An Adrenalectomy Mouse Model Reflecting Clinical Features for Chronic Fatigue Syndrome


Abstract
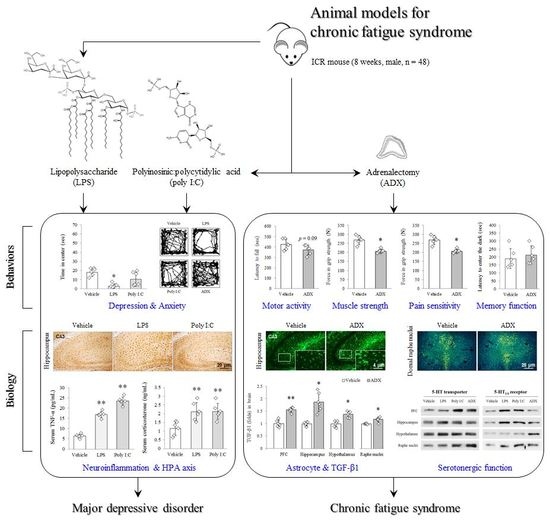
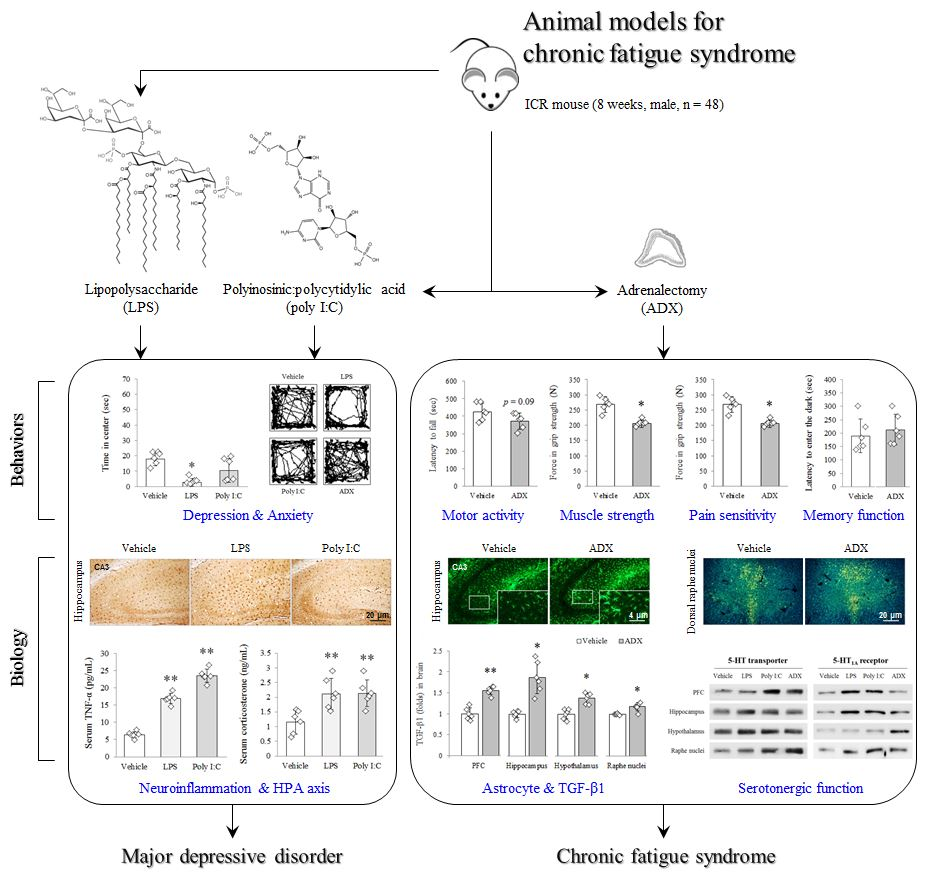{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Animals and Experimental Design
2.3. Evaluation of Anxiety and Locomotor Behavior, Open Field Test
2.4. Evaluation of Motor Activity, Rotarod Test
2.5. Evaluation of Muscular Strength, Grip Strength Test
2.6. Evaluation of Pain Sensitivity, Plantar Test
2.7. Evaluation of Long-Term Memory, Passive Avoidance Test
2.8. Sample Preparation
2.9. Determination of Corticosterone and TNF-α
2.10. Determination of TGF-β1
2.11. Western Blotting Analysis
2.12. Immunohistological Staining Analysis
2.13. Statistical Analysis
3. Results
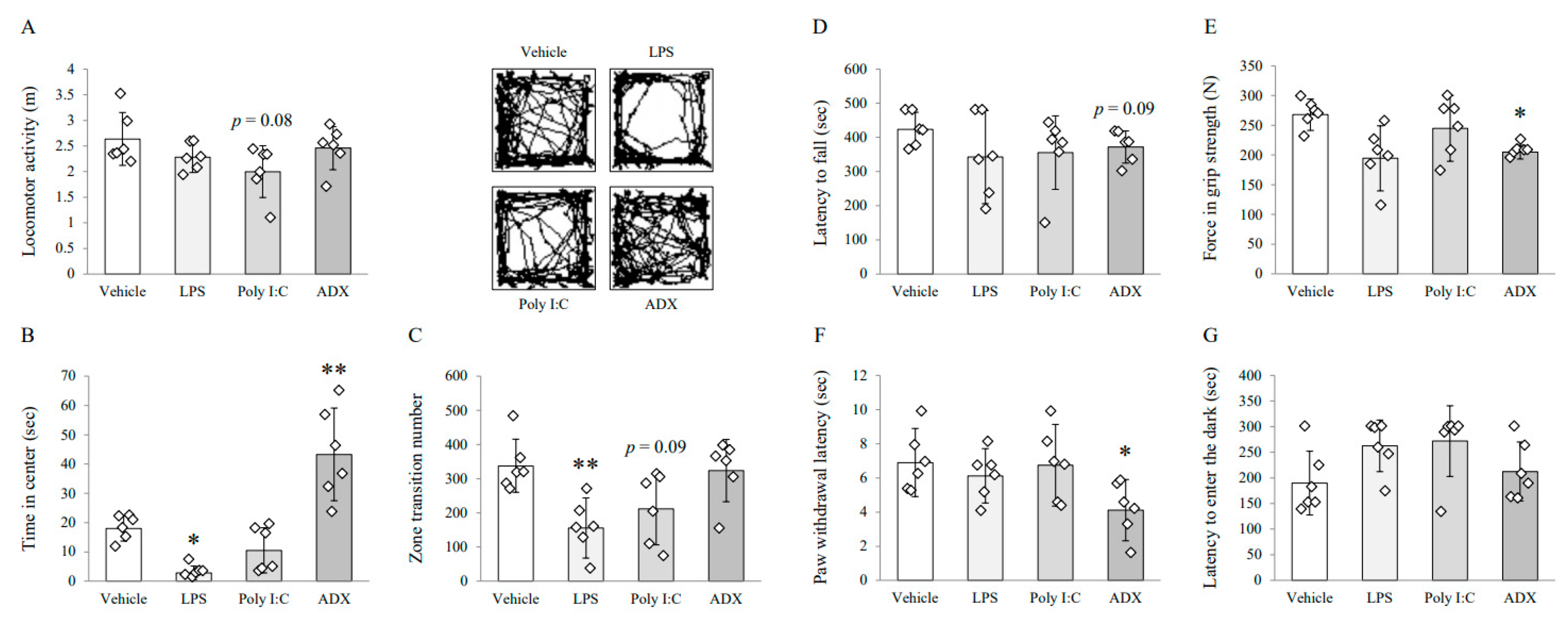
3.1. Changes on Depressive/Anxious-Like Behaviors: Open Field Test
3.2. Changes on Fatigue-Like Behaviors: Rotarod Test
3.3. Changes on Muscular Strength and Pain Sensitivity: Grip Strength Test and Plantar Test
3.4. Changes on Long-Term Memory: Passive Avoidance Test
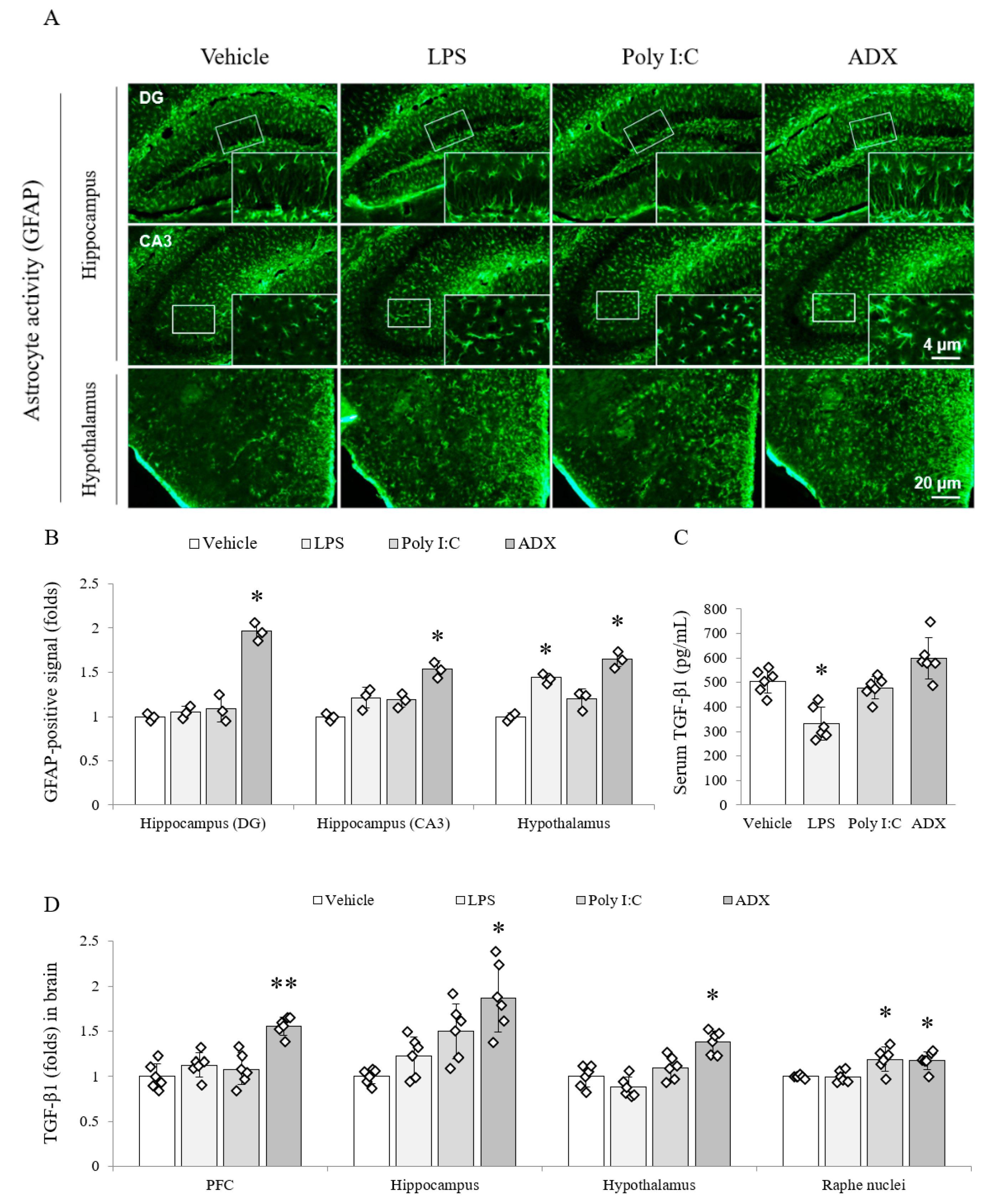
3.5. Changes of the Astrocyte Activity in Brain Tissues
3.6. Changes of TGF-β1 in Serum and Brain Tissues
3.7. Changes of the Microglia Activity in Brain Tissues
3.8. Changes of Corticosterone and TNF-α in Serum
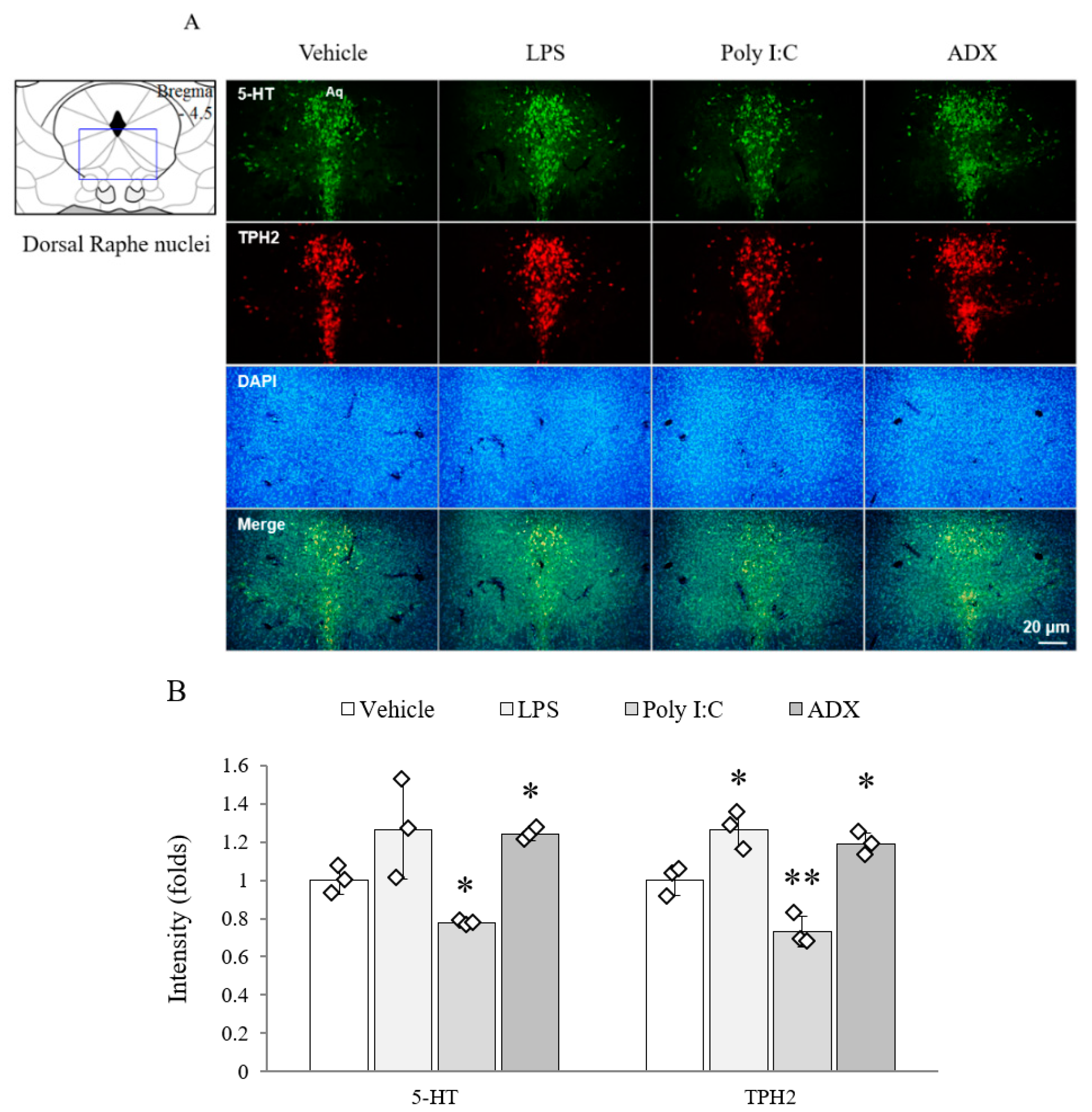
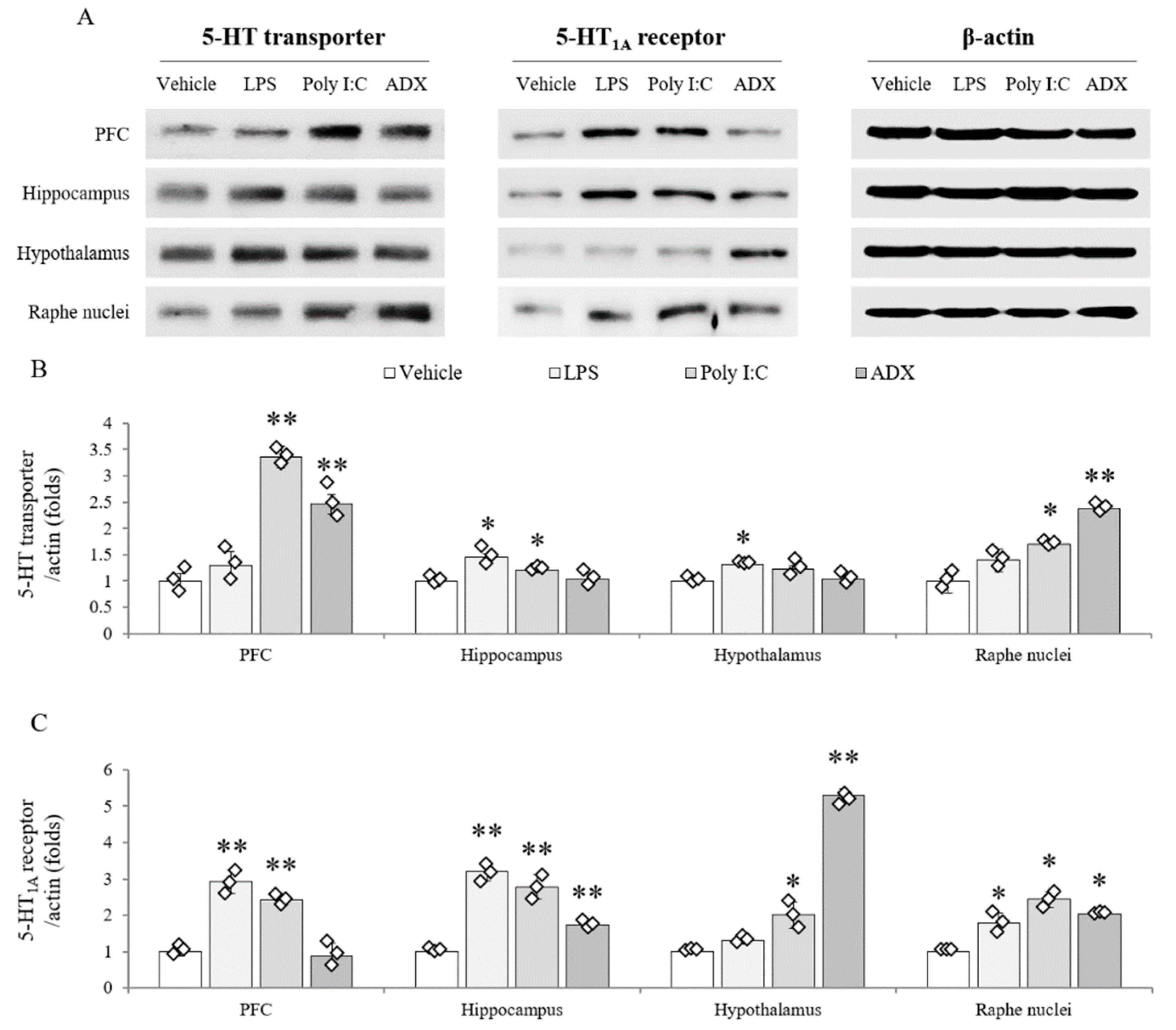
3.9. Changes of Serotonergic System in Brain Tissues
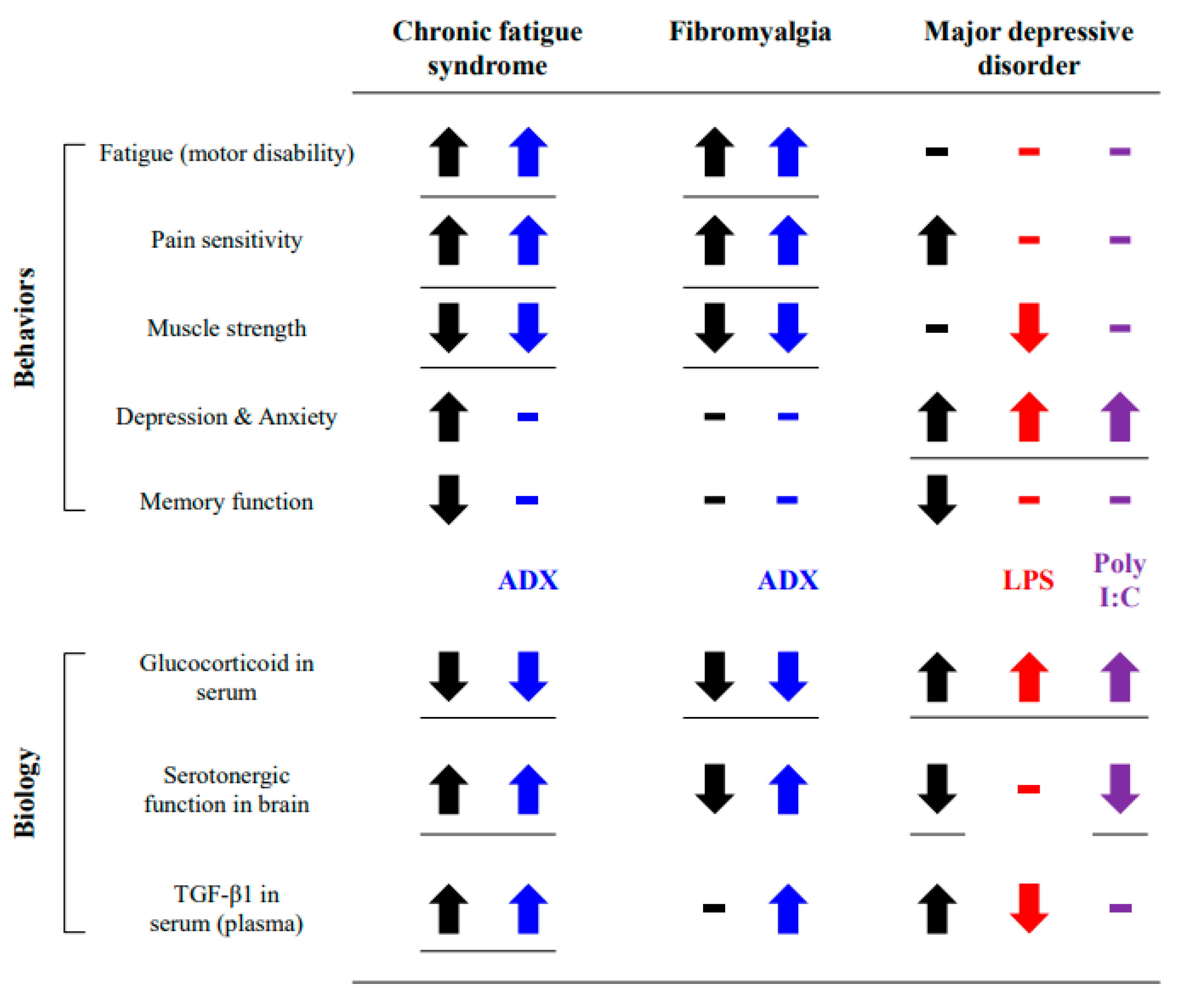
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pendergrast, T.; Brown, A.; Sunnquist, M.; Jantke, R.; Newton, J.L.; Strand, E.B.; Jason, L.A. Housebound versus nonhousebound patients with myalgic encephalomyelitis and chronic fatigue syndrome. Chronic Illn. 2016, 12, 292–307. [Google Scholar] [CrossRef]
- Falk Hvidberg, M.; Brinth, L.S.; Olesen, A.V.; Petersen, K.D.; Ehlers, L. The health-related quality of life for patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). PLoS ONE 2015, 10, e0132421. [Google Scholar] [CrossRef]
