Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine



Abstract
1. Introduction
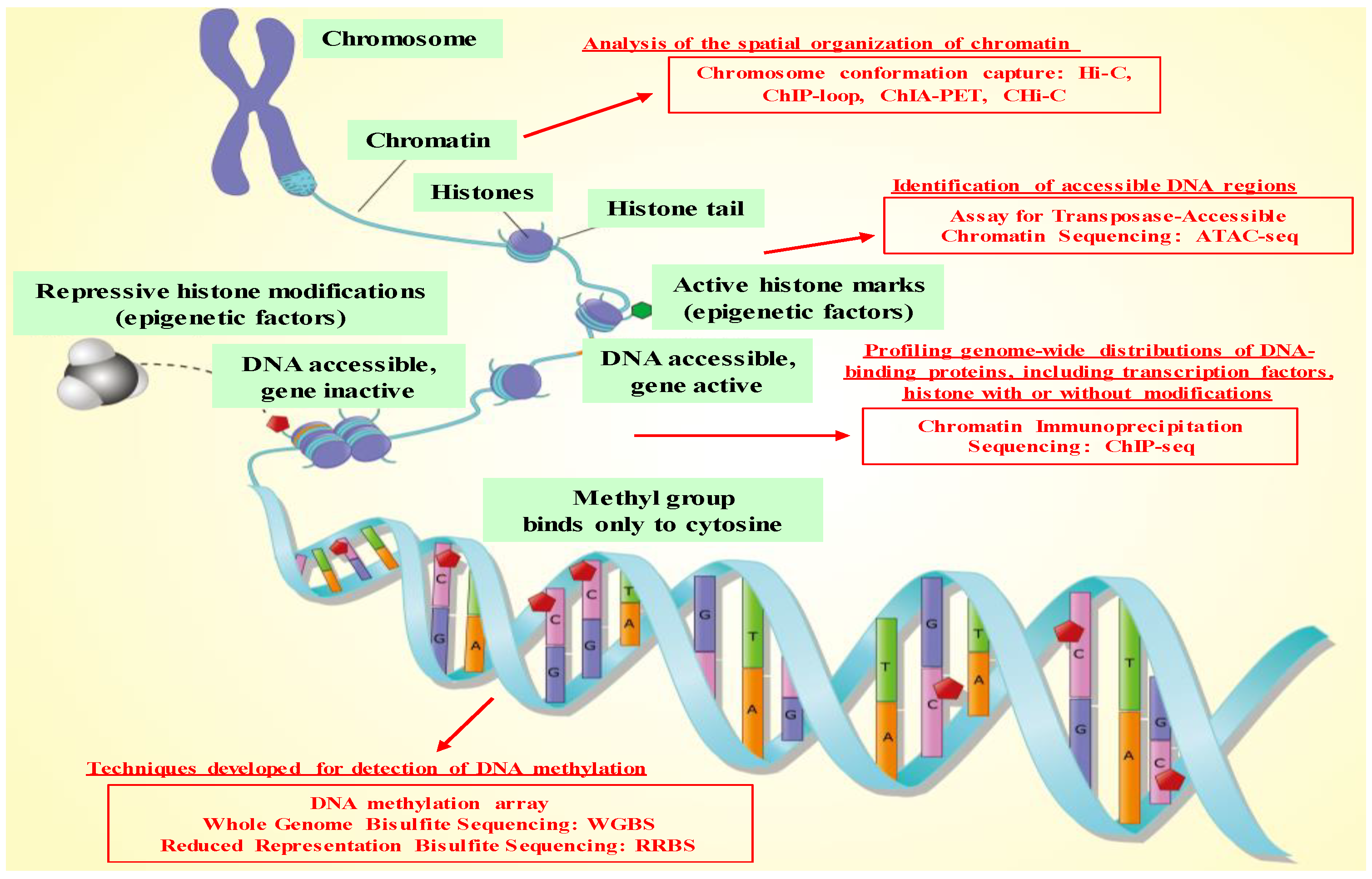
2. Characteristics of Epigenetics and Technologies for Epigenetics Analysis
2.1. General Characteristics of Epigenetics
2.2. Technologies for Epigenetics Analysis before the NGS Era
2.3. Technologies for Epigenetics Analysis in the NGS Era and Genome-Wide Epigenetics Analysis
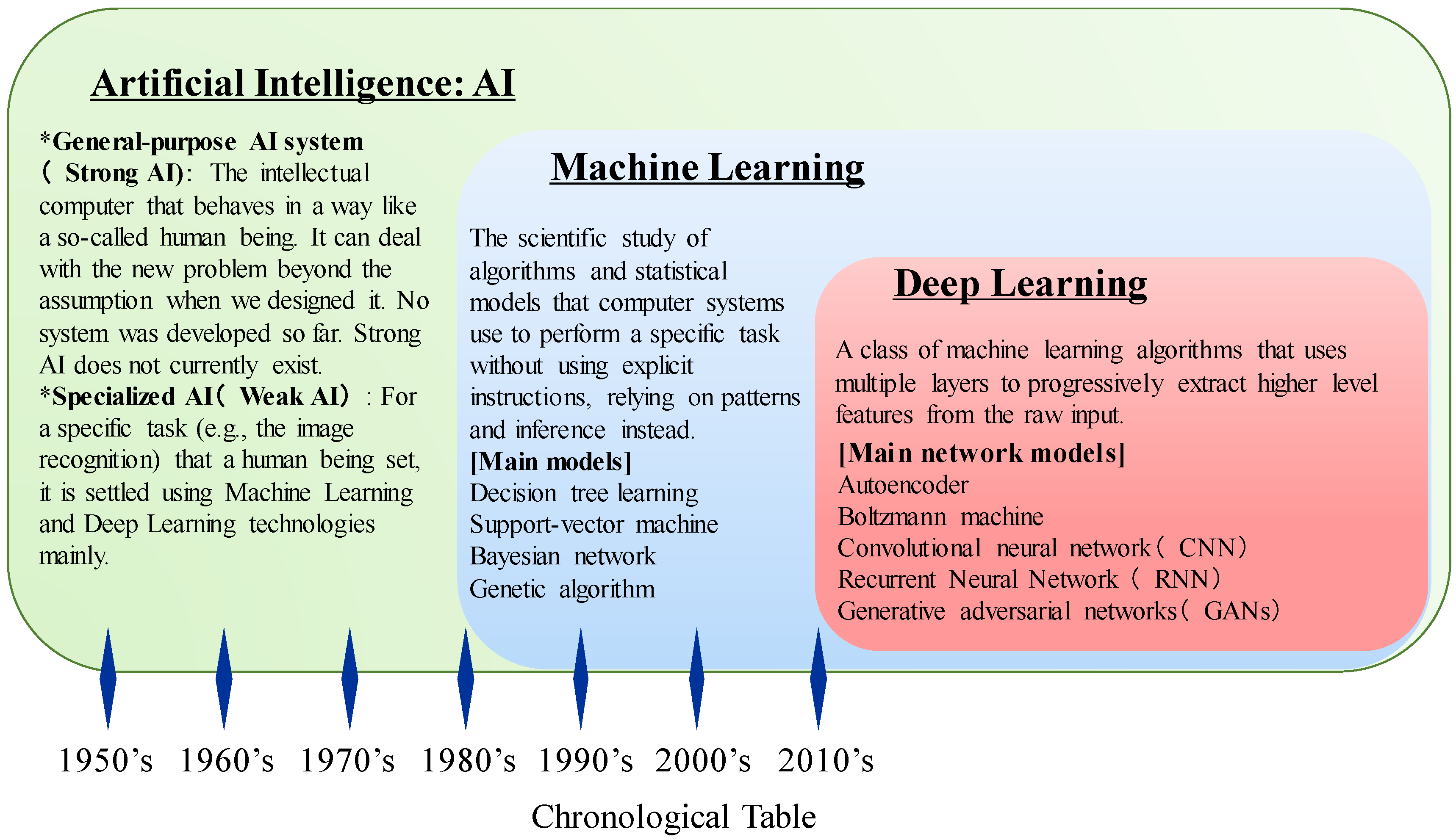
3. Development of Artificial Intelligence (AI)
3.1. Machine Learning Techniques and Evolution of AI Technologies
3.2. AI Revolution Using Deep Learning in the Big Data Era
4. Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using AI Technologies in the Medical Field
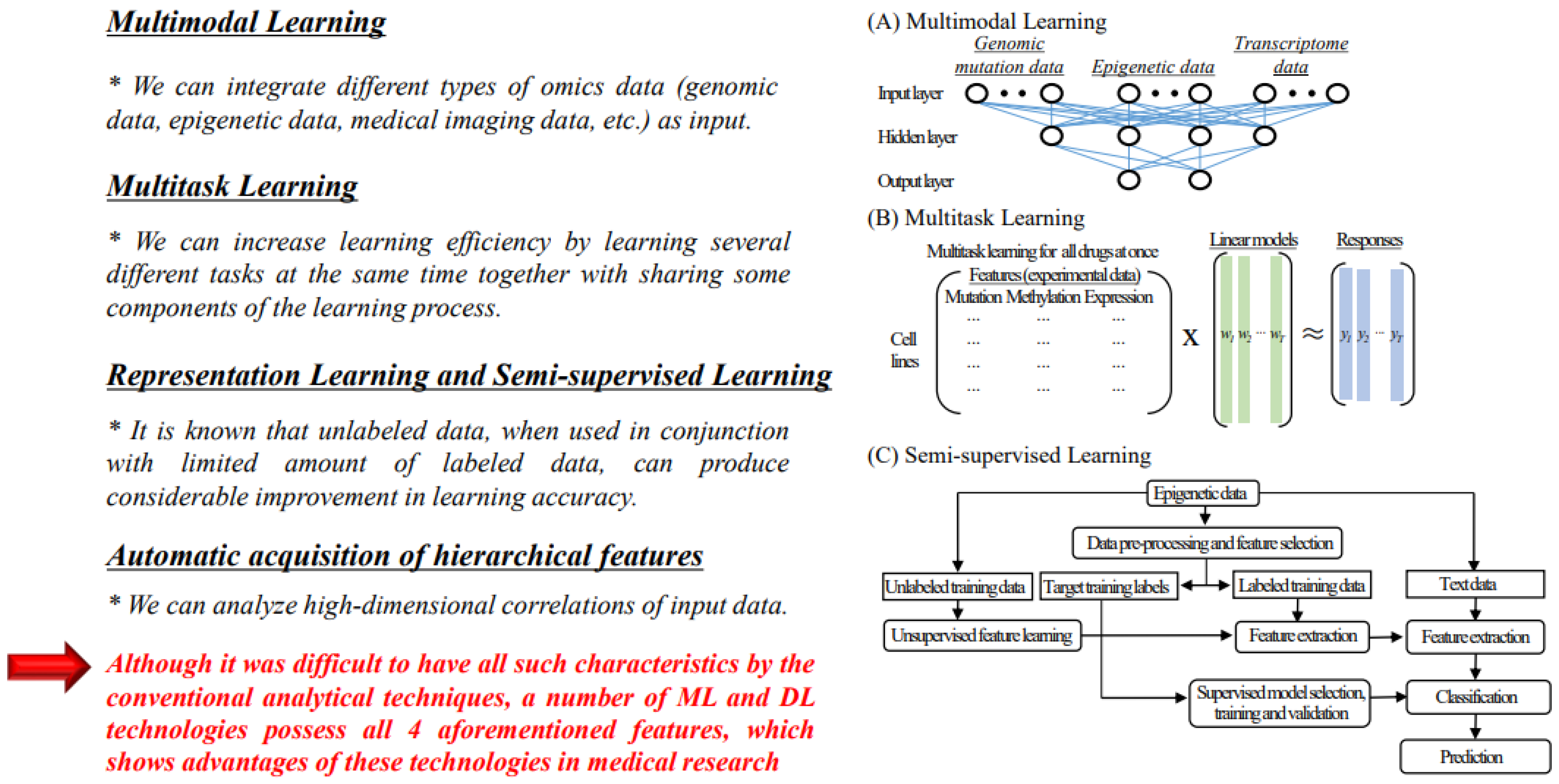
4.1. Advantages of Machine Learning and Deep Learning Technologies for Analysis of Medical Big Data
4.1.1. Multimodal Learning
4.1.2. Multitask Learning
4.1.3. Representation Learning and Semi-Supervised Learning
4.1.4. Automatic Acquisition of Hierarchical Characteristics
4.2. Analysis of Epigenetic Data and Integrated Analysis of Epigenetic Data and Other Omics Data Using AI Technologies
4.3. Issues of AI Technologies for Omics Analysis
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoosain, N.; Pearce, B.; Jacobs, C.; Benjeddou, M. Mapping SLCO1B1 Genetic Variation for Global Precision Medicine in Understudied Regions in Africa: A Focus on Zulu and Cape Admixed Populations. OMICS 2016, 20, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.R. Scientific and Technical Terms in Bioengineering and Biological Engineering; Apple Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Kasztura, M.; Richard, A.; Bempong, N.E.; Loncar, D.; Flahault, A. Cost-effectiveness of precision medicine: A scoping review. Int. J. Public Health 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, H.; Zhang, R. Challenges and future of precision medicine strategies for breast cancer based on a database on drug reactions. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Brusco, L.; Shaw, K.; Horombe, C.; Kopetz, S.; Davies, M.A.; Routbort, M.; Piha-Paul, S.A.; Janku, F.; Ueno, N.; et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J. Clin. Oncol. 2015, 33, 2753–2762. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Rozek, L.S.; Dolinoy, D.C.; Sartor, M.A.; Omenn, G.S. Epigenetics: Relevance and implications for public health. Annu. Rev. Public Health 2014, 35, 105–122. [Google Scholar] [CrossRef]
- Baylin, S.B. Resistance, epigenetics and the cancer ecosystem. Nat. Med. 2011, 17, 288–289. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Baylin, S.B. Linking cell signaling and the epigenetic machinery. Nat. Biotechnol. 2010, 28, 1033–1038. [Google Scholar] [CrossRef]
- Ezponda, T.; Popovic, R.; Shah, M.Y.; Martinez-Garcia, E.; Zheng, Y.; Min, D.J.; Will, C.; Neri, A.; Kelleher, N.L.; Yu, J.; et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 2013, 32, 2882–2890. [Google Scholar] [CrossRef]
- Cho, H.S.; Hayami, S.; Toyokawa, G.; Maejima, K.; Yamane, Y.; Suzuki, T.; Dohmae, N.; Kogure, M.; Kang, D.; Neal, D.E.; et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia 2012, 14, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Suzuki, T.; Dohmae, N.; Hayami, S.; Unoki, M.; Yoshimatsu, M.; Toyokawa, G.; Takawa, M.; Chen, T.; Kurash, J.K.; et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011, 71, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Yoshimatsu, M.; Veerakumarasivam, A.; Unoki, M.; Iwai, Y.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Yamaue, H.; et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: Involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol. Cancer 2010, 9, 59. [Google Scholar] [CrossRef]
- Saloura, V.; Cho, H.S.; Kyiotani, K.; Alachkar, H.; Zuo, Z.; Nakakido, M.; Tsunoda, T.; Seiwert, T.; Lingen, M.; Licht, J.; et al. WHSC1 Promotes Oncogenesis through Regulation of NIMA-related-kinase-7 in Squamous Cell Carcinoma of the Head and Neck. Mol. Cancer Res. 2015, 13, 293–304. [Google Scholar] [CrossRef]
