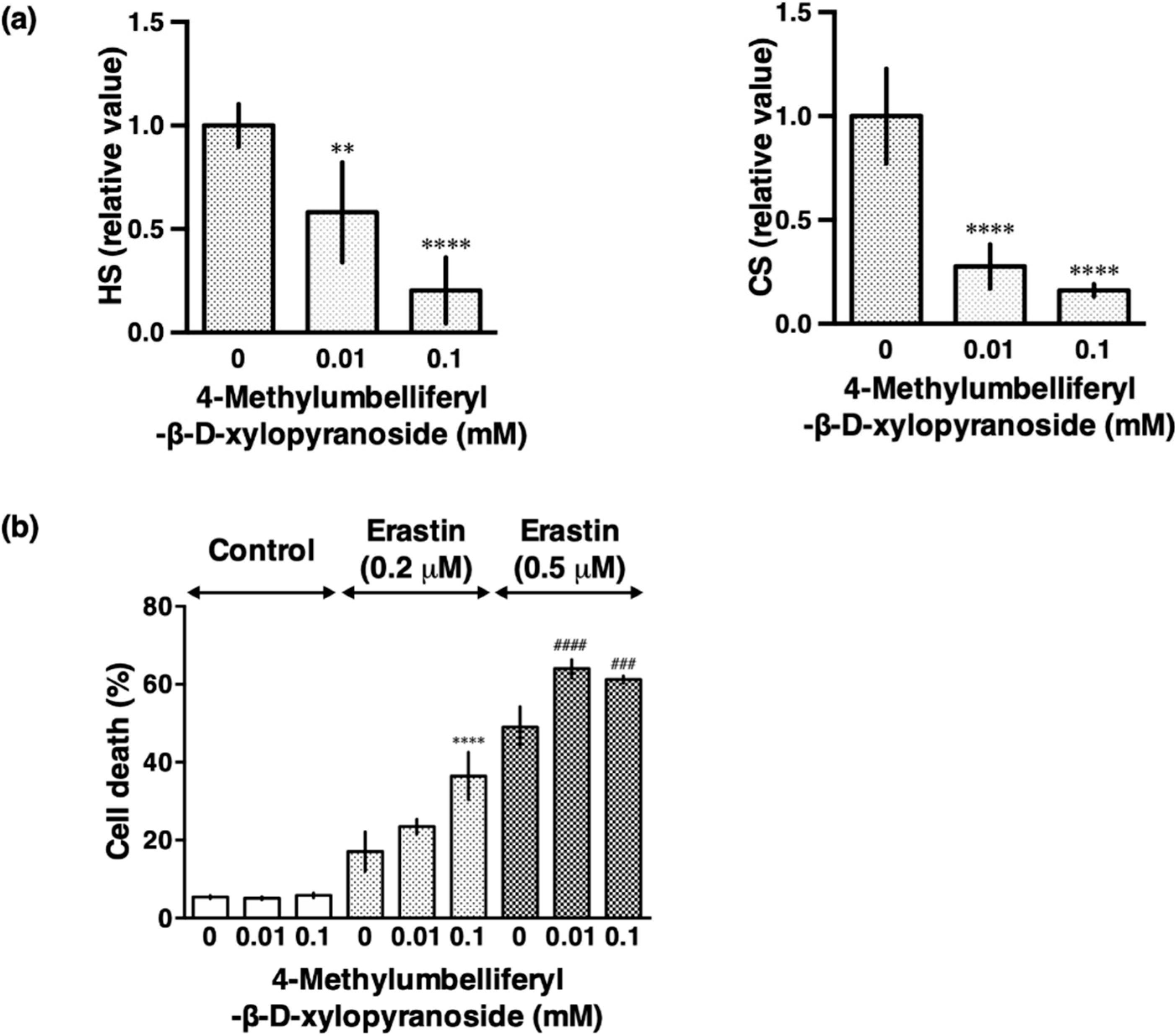
3.2. Sodium Chlorate Reduced the Sulfation of GAGs
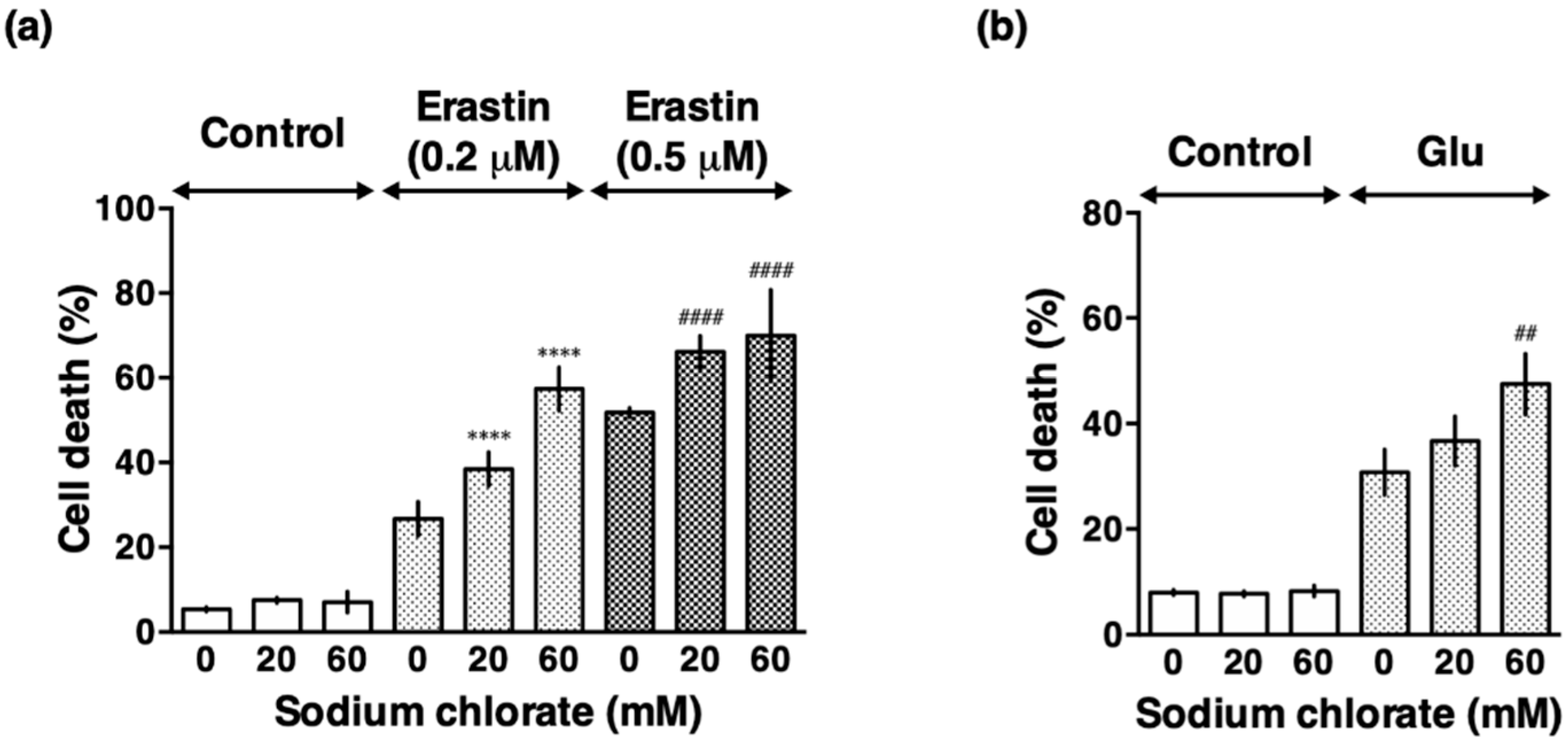
3.3. Sodium Chlorate Treatment Enhanced Extracellular Glutamate- and Erastin-Induced Cell Death in HT22 Cells
3.4. β-d-Xyloside Enhanced Extracellular Glutamate- and Erastin-Induced Cell Death in HT22 Cells
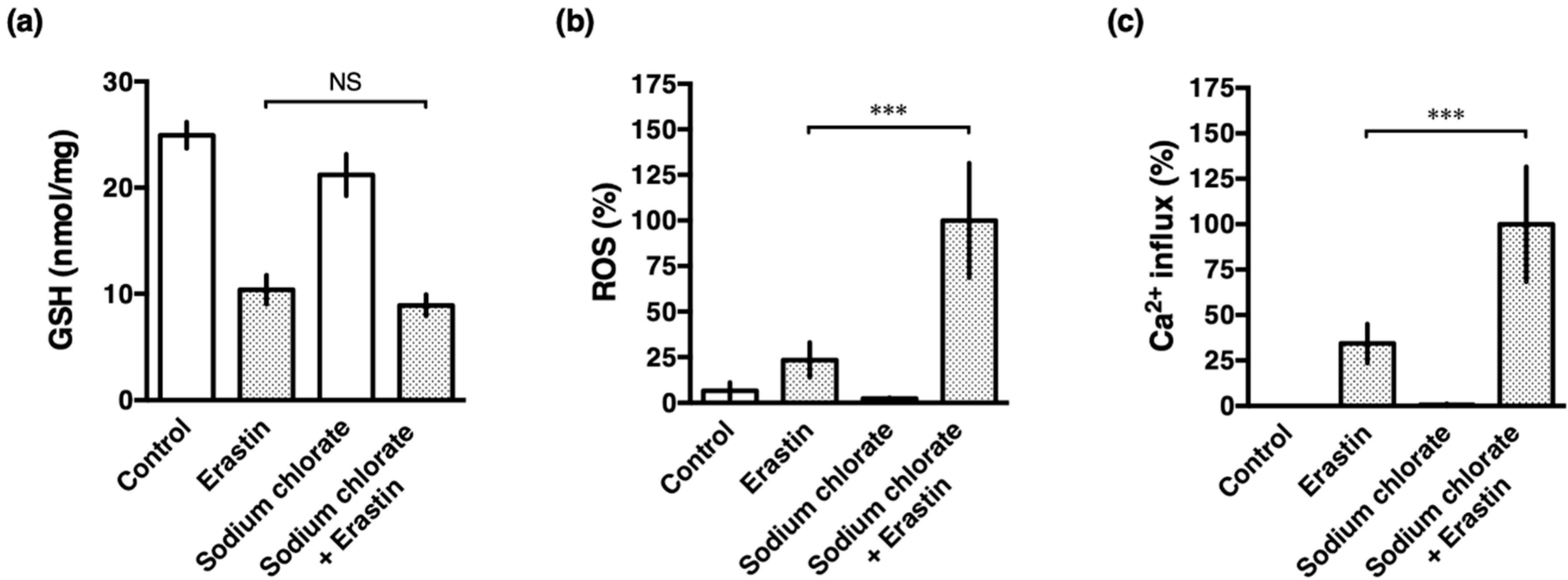
3.5. Sodium Chlorate Treatment Enhanced Erastin-Induced ROS Production but Did Not Affect GSH Depletion in HT22 Cells
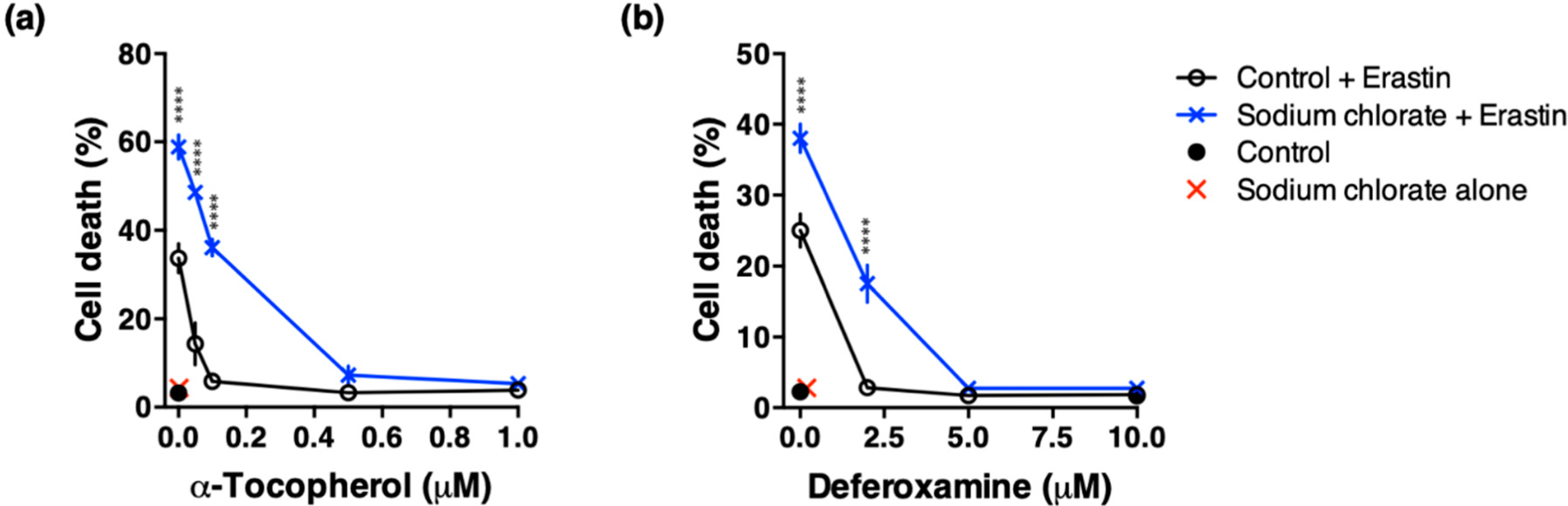
3.6. Effect of ROS Scavenging and Iron Chelating Chemicals on Sodium Chlorate-Enhanced Ferroptotic Cell Death
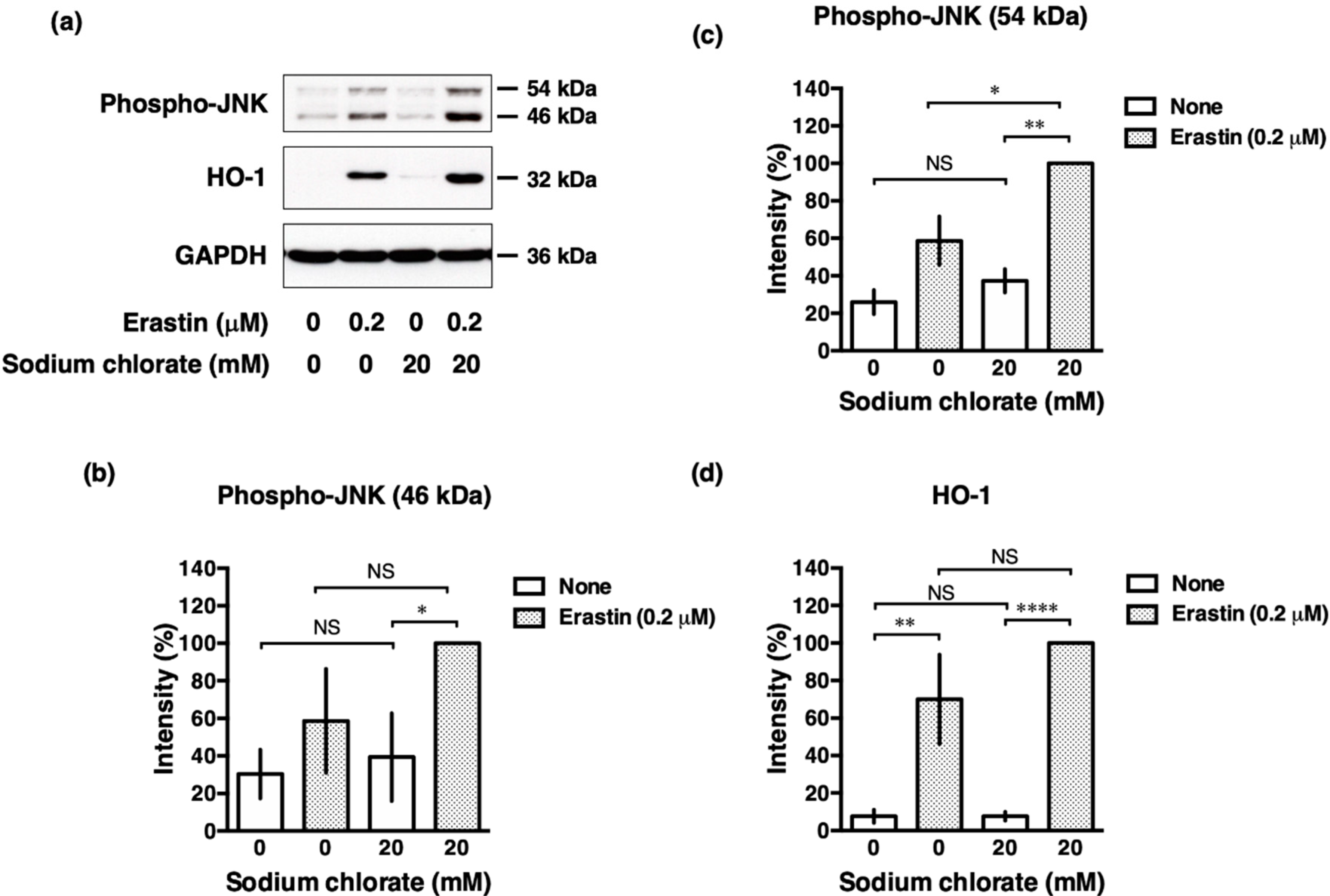
3.7. Sodium Chlorate Treatment Enhanced Erastin-Induced Phosphorylation of c-Jun N-Terminal Kinase (JNK) in HT22 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/Ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Davis, J.B. The role of monoamine metabolism in oxidative glutamate toxicity. J. Neurosci. 1996, 16, 6394–6401. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Ishige, K.; Schubert, D.; Sagara, Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001, 30, 433–446. [Google Scholar] [CrossRef]
- Aoun, P.; Watson, D.G.; Simpkins, J.W. Neuroprotective effects of PPARgamma agonists against oxidative insults in HT-22 cells. Eur. J. Pharm. 2003, 472, 65–71. [Google Scholar] [CrossRef]