- Castro-Marrero, J.; Zaragoza, M.C.; Gonzalez-Garcia, S.; Aliste, L.; Saez-Francas, N.; Romero, O.; Ferre, A.; Fernandez de Sevilla, T.; Alegre, J. Poor self-reported sleep quality and health-related quality of life in patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Sleep Res. 2018, 27, e12703. [Google Scholar] [CrossRef]
- Johnston, S.; Brenu, E.W.; Staines, D.; Marshall-Gradisnik, S. The prevalence of chronic fatigue syndrome/ myalgic encephalomyelitis: A meta-analysis. Clin. Epidemiol. 2013, 5, 105–110. [Google Scholar] [CrossRef]
- Monro, J.A.; Puri, B.K. A molecular neurobiological approach to understanding the aetiology of chronic fatigue syndrome (myalgic encephalomyelitis or systemic exertion intolerance disease) with treatment implications. Mol. Neurobiol. 2018, 55, 7377–7388. [Google Scholar] [CrossRef]
- Friedman, K.J. Advances in ME/CFS: Past, present, and future. Front. Pediatr. 2019, 7, 131. [Google Scholar] [CrossRef]
- Watanabe, Y. Brain science on Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Brain Nerve 2018, 70, 1193–1201. [Google Scholar] [CrossRef]
- Missailidis, D.; Annesley, S.J.; Fisher, P.R. Pathological mechanisms underlying Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Diagnostics 2019, 9, 80. [Google Scholar] [CrossRef]
- Rasa, S.; Nora-Krukle, Z.; Henning, N.; Eliassen, E.; Shikova, E.; Harrer, T.; Scheibenbogen, C.; Murovska, M.; Prusty, B.K. Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J. Transl. Med. 2018, 16, 268. [Google Scholar] [CrossRef]
- Cleare, A.J. The neuroendocrinology of chronic fatigue syndrome. Endocr. Rev. 2003, 24, 236–252. [Google Scholar] [CrossRef]
- Lorusso, L.; Mikhaylova, S.V.; Capelli, E.; Ferrari, D.; Ngonga, G.K.; Ricevuti, G. Immunological aspects of chronic fatigue syndrome. Autoimmun. Rev. 2009, 8, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Ouchi, Y.; Onoe, H.; Yoshikawa, E.; Tsukada, H.; Takahashi, H.; Iwase, M.; Yamaguti, K.; Kuratsune, H.; Watanabe, Y. Reduction of serotonin transporters of patients with chronic fatigue syndrome. Neuroreport 2004, 15, 2571–2574. [Google Scholar] [CrossRef] [PubMed]