- Tomasi, T.B.; Magner, W.J.; Khan, A.N. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol. Immunother. 2006, 55, 1159–1184. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Kelly, J.D.; Hayami, S.; Toyokawa, G.; Takawa, M.; Yoshimatsu, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Ponder, B.A.; et al. Enhanced expression of EHMT2 is involved in the proliferation of cancer cells through negative regulation of SIAH1. Neoplasia 2011, 13, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Toyokawa, G.; Daigo, Y.; Hayami, S.; Masuda, K.; Ikawa, N.; Yamane, Y.; Maejima, K.; Tsunoda, T.; Field, H.I.; et al. The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G1/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. Int. J. Cancer 2012, 131, E179–E189. [Google Scholar] [CrossRef]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef]
- Hamamoto, R.; Nakamura, Y. Dysregulation of protein methyltransferases in human cancer: An emerging target class for anticancer therapy. Cancer Sci. 2016. [Google Scholar] [CrossRef]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124. [Google Scholar] [CrossRef]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Cho, H.S.; Toyokawa, G.; Kogure, M.; Yamane, Y.; Iwai, Y.; Hayami, S.; Tsunoda, T.; Field, H.I.; Matsuda, K.; et al. The histone methyltransferase Wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosom. Cancer 2013, 52, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Kogure, M.; Takawa, M.; Cho, H.S.; Toyokawa, G.; Hayashi, K.; Tsunoda, T.; Kobayashi, T.; Daigo, Y.; Sugiyama, M.; Atomi, Y.; et al. Deregulation of the histone demethylase JMJD2A is involved in human carcinogenesis through regulation of the G(1)/S transition. Cancer Lett. 2013, 336, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Kogure, M.; Takawa, M.; Saloura, V.; Sone, K.; Piao, L.; Ueda, K.; Ibrahim, R.; Tsunoda, T.; Sugiyama, M.; Atomi, Y.; et al. The oncogenic polycomb histone methyltransferase EZH2 methylates lysine 120 on histone H2B and competes ubiquitination. Neoplasia 2013, 15, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Suzuki, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. SUV39H2 methylates and stabilizes LSD1 by inhibiting polyubiquitination in human cancer cells. Oncotarget 2015, 6, 16939–16950. [Google Scholar] [CrossRef]
- Silva, F.P.; Hamamoto, R.; Kunizaki, M.; Tsuge, M.; Nakamura, Y.; Furukawa, Y. Enhanced methyltransferase activity of SMYD3 by the cleavage of its N-terminal region in human cancer cells. Oncogene 2008, 27, 2686–2692. [Google Scholar] [CrossRef][Green Version]
- Takawa, M.; Masuda, K.; Kunizaki, M.; Daigo, Y.; Takagi, K.; Iwai, Y.; Cho, H.S.; Toyokawa, G.; Yamane, Y.; Maejima, K.; et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011, 102, 1298–1305. [Google Scholar] [CrossRef]
- Toyokawa, G.; Cho, H.S.; Iwai, Y.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Shimizu, N.; Tanaka, H.; Tsunoda, T.; et al. The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev. Res. 2011, 4, 2051–2061. [Google Scholar] [CrossRef]
- Toyokawa, G.; Cho, H.S.; Masuda, K.; Yamane, Y.; Yoshimatsu, M.; Hayami, S.; Takawa, M.; Iwai, Y.; Daigo, Y.; Tsuchiya, E.; et al. Histone Lysine Methyltransferase Wolf-Hirschhorn Syndrome Candidate 1 Is Involved in Human Carcinogenesis through Regulation of the Wnt Pathway. Neoplasia 2011, 13, 887–898. [Google Scholar] [CrossRef]
- Tsuge, M.; Hamamoto, R.; Silva, F.P.; Ohnishi, Y.; Chayama, K.; Kamatani, N.; Furukawa, Y.; Nakamura, Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5′ flanking region of SMYD3 is a risk factor for human cancers. Nat. Genet. 2005, 37, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, M.; Toyokawa, G.; Hayami, S.; Unoki, M.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Maehara, Y.; Ponder, B.A.; et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 2011, 128, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Sone, K.; Oda, K.; Hamamoto, R.; Kaneko, S.; Oki, S.; Kukita, A.; Machino, H.; Honjoh, H.; Kawata, Y.; et al. The histone methyltransferase WHSC1 is regulated by EZH2 and is important for ovarian clear cell carcinoma cell proliferation. BMC Cancer 2019, 19, 455. [Google Scholar] [CrossRef] [PubMed]