- Hirata, Y.; Yamada, C.; Ito, Y.; Yamamoto, S.; Nagase, H.; Oh-Hashi, K.; Kiuchi, K.; Suzuki, H.; Sawada, M.; Furuta, K. Novel oxindole derivatives prevent oxidative stress-induced cell death in mouse hippocampal HT22cells. Neuropharmacology 2018, 135, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.M.; Avenoso, A.; Campo, S.; Ferlazzo, A.M.; Calatroni, A. Antioxidant activity of chondroitin sulfate. Adv. Pharm. 2006, 53, 417–431. [Google Scholar]
- Lucena, S.V.; Moura, G.; Rodrigues, T.; Watashi, C.M.; Melo, F.H.; Icimoto, M.Y.; Viana, G.M.; Nader, H.B.; Monteiro, H.P.; Tersariol, I.L.S.; et al. Heparan sulfate proteoglycan deficiency up-regulates the intracellular production of nitric oxide in Chinese hamster ovary cell lines. J. Cell. Physiol. 2018, 233, 3176–3194. [Google Scholar] [CrossRef]
- Campo, G.M.; D’Ascola, A.; Avenoso, A.; Campo, S.; Ferlazzo, A.M.; Micali, C.; Zanghi, L.; Calatroni, A. Glycosaminoglycans reduce oxidative damage induced by copper (Cu+2), iron (Fe+2) and hydrogen peroxide (H2O2) in human fibroblast cultures. Glycoconj. J. 2004, 20, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Dudas, B.; Rose, M.; Cornelli, U.; Pavlovich, A.; Hanin, I. Neuroprotective properties of glycosaminoglycans: Potential treatment for neurodegenerative disorders. Neurodegener. Dis. 2008, 5, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Suttkus, A.; Rohn, S.; Jager, C.; Arendt, T.; Morawski, M. Neuroprotection against iron-induced cell death by perineuronal nets—An in vivo analysis of oxidative stress. Am. J. Neurodegener. Dis. 2012, 1, 122–129. [Google Scholar] [PubMed]
- Fadel, S.; Eley, A. Chlorate: A reversible inhibitor of proteoglycan sulphation in Chlamydia trachomatis-infected cells. J. Med. Microbiol. 2004, 53, 93–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Humphries, D.E.; Silbert, J.E. Chlorate: A reversible inhibitor of proteoglycan sulfation. Biochem. Biophys. Res. Commun. 1988, 154, 365–371. [Google Scholar] [CrossRef]
- Galligani, L.; Hopwood, J.; Schwartz, N.B.; Dorfman, A. Stimulation of synthesis of free chondroitin sulfate chains by β-d-xylosides in cultured cells. J. Biol. Chem. 1975, 250, 5400–5406. [Google Scholar]
- Schwartz, N.B.; Domowicz, M.S. Proteoglycans in brain development and pathogenesis. FEBS Lett. 2018, 592, 3791–3805. [Google Scholar] [CrossRef]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- van den Born, J.; Salmivirta, K.; Henttinen, T.; Ostman, N.; Ishimaru, T.; Miyaura, S.; Yoshida, K.; Salmivirta, M. Novel heparan sulfate structures revealed by monoclonal antibodies. J. Biol. Chem. 2005, 280, 20516–20523. [Google Scholar] [CrossRef]