- Cleare, A.J.; Messa, C.; Rabiner, E.A.; Grasby, P.M. Brain 5-HT1A receptor binding in chronic fatigue syndrome measured using positron emission tomography and [11C]WAY-100635. Biol. Psychiatry 2005, 57, 239–246. [Google Scholar] [CrossRef]
- Dinan, T.G.; Majeed, T.; Lavelle, E.; Scott, L.V.; Berti, C.; Behan, P. Blunted serotonin-mediated activation of the hypothalamic-pituitary-adrenal axis in chronic fatigue syndrome. Psychoneuroendocrinology 1997, 22, 261–267. [Google Scholar] [CrossRef]
- Wen, F.Q.; Kohyama, T.; Skold, C.M.; Zhu, Y.K.; Liu, X.; Romberger, D.J.; Stoner, J.; Rennard, S.I. Glucocorticoids modulate TGF-beta production. Inflammation 2002, 26, 279–290. [Google Scholar] [CrossRef]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hasson, Y.; Valencia, I.J.; Chu, L.; Younger, J.W.; Tato, C.M.; Davis, M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. USA 2017, 114, 7150–7158. [Google Scholar] [CrossRef]
- Blundell, S.; Ray, K.K.; Buckland, M.; White, P.D. Chronic fatigue syndrome and circulating cytokines: A systematic review. Brain Behav. Immun. 2015, 50, 186–195. [Google Scholar] [CrossRef]
- Singh, S.; Vandana, G.H.K.; Bansal, P.K. Animal models of depression and Chronic Fatigue Syndrome. In Animal Models of Neurological; Springer: Berlin, Germany, 2017; pp. 159–179. [Google Scholar]
- Gupta, A.; Vij, G.; Sharma, S.; Tirkey, N.; Rishi, P.; Chopra, K. Curcumin, a polyphenolic antioxidant, attenuates chronic fatigue syndrome in murine water immersion stress model. Immunobiology 2009, 214, 33–39. [Google Scholar] [CrossRef]
- Katafuchi, T.; Kondo, T.; Yasaka, T.; Kubo, K.; Take, S.; Yoshimura, M. Prolonged effects of polyriboinosinic:polyribocytidylic acid on spontaneous running wheel activity and brain interferon-alpha mRNA in rats: A model for immunologically induced fatigue. Neuroscience 2003, 120, 837–845. [Google Scholar] [CrossRef]