- Kukita, A.; Sone, K.; Oda, K.; Hamamoto, R.; Kaneko, S.; Komatsu, M.; Wada, M.; Honjoh, H.; Kawata, Y.; Kojima, M.; et al. Histone methyltransferase SMYD2 selective inhibitor LLY-507 in combination with poly ADP ribose polymerase inhibitor has therapeutic potential against high-grade serous ovarian carcinomas. Biochem. Biophys. Res. Commun. 2019, 513, 340–346. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, K.; Ryu, J.W.; Ryu, T.Y.; Lim, J.H.; Oh, J.H.; Min, J.K.; Jung, C.R.; Hamamoto, R.; Son, M.Y.; et al. The novel prognostic marker, EHMT2, is involved in cell proliferation via HSPD1 regulation in breast cancer. Int. J. Oncol. 2019, 54, 65–76. [Google Scholar] [CrossRef]
- Shigekawa, Y.; Hayami, S.; Ueno, M.; Miyamoto, A.; Suzaki, N.; Kawai, M.; Hirono, S.; Okada, K.I.; Hamamoto, R.; Yamaue, H. Overexpression of KDM5B/JARID1B is associated with poor prognosis in hepatocellular carcinoma. Oncotarget 2018, 9, 34320–34335. [Google Scholar] [CrossRef]
- Ryu, J.W.; Kim, S.K.; Son, M.Y.; Jeon, S.J.; Oh, J.H.; Lim, J.H.; Cho, S.; Jung, C.R.; Hamamoto, R.; Kim, D.S.; et al. Novel prognostic marker PRMT1 regulates cell growth via downregulation of CDKN1A in HCC. Oncotarget 2017, 8, 115444–115455. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, D.S.; Lis, J.T. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol. Cell Biol. 1985, 5, 2009–2018. [Google Scholar] [CrossRef]
- Collas, P. The current state of chromatin immunoprecipitation. Mol. Biotechnol. 2010, 45, 87–100. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Fcraga, M.F.; Esteller, M. DNA methylation: A profile of methods and applications. Biotechniques 2002, 33, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Allis, C.D. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc. Natl. Acad. Sci. USA 1995, 92, 6364–6368. [Google Scholar] [CrossRef]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Huang, T.H.; Perry, M.R.; Laux, D.E. Methylation profiling of CpG islands in human breast cancer cells. Hum. Mol. Genet. 1999, 8, 459–470. [Google Scholar] [CrossRef]
- Zuo, T.; Tycko, B.; Liu, T.M.; Lin, J.J.; Huang, T.H. Methods in DNA methylation profiling. Epigenomics 2009, 1, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Blat, Y.; Kleckner, N. Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell 1999, 98, 249–259. [Google Scholar] [CrossRef]
- Lieb, J.D.; Liu, X.; Botstein, D.; Brown, P.O. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 2001, 28, 327–334. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Sone, K.; Piao, L.; Nakakido, M.; Ueda, K.; Jenuwein, T.; Nakamura, Y.; Hamamoto, R. Critical role of lysine 134 methylation on histone H2AX for gamma-H2AX production and DNA repair. Nat. Commun. 2014, 5, 5691. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Stockwell, P.A.; Weeks, R.J.; Morison, I.M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. J. Biomed. Biotechnol. 2012, 2012, 741542. [Google Scholar] [CrossRef] [PubMed]
- Hakim, O.; Misteli, T. SnapShot: Chromosome confirmation capture. Cell 2012, 148, 1068-e1. [Google Scholar] [CrossRef]
- Gavrilov, A.; Eivazova, E.; Priozhkova, I.; Lipinski, M.; Razin, S.; Vassetzky, Y. Chromosome conformation capture (from 3C to 5C) and its ChIP-based modification. Methods Mol. Biol. 2009, 567, 171–188. [Google Scholar]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Schmid, C.D.; Bucher, P. ChIP-Seq data reveal nucleosome architecture of human promoters. Cell 2007, 131, 831–832. [Google Scholar] [CrossRef][Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Stevens, M.; Cheng, J.B.; Li, D.; Xie, M.; Hong, C.; Maire, C.L.; Ligon, K.L.; Hirst, M.; Marra, M.A.; Costello, J.F.; et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 2013, 23, 1541–1553. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fullwood, M.J.; Xu, H.; Mulawadi, F.H.; Velkov, S.; Vega, V.; Ariyaratne, P.N.; Mohamed, Y.B.; Ooi, H.S.; Tennakoon, C.; et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010, 11, R22. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Kumasaka, N.; Knights, A.J.; Gaffney, D.J. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 2016, 48, 206–213. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Hofmeister, B.T.; Vollmers, C.; DuBois, R.M.; Schmitz, R.J. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017, 45, e41. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Blalock, E.L.; Barwick, B.G.; Haines, R.R.; Wei, C.; Sanz, I.; Boss, J.M. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naive SLE B cells. Sci. Rep. 2016, 6, 27030. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. Single-cell ATAC-seq: Strength in numbers. Genome Biol. 2015, 16, 172. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Saligrama, N.; Buenrostro, J.D.; Wei, Y.; Wu, B.; Rubin, A.J.; Granja, J.M.; Lareau, C.A.; Li, R.; Qi, Y.; et al. Transcript-indexed ATAC-seq for precision immune profiling. Nat. Med. 2018, 24, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zibetti, C.; Shang, P.; Sripathi, S.R.; Zhang, P.; Cano, M.; Hoang, T.; Xia, S.; Ji, H.; Merbs, S.L.; et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 2018, 9, 1364. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Preussner, J.; Chen, X.; Guenther, S.; Yuan, X.; Yekelchyk, M.; Kuenne, C.; Looso, M.; Zhou, Y.; Teichmann, S.; et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun. 2018, 9, 4877. [Google Scholar] [CrossRef] [PubMed]
- Dryden, N.H.; Broome, L.R.; Dudbridge, F.; Johnson, N.; Orr, N.; Schoenfelder, S.; Nagano, T.; Andrews, S.; Wingett, S.; Kozarewa, I.; et al. Unbiased Analysis of Potential Targets of Breast Cancer Susceptibility Loci by Capture Hi-C. Genome Res. 2014, 24, 1854–1868.77. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. The CBP co-activator is a histone acetyltransferase. Nature 1996, 384, 641–643. [Google Scholar] [CrossRef]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef]
- Zhou, L.; Ng, H.K.; Drautz-Moses, D.I.; Schuster, S.C.; Beck, S.; Kim, C.; Chambers, J.C.; Loh, M. Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing. Sci. Rep. 2019, 9, 10383. [Google Scholar] [CrossRef]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef]
- Yan, H.; Tian, S.; Slager, S.L.; Sun, Z. ChIP-seq in studying epigenetic mechanisms of disease and promoting precision medicine: Progresses and future directions. Epigenomics 2016, 8, 1239–1258. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Schulz, M.H.; Look, T.; Begemann, M.; Zenke, M.; Costa, I.G. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Hebb, D.O. The Organization of Behavior: A Neuropsychological Theory; Wiley & Sons: New York, NY, USA, 1949. [Google Scholar]
- Liu, J.; Gong, M.; Miao, Q. Modeling Hebb Learning Rule for Unsupervised Learning. In Proceedings of the Twenty-Sixth International Joint Conference on Artificial Intelligence (IJCAI-17), Melbourne, Australia, 19–25 August 2017; pp. 2315–2321. [Google Scholar]
- Kuriscak, E.; Marsalek, P.; Stroffek, J.; Toth, P. Biological context of Hebb learning in artificial nural networks, a review. Neurocomputing 2015, 152, 27–35. [Google Scholar] [CrossRef]
- Rosenblatt, F. The Perceptron: A Probabilistic Model for Information Storage and Organization in the Brain. Psychol. Rev. 1958, 65, 386–408. [Google Scholar] [CrossRef]
- Fukushima, K. Neocognitron: A self-organizing neural network model for a mechanism of pattern recognition unaffected by shift in position. Biol. Cybern. 1980, 36, 193–202. [Google Scholar] [CrossRef]
- Nebauer, C. Evaluation of convolutional neural networks for visual recognition. IEEE Trans. Neural Netw. 1998, 9, 685–696. [Google Scholar] [CrossRef]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning reprensations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Watkins, C.J.C.H.; Dayan, P. Q-learning. Mach. Learn. 1992, 8, 279–292. [Google Scholar] [CrossRef]
- Watkins, C.J.C.H. Learning from Delayed Rewards. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 1989. [Google Scholar]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. A Training Algorithm for Optimal Margin Classifiers. In Proceedings of the 5th Annual Workshop on Computational Learning Theory (COLT’92), Pittsburgh, PA, USA, 27–29 July 1992; pp. 144–152. [Google Scholar]
- Vapnik, V.; Lerner, A. Pattern Recognition Using Generalized Portrait Method. Autom. Remote Control 1963, 24, 774–780. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Ben-Hur, A.; Horn, D.; Siegelmann, H.; Vapnilk, V. Support vector clustering. J. Mach. Learn. Res. 2001, 2, 125–137. [Google Scholar] [CrossRef]
- Ho, T.K. Random Decision Forests. In Proceedings of the IEEE Third International Conference on Document Analysis and Recognition, Montreal, QC, USA, 14–16 August 1995; Volume 1, pp. 278–282. [Google Scholar]