- Carrino, D.A.; Caplan, A.I. The effects of β-d-xyloside on the synthesis of proteoglycans by skeletal muscle: Lack of effect on decorin and differential polymerization of core protein-bound and xyloside-linked chondroitin sulfate. Matrix Biol. 1994, 14, 121–133. [Google Scholar] [CrossRef]
- Behl, C.; Widmann, M.; Trapp, T.; Holsboer, F. 17-β estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem. Biophys. Res. Commun. 1995, 216, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Ito, Y.; Takashima, M.; Yagyu, K.; Oh-Hashi, K.; Suzuki, H.; Ono, K.; Furuta, K.; Sawada, M. Novel oxindole-curcumin hybrid compound for antioxidative stress and neuroprotection. ACS Chem. Neurosci. 2020, 11, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Hirata, Y.; Iwasaki, T.; Makimura, Y.; Okajima, S.; Oh-Hashi, K.; Takemori, H. Inhibition of double-stranded RNA-dependent protein kinase prevents oxytosis and ferroptosis in mouse hippocampal HT22 cells. Toxicology 2019, 418, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Safaiyan, F.; Kolset, S.O.; Prydz, K.; Gottfridsson, E.; Lindahl, U.; Salmivirta, M. Selective effects of sodium chlorate treatment on the sulfation of heparan sulfate. J. Biol. Chem. 1999, 274, 36267–36273. [Google Scholar] [CrossRef]