- Zhang, Z.T.; Du, X.M.; Ma, X.J.; Zong, Y.; Chen, J.K.; Yu, C.L.; Liu, Y.G.; Chen, Y.C.; Zhao, L.J.; Lu, G.C. Activation of the NLRP3 inflammasome in lipopolysaccharide-induced mouse fatigue and its relevance to chronic fatigue syndrome. J. Neuroinflamm. 2016, 13, 71. [Google Scholar] [CrossRef]
- Shao, C.; Ren, Y.; Wang, Z.; Kang, C.; Jiang, H.; Chi, A. Detection of Urine Metabolites in a Rat Model of Chronic Fatigue Syndrome before and after Exercise. Biomed Res. Int. 2017, 2017, 8182020. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, R. Adrenalectomy in rats. Am. J. Physiol. 1933, 103, 494–510. [Google Scholar] [CrossRef]
- Hirohama, D.; Ayuzawa, N.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Watanabe, A.; Shimosawa, T.; Marumo, T.; Shibata, S.; Fujita, T. Aldosterone Is Essential for Angiotensin II-Induced Upregulation of Pendrin. J. Am. Soc. Nephrol. 2018, 29, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Ieraci, A.; Herrera, D.G. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med. 2006, 3, 101. [Google Scholar] [CrossRef]
- Maxmen, A. A reboot for chronic fatigue syndrome research. Nature 2018, 553, 14–17. [Google Scholar] [CrossRef]
- Scheibenbogen, C.; Freitag, H.; Blanco, J.; Capelli, E.; Lacerda, E.; Authier, J.; Meeus, M.; Castro Marrero, J.; Nora-Krukle, Z.; Oltra, E.; et al. The European ME/CFS Biomarker Landscape project: An initiative of the European network EUROMENE. J. Transl. Med. 2017, 15, 162. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yamato, M.; Miyashige, Y.; Tamura, Y.; Cui, Y. Neuroinflammation in Animal Models of Fatigue. Adv. Neuroimmune Biol. 2013, 4, 237–244. [Google Scholar] [CrossRef]
- Mikolajczyk, A.; Kozlowska, A.; Gonkowski, S. Distribution and Neurochemistry of the Porcine Ileocaecal Valve Projecting Sensory Neurons in the Dorsal Root Ganglia and the Influence of Lipopolysaccharide from Different Serotypes of Salmonella spp. on the Chemical Coding of DRG Neurons in the Cell Cultures. Int. J. Mol. Sci. 2018, 19, 2551. [Google Scholar] [CrossRef]
- Dogan, M.D.; Ataoglu, H.; Akarsu, E.S. Effects of different serotypes of Escherichia coli lipopolysaccharides on body temperature in rats. Life Sci. 2000, 67, 2319–2329. [Google Scholar] [CrossRef]