- Hastie, T.; TIbshirani, R.; Friedman, J. The Elements of Statistical Learning, 2nd ed.; Springer: Berlin, Germany, 2008; pp. 587–588. [Google Scholar]
- Lohr, S. IBM Is Counting on Its Bet on Watson, and Paying Big Money for It; The New York Times: New York, NY, USA, 2016. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Marblestone, A.H.; Wayne, G.; Kording, K.P. Toward an Integration of Deep Learning and Neuroscience. Front. Comput. Neurosci. 2016, 10, 94. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 1, 1097–1105. [Google Scholar] [CrossRef]
- Alom, M.Z.; Taha, T.M.; Yakopcic, C.; Westberg, S.; Sidike, P.; Nasrin, M.S.; Hasan, M.; Van Essen, B.C.; Awwal, A.; Asari, V.K. A State-of-the-Art Survey on Deep Learning Theory and Architectures. Electronics 2019, 8, 292. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Silver, D.; Schrittwieser, J.; Simonyan, K.; Antonoglou, I.; Huang, A.; Guez, A.; Hubert, T.; Baker, L.; Lai, M.; Bolton, A.; et al. Mastering the game of Go without human knowledge. Nature 2017, 550, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Saito, Y.; Imaoka, H.; Saiko, M.; Yamada, S.; Kondo, H.; Takamaru, H.; Sakamoto, T.; Sese, J.; Kuchiba, A.; et al. Development of a real-time endoscopic image diagnosis support system using deep learning technology in colonoscopy. Sci. Rep. 2019, 9, 14465. [Google Scholar] [CrossRef]
- Yasutomi, S.; Arakaki, T.; Hamamoto, R. Shadow Detection for Ultrasound Images Using Unlabeled Data and Synthetic Shadows. arXiv 2019, arXiv:1908.01439. [Google Scholar]
- Yasutomi, S.; Sakai, A.; Komatsu, M.; Matsuoka, R.; Komatsu, R.; Arakaki, T.; Tokunaka, M.; Kobayashi, K.; Asada, K.; Kaneko, S.; et al. Unsupervised Shadow Detection for Ultrasound Images by Deep Learning. IEICE Tech. Rep. 2019, 118, 151–156. [Google Scholar]
- Srivastava, N.; Salakhutdinov, R. Multimodal Learning with Deep Boltzmann Machines. J. Mach. Learn. Res. 2014, 15, 2949–2980. [Google Scholar]
- Zhu, B.; Song, N.; Shen, R.; Arora, A.; Machiela, M.J.; Song, L.; Landi, M.T.; Ghosh, D.; Chatterjee, N.; Baladandayuthapani, V.; et al. Integrating Clinical and Multiple Omics Data for Prognostic Assessment across Human Cancers. Sci. Rep. 2017, 7, 16954. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Celik, S.; Logsdon, B.A.; Lundberg, S.M.; Martins, T.J.; Oehler, V.G.; Estey, E.H.; Miller, C.P.; Chien, S.; Dai, J.; et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat. Commun. 2018, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Paskov, I.; Paskov, H.; Gonzalez, A.J.; Leslie, C.S. Multitask learning improves prediction of cancer drug sensitivity. Sci. Rep. 2016, 6, 31619. [Google Scholar] [CrossRef]
- Xiao, Y.; Wu, J.; Lin, Z.; Zhao, X. A semi-supervised deep learning method based on stacked sparse auto-encoder for cancer prediction using RNA-seq data. Comput. Methods Programs Biomed. 2018, 166, 99–105. [Google Scholar] [CrossRef]
- Strezoski, G.; Van Noord, N.; Worring, M. Learning Task Relatedness in Multi-Task Learning for Images in Context. arXiv 2019, arXiv:1904.03011. [Google Scholar]
- Baxter, J. A model of inductive bias learning. J. Artif. Intell. Res. 2000, 12, 149–198. [Google Scholar] [CrossRef]
- Caruana, R. Multitask Learning. Mach. Learn. 1997, 28, 41–75. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, Q. A Survey on Multi-Task Learning. arXiv 2018, arXiv:1707.08114v2.119. [Google Scholar]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gonen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-ud-din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef]
- Gonen, M.; Margolin, A.A. Drug susceptibility prediction against a panel of drugs using kernelized Bayesian multitask learning. Bioinformatics 2014, 30, i556–i563. [Google Scholar] [CrossRef] [PubMed]
- Heider, D.; Senge, R.; Cheng, W.; Hullermeier, E. Multilabel classification for exploiting cross-resistance information in HIV-1 drug resistance prediction. Bioinformatics 2013, 29, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Margolin, A.A.; Haery, L.; Brown, E.; Cucolo, L.; Julian, B.; Shehata, S.; Kung, A.L.; Beroukhim, R.; Golub, T.R. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell 2012, 21, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, H.; Fang, Y.; Wang, J.; Zheng, X.; Liu, X.S. Predicting Anticancer Drug Responses Using a Dual-Layer Integrated Cell Line-Drug Network Model. PLoS Comput. Biol. 2015, 11, e1004498. [Google Scholar] [CrossRef]