- Ali, S.N.; Ahmad, M.K.; Mahmood, R. Sodium chlorate, a herbicide and major water disinfectant byproduct, generates reactive oxygen species and induces oxidative damage in human erythrocytes. Env. Sci. Pollut. Res. Int. 2017, 24, 1898–1909. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Huttner, W.B. Chlorate—A potent inhibitor of protein sulfation in intact cells. Biochem. Biophys. Res. Commun. 1986, 141, 870–877. [Google Scholar] [CrossRef]
- Okamoto, M.; Mori, S.; Endo, H. A protective action of chondroitin sulfate proteoglycans against neuronal cell death induced by glutamate. Brain Res. 1994, 637, 57–67. [Google Scholar] [CrossRef]
- Okamoto, M.; Mori, S.; Ichimura, M.; Endo, H. Chondroitin sulfate proteoglycans protect cultured rat’s cortical and hippocampal neurons from delayed cell death induced by excitatory amino acids. Neurosci. Lett. 1994, 172, 51–54. [Google Scholar] [CrossRef]
- Naylor, M.C.; Negia, M.; Noetzel, M.; Burns, T.C.; Demorest, Z.L.; Low, W.C. Heparan sulfate mediates neuroprotection from degeneration in experimental glutaric aciduria. Cell Transpl. 2007, 16, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Hrabetova, S.; Masri, D.; Tao, L.; Xiao, F.; Nicholson, C. Calcium diffusion enhanced after cleavage of negatively charged components of brain extracellular matrix by chondroitinase ABC. J. Physiol. 2009, 587, 4029–4049. [Google Scholar] [CrossRef] [PubMed]