- Mikolajczyk, A.; Zlotkowska, D. Neuroimmunological Implications of Subclinical Lipopolysaccharide from Salmonella Enteritidis. Int. J. Mol. Sci. 2018, 19, 3274. [Google Scholar] [CrossRef]
- Fink, M.P. Animal models of sepsis. Virulence 2014, 5, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Meneses, G.; Rosetti, M.; Espinosa, A.; Florentino, A.; Bautista, M.; Diaz, G.; Olvera, G.; Barcena, B.; Fleury, A.; Adalid-Peralta, L.; et al. Recovery from an acute systemic and central LPS-inflammation challenge is affected by mouse sex and genetic background. PLoS ONE 2018, 13, e0201375. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; Lawson, M.A.; Andre, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef]
- Gibney, S.M.; McGuinness, B.; Prendergast, C.; Harkin, A.; Connor, T.J. Poly I:C-induced activation of the immune response is accompanied by depression and anxiety-like behaviours, kynurenine pathway activation and reduced BDNF expression. Brain Behav. Immun. 2013, 28, 170–181. [Google Scholar] [CrossRef]
- Son, C.G. Differential diagnosis between “chronic fatigue” and “chronic fatigue syndrome”. Integr. Med. Res. 2019, 8, 89–91. [Google Scholar] [CrossRef]
- Darbishire, L.; Ridsdale, L.; Seed, P.T. Distinguishing patients with chronic fatigue from those with chronic fatigue syndrome: A diagnostic study in UK primary care. Br. J. Gen. Pr. 2003, 53, 441–445. [Google Scholar]
- National Academies Press. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness; National Academies Press: Washington, DC, USA, 2015. [Google Scholar]
- Nijhof, S.L.; Rutten, J.M.; Uiterwaal, C.S.; Bleijenberg, G.; Kimpen, J.L.; Putte, E.M. The role of hypocortisolism in chronic fatigue syndrome. Psychoneuroendocrinology 2014, 42, 199–206. [Google Scholar] [CrossRef]
- Castro-Marrero, J.; Faro, M.; Aliste, L.; Saez-Francas, N.; Calvo, N.; Martinez-Martinez, A.; de Sevilla, T.F.; Alegre, J. Comorbidity in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: A nationwide population-based cohort study. Psychosomatics 2017, 58, 533–543. [Google Scholar] [CrossRef]