- Bengio, Y.; Courville, A.; Vincent, P. Representation learning: A review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intel. 2013, 35, 1798–1828. [Google Scholar] [CrossRef]
- Oliver, A.; Odena, A.; Raffel, C.; Cubuk, E.D.; Goodfellow, I.J. Realistic Evaluation of Deep Semi-Supervised Learning Algorithms. arXiv 2018, arXiv:1804.09170. [Google Scholar]
- Shi, M.; Zhang, B. Semi-supervised learning improves gene expression-based prediction of cancer recurrence. Bioinformatics 2011, 27, 3017–3023. [Google Scholar] [CrossRef]
- Chapelle, O.; Sindhwani, V.; Keerthi, S.S. Optimization Techniques for Semi-Supervised Support Vector Machines. J. Mach. Learn. Res. 2008, 9, 203–233. [Google Scholar]
- Bengio, Y. Learning Deep Architectures for AI. Found. Trends® Mach. Learn. 2009, 2, 1–127. [Google Scholar] [CrossRef]
- Leygo, C.; Williams, M.; Jin, H.C.; Chan, M.W.Y.; Chu, W.K.; Grusch, M.; Cheng, Y.Y. DNA Methylation as a Noninvasive Epigenetic Biomarker for the Detection of Cancer. Dis. Mark. 2017, 2017, 3726595. [Google Scholar] [CrossRef]
- Elliott, G.O.; Johnson, I.T.; Scarll, J.; Dainty, J.; Williams, E.A.; Garg, D.; Coupe, A.; Bradburn, D.M.; Mathers, J.C.; Belshaw, N.J. Quantitative profiling of CpG island methylation in human stool for colorectal cancer detection. Int. J. Colorectal Dis. 2013, 28, 35–42. [Google Scholar] [CrossRef]
- Linton, A.; Cheng, Y.Y.; Griggs, K.; Schedlich, L.; Kirschner, M.B.; Gattani, S.; Srikaran, S.; Chuan-Hao Kao, S.; McCaughan, B.C.; Klebe, S.; et al. An RNAi-based screen reveals PLK1, CDK1 and NDC80 as potential therapeutic targets in malignant pleural mesothelioma. Br. J. Cancer 2014, 110, 510–519. [Google Scholar] [CrossRef]
- Yang, X.; Dai, W.; Kwong, D.L.; Szeto, C.Y.; Wong, E.H.; Ng, W.T.; Lee, A.W.; Ngan, R.K.; Yau, C.C.; Tung, S.Y.; et al. Epigenetic markers for noninvasive early detection of nasopharyngeal carcinoma by methylation-sensitive high resolution melting. Int. J. Cancer 2015, 136, E127–E135. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Merve, A.; Millner, T.O.; Marino, S. Integrated phenotype-genotype approach in diagnosis and classification of common central nervous system tumours. Histopathology 2019, 75, 299–311. [Google Scholar] [CrossRef]
- Van den Bent, M.J. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: A clinician’s perspective. Acta Neuropathol. 2010, 120, 297–304. [Google Scholar] [CrossRef]
- Ellison, D.W.; Kocak, M.; Figarella-Branger, D.; Felice, G.; Catherine, G.; Pietsch, T.; Frappaz, D.; Massimino, M.; Grill, J.; Boyett, J.M.; et al. Histopathological grading of pediatric ependymoma: Reproducibility and clinical relevance in European trial cohorts. J. Negat. Results Biomed. 2011, 10, 7. [Google Scholar] [CrossRef]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef]
- Hovestadt, V.; Remke, M.; Kool, M.; Pietsch, T.; Northcott, P.A.; Fischer, R.; Cavalli, F.M.; Ramaswamy, V.; Zapatka, M.; Reifenberger, G.; et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013, 125, 913–916. [Google Scholar] [CrossRef]
- Reuss, D.E.; Kratz, A.; Sahm, F.; Capper, D.; Schrimpf, D.; Koelsche, C.; Hovestadt, V.; Bewerunge-Hudler, M.; Jones, D.T.; Schittenhelm, J.; et al. Adult IDH wild type astrocytomas biologically and clinically resolve into other tumor entities. Acta Neuropathol. 2015, 130, 407–417. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.R.; Witt, H.; Hovestadt, V.; Zucknick, M.; Kool, M.; Pearson, D.M.; Korshunov, A.; Ryzhova, M.; Ichimura, K.; Jabado, N.; et al. Differential expression and methylation of brain developmental genes define location-specific subsets of pilocytic astrocytoma. Acta Neuropathol. 2013, 126, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stutz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Wiestler, B.; Capper, D.; Sill, M.; Jones, D.T.; Hovestadt, V.; Sturm, D.; Koelsche, C.; Bertoni, A.; Schweizer, L.; Korshunov, A.; et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol. 2014, 128, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, S.S.; Boyle, A.P. Mining the Unknown: Assigning Function to Noncoding Single Nucleotide Polymorphisms. Trends Genet. 2017, 33, 34–45. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol. 2016, 17, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wong, W.H.; Jiang, R. DeepTACT: Predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Res. 2019, 47, e60. [Google Scholar] [CrossRef]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef]