- Takashima, M.; Ichihara, K.; Hirata, Y. Neuroprotective effects of Brazilian green propolis on oxytosis/ferroptosis in mouse hippocampal HT22 cells. Food Chem. Toxicol. 2019, 132, 110669. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Liu, Z.G. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2006, 40, 928–939. [Google Scholar] [CrossRef]
- Hahn, C.; Orr, A.W.; Sanders, J.M.; Jhaveri, K.A.; Schwartz, M.A. The subendothelial extracellular matrix modulates JNK activation by flow. Circ. Res. 2009, 104, 995–1003. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagase, H.; Katagiri, Y.; Oh-hashi, K.; Geller, H.M.; Hirata, Y. Reduced Sulfation Enhanced Oxytosis and Ferroptosis in Mouse Hippocampal HT22 Cells. Biomolecules 2020, 10, 92. https://doi.org/10.3390/biom10010092
Nagase H, Katagiri Y, Oh-hashi K, Geller HM, Hirata Y. Reduced Sulfation Enhanced Oxytosis and Ferroptosis in Mouse Hippocampal HT22 Cells. Biomolecules. 2020; 10(1):92. https://doi.org/10.3390/biom10010092
Chicago/Turabian StyleNagase, Haruna, Yasuhiro Katagiri, Kentaro Oh-hashi, Herbert M. Geller, and Yoko Hirata. 2020. "Reduced Sulfation Enhanced Oxytosis and Ferroptosis in Mouse Hippocampal HT22 Cells" Biomolecules 10, no. 1: 92. https://doi.org/10.3390/biom10010092
APA StyleNagase, H., Katagiri, Y., Oh-hashi, K., Geller, H. M., & Hirata, Y. (2020). Reduced Sulfation Enhanced Oxytosis and Ferroptosis in Mouse Hippocampal HT22 Cells. Biomolecules, 10(1), 92. https://doi.org/10.3390/biom10010092