- Daniels, J.; Brigden, A.; Kacorova, A. Anxiety and depression in chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME): Examining the incidence of health anxiety in CFS/ME. Psychol. Psychother. 2017, 90, 502–509. [Google Scholar] [CrossRef]
- Vercoulen, J.H.; Swanink, C.M.; Zitman, F.G.; Vreden, S.G.; Hoofs, M.P.; Fennis, J.F.; Galama, J.M.; van der Meer, J.W.; Bleijenberg, G. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996, 347, 858–861. [Google Scholar] [CrossRef]
- Pardini, M.; Guida, S.; Primavera, A.; Krueger, F.; Cocito, L.; Gialloreti, L.E. Amisulpride vs. fluoxetine treatment of chronic fatigue syndrome: A pilot study. Eur. Neuropsychopharmacol. 2011, 21, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.A.; Janal, M.N.; Aktan, N.; Ottenweller, J.E.; Natelson, B.H. Sex differences in plasma prolactin response to tryptophan in chronic fatigue syndrome patients with and without comorbid fibromyalgia. J. Women’s Health 2010, 19, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Revisiting the serotonin hypothesis: Implications for major depressive disorders. Mol. Neurobiol. 2016, 53, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.; DeLorenzo, C.; Choudhury, S.; Parsey, R.V. The 5-HT1A receptor in major depressive disorder. Eur. Neuropsychopharmacol. 2016, 26, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Aaron, L.A.; Herrell, R.; Ashton, S.; Belcourt, M.; Schmaling, K.; Goldberg, J.; Buchwald, D. Comorbid clinical conditions in chronic fatigue: A co-twin control study. J. Gen. Intern. Med. 2001, 16, 24–31. [Google Scholar] [CrossRef]
- Kishi, A.; Natelson, B.H.; Togo, F.; Struzik, Z.R.; Rapoport, D.M.; Yamamoto, Y. Sleep-stage dynamics in patients with chronic fatigue syndrome with or without fibromyalgia. Sleep 2011, 34, 1551–1560. [Google Scholar] [CrossRef]
- Bailey, S.P.; Davis, J.M.; Ahlborn, E.N. Serotonergic agonists and antagonists affect endurance performance in the rat. Int. J. Sports Med. 1993, 14, 330–333. [Google Scholar] [CrossRef]