- Zhang, Y.; An, L.; Xu, J.; Zhang, B.; Zheng, W.J.; Hu, M.; Tang, J.; Yue, F. Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat. Commun. 2018, 9, 750. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, Z.; Zhang, K.; Wang, M.; Medovoy, D.; Whitaker, J.W.; Ding, B.; Li, N.; Zheng, L.; Wang, W. Constructing 3D interaction maps from 1D epigenomes. Nat. Commun. 2016, 7, 10812. [Google Scholar] [CrossRef]
- Al Bkhetan, Z.; Plewczynski, D. Three-dimensional Epigenome Statistical Model: Genome-wide Chromatin Looping Prediction. Sci. Rep. 2018, 8, 5217. [Google Scholar] [CrossRef]
- Whalen, S.; Truty, R.M.; Pollard, K.S. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016, 48, 488–496. [Google Scholar] [CrossRef]
- Wallace, B.C.; Small, K.; Brodley, C.E.; Trikalinos, T.A. Class Imbalance, Redux. In Proceedings of the 2011 IEEE ICDM 11th International Conference on Data Mining, Vancouver, BC, Canada, 11–14 December 2011; pp. 754–763. [Google Scholar]
- Sirmacek, B.; Kivits, M. Semantic Segmentation of Skin Lesions using a Small Data Set. arXiv 2019, arXiv:1910.10534. [Google Scholar]
- Salman, S.; Liu, X. Overfitting Mechanism and Avoidance in Deep Neural Networks. arXiv 2019, arXiv:1901.06566. [Google Scholar]
- Zhang, C.; Bengio, S.; Hardt, M.; Recht, B.; Vinyals, O. Understanding deep learning requires rethinking generalization. arXiv 2017, arXiv:1611.03530. [Google Scholar]
- Arpit, D.; Jastrzębski, S.; Ballas, N.; Krueger, D.; Bengio, E.; Kanwal, M.S.; Maharaj, T.; Fischer, A.; Courville, A.; Bengio, Y.; et al. A Closer Look at Memorization in Deep Networks. arXiv 2017, arXiv:1706.05394. [Google Scholar]
- Rand-Hendriksen, K.; Ramos-Goni, J.M.; Augestad, L.A.; Luo, N. Less Is More: Cross-Validation Testing of Simplified Nonlinear Regression Model Specifications for EQ-5D-5L Health State Values. Value Health 2017, 20, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Lever, J.; Krzywinski, M.; Altman, N. Regularization. Nat. Methods 2016, 13, 803–804. [Google Scholar] [CrossRef]
- Murugan, P.; Durairaj, S. Regularization and Optimization strategies in Deep Convolutional Neural Network. arXiv 2017, arXiv:1712.04711. [Google Scholar]
- Collins, A.; Yao, Y. Machine Learning Approaches: Data Integration for Disease Prediction and Prognosis. In Applied Computational Genomics; Springer: Berlin, Germany, 2018. [Google Scholar]
- Wu, Q.; Boueiz, A.; Bozkurt, A.; Masoomi, A.; Wang, A.; DeMeo, D.L.; Weiss, S.T.; Qiu, W. Deep Learning Methods for Predicting Disease Status Using Genomic Data. J. Biometr. Biostat. 2018, 9, 417. [Google Scholar]
- Wu, S.; Jiang, H.; Shen, H.; Yang, Z. Gene Selection in Cancer Classification Using Sparse Logistic Regression with L1/2 Regularization. Appl. Sci. 2018, 8, 1569. [Google Scholar] [CrossRef]
- Romero, A.; Carrier, P.L.; Erraqabi, A.; Sylvain, T.; Auvolat, A.; Dejoie, E.; Legault, M.A.; Dubé, M.P.; Hussin, J.G.; Bengio, Y. Diet Networks: Thin Parameters for Fat Genomics. arXiv 2016, arXiv:1611.09340. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Method Name | Purpose | Methodology | Era | Ref. |
|---|---|---|---|---|
| Chromatin immunoprecipitation (ChIP) assay | Analysis of histone modification and transcription factor binding status | A type of immunoprecipitation experimental technique used to investigate the interaction between proteins and DNA in the cell. It aims to determine whether specific proteins are associated with specific genomic regions, and also aims to determine the specific location in the genome that various histone modifications are associated with. | 1985 | [39,40] |
| Bisulfite sequencing (BS-Seq) | DNA methylation analysis | Treatment of DNA with bisulfite converts cytosine residues to uracil, but leaves 5-methylcytosine residues unaffected. Hence, DNA that has been treated with bisulfite retains only methylated cytosines. | 1992 | [41,42] |