- Fernstrom, J.D.; Fernstrom, M.H. Exercise, serum free tryptophan, and central fatigue. J. Nutr. 2006, 136, 553–559. [Google Scholar] [CrossRef]
- Cordeiro, L.M.S.; Rabelo, P.C.R.; Moraes, M.M.; Teixeira-Coelho, F.; Coimbra, C.C.; Wanner, S.P.; Soares, D.D. Physical exercise-induced fatigue: The role of serotonergic and dopaminergic systems. Braz. J. Med. Biol. Res. 2017, 50, 6432. [Google Scholar] [CrossRef]
- Cotel, F.; Exley, R.; Cragg, S.J.; Perrier, J.F. Serotonin spillover onto the axon initial segment of motoneurons induces central fatigue by inhibiting action potential initiation. Proc. Natl. Acad. Sci. USA 2013, 110, 4774–4779. [Google Scholar] [CrossRef]
- Morris, G.; Anderson, G.; Maes, M. Hypothalamic-pituitary-adrenal hypofunction in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a consequence of activated immune-inflammatory and oxidative and nitrosative pathways. Mol. Neurobiol. 2017, 54, 6806–6819. [Google Scholar] [CrossRef] [PubMed]
- Fries, E.; Hesse, J.; Hellhammer, J.; Hellhammer, D.H. A new view on hypocortisolism. Psychoneuroendocrinology 2005, 30, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Torres-Harding, S.; Sorenson, M.; Jason, L.; Maher, K.; Fletcher, M.A.; Reynolds, N.; Brown, M. The associations between basal salivary cortisol and illness symptomatology in chronic fatigue syndrome. J. Appl. Biobehav. Res. 2008, 13, 157–180. [Google Scholar] [CrossRef]
- Hall, D.L.; Lattie, E.G.; Antoni, M.H.; Fletcher, M.A.; Czaja, S.; Perdomo, D.; Klimas, N.G. Stress management skills, cortisol awakening response, and post-exertional malaise in Chronic Fatigue Syndrome. Psychoneuroendocrinology 2014, 49, 26–31. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Gray, T.S.; D’Souza, D.N.; Carrasco, G.A.; Damjanoska, K.J.; Dudas, B.; Garcia, F.; Zainelli, G.M.; Sullivan Hanley, N.R.; Battaglia, G.; et al. Desensitization of 5-HT1A receptors by 5-HT2A receptors in neuroendocrine neurons in vivo. J. Pharm. Exp. 2004, 310, 59–66. [Google Scholar] [CrossRef]
- Gong, H.; Liu, L.; Su, W.J.; Zhu, Z.; Liu, Y.Z.; Lian, Y.J.; Peng, W.; Cao, Z.Y.; Zhang, T.; Jiang, C.L. Corticosterone rapidly improves the endurance of high-intensity exercise (swimming) via nongenomic mechanisms in mice. J. Sports Med. Phys. Fit. 2019, 59, 886–891. [Google Scholar] [CrossRef]
- Inoue, K.; Yamazaki, H.; Manabe, Y.; Fukuda, C.; Hanai, K.; Fushiki, T. Transforming growth factor-beta activated during exercise in brain depresses spontaneous motor activity of animals. Relevance To central fatigue. Brain Res. 1999, 846, 145–153. [Google Scholar] [CrossRef]
- Matsumura, S.; Shibakusa, T.; Fujikawa, T.; Yamada, H.; Inoue, K.; Fushiki, T. Increase in transforming growth factor-beta in the brain during infection is related to fever, not depression of spontaneous motor activity. Neuroscience 2007, 144, 1133–1140. [Google Scholar] [CrossRef]
- Radtke, F.A.; Chapman, G.; Hall, J.; Syed, Y.A. Modulating neuroinflammation to treat neuropsychiatric disorders. Biomed Res. Int. 2017, 2017, 5071786. [Google Scholar] [CrossRef]
- Yang, T.; Yang, Y.; Wang, D.; Li, C.; Qu, Y.; Guo, J.; Shi, T.; Bo, W.; Sun, Z.; Asakawa, T. The clinical value of cytokines in chronic fatigue syndrome. J. Transl. Med. 2019, 17, 213. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-S.; Jeon, Y.-J.; Park, S.-Y.; Son, C.-G. An Adrenalectomy Mouse Model Reflecting Clinical Features for Chronic Fatigue Syndrome. Biomolecules 2020, 10, 71. https://doi.org/10.3390/biom10010071
Lee J-S, Jeon Y-J, Park S-Y, Son C-G. An Adrenalectomy Mouse Model Reflecting Clinical Features for Chronic Fatigue Syndrome. Biomolecules. 2020; 10(1):71. https://doi.org/10.3390/biom10010071
Chicago/Turabian StyleLee, Jin-Seok, Yoo-Jin Jeon, Samuel-Young Park, and Chang-Gue Son. 2020. "An Adrenalectomy Mouse Model Reflecting Clinical Features for Chronic Fatigue Syndrome" Biomolecules 10, no. 1: 71. https://doi.org/10.3390/biom10010071
APA StyleLee, J.-S., Jeon, Y.-J., Park, S.-Y., & Son, C.-G. (2020). An Adrenalectomy Mouse Model Reflecting Clinical Features for Chronic Fatigue Syndrome. Biomolecules, 10(1), 71. https://doi.org/10.3390/biom10010071