| Histone acetyltransferase (HAT) assay | Assay for histone acetyltransferase activity | Multiple biochemical HAT assays have been described; these assays measure HAT activity by detecting either the acetylated histone-based product (direct) or the free CoA product (indirect). | 1995 | [43,44] |
| DNA methylation array: differential methylation hybridization (DMH) | DNA methylation analysis | A DNA array-based method, called differential methylation hybridization (DMH), to identify hypermethylated sequences in tumor cells by simultaneously screening many CpG island loci derived from a genomic library, CGI. | 1999 | [45,46] |
| ChIP-on-chip | Genome-wide analysis of histone modification and transcription factor binding status | A technology that combines chromatin immunoprecipitation (ChIP) with DNA microarray (chip). It allows the identification of the cistrome, the sum of binding sites, for DNA-binding proteins on a genome-wide basis. | 1999 | [47,48] |
| Histone methyltransferase (HMT) assay | Assay for histone methyltransferase activity | Radiometric Assays, Mass Spectrometry, Anti-Methylation Antibody-Based Detection, Enzyme-Coupled SAH Detection, Protease-Coupled Detection, Competition Binding. | 2000 | [49,50] |
| Histone demethylase (HDMT) assay | Assay for histone demethylase activity | Measuring the release of radiolabeled formaldehyde from 3H-labeled methylated histone substrates, by monitoring the change in methylation levels of histone substrates by immunoblotting with site-specific methyl-histone antibodies, or by using mass spectrometry to detect reductions in histone peptide masses that correspond to methyl groups. | 2004 | [51,52] |
| Reduced Representation Bisulfite Sequencing (RRBS) | Genome-wide DNA methylation analysis | An efficient and high-throughput technique for analyzing the genome-wide methylation profiles on a single nucleotide level; it combines restriction enzymes and bisulfite sequencing to enrich for areas of the genome with a high CpG content. | 2005 | [53,54] |
| ChIP-loop | Chromosome conformation capture technique | This method combines the standard 3C protocol with a routine ChIP protocol; it allows the selective identification of long-range chromatin interactions between loci that are bound to specific proteins of interest. | 2005 | [55,56] |
| ChIP-sequencing (ChIP-seq) | Genome-wide analysis of histone modification and transcription factor binding status | By combining chromatin immunoprecipitation (ChIP) assays with next-generation sequencing (NGS), ChIP sequencing (ChIP-seq) is a powerful method for identifying genome-wide DNA binding sites for transcription factors and other proteins. | 2007 | [57,58] |
| Whole Genome Bisulfite Sequencing (WGBS) | Genome-wide DNA methylation analysis | A NGS technology used to determine the DNA methylation status of single cytosines by treating the DNA with sodium bisulfite before sequencing. | 2009 | [59,60] |
| Hi-C | Chromosome conformation capture technique | A genome-wide chromatin conformation capture protocol using proximity ligation. The technology is of special interest for three-dimensional genome organization in the nucleus and de novo genome assemblies. | 2009 | [61,62] |
| ChIA-PET | Determination of de novo long-range chromatin interactions genome-wide | The ChIA-PET method combines ChIP-based methods, and Chromosome conformation capture (3C), to extend the capabilities of both approaches. | 2009 | [63,64] |
| ATAC-seq | Identification of accessible DNA regions | This method relies on NGS library construction using the hyperactive transposase Tn5. NGS adapters are loaded onto the transposase, which allows simultaneous fragmentation of chromatin and integration of those adapters into open chromatin regions. | 2013 | [65,66,67,68,69,70,71,72,73,74] |
| Capture Hi-C (CHi-C) | Identification of higher resolution mapping of chromatin interactions | The CHi-C is a new technique for assessing genome organization based on chromosome conformation capture coupled to oligonucleotide capture of regions of interest like gene promoters. | 2014 | [75] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamamoto, R.; Komatsu, M.; Takasawa, K.; Asada, K.; Kaneko, S. Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. Biomolecules 2020, 10, 62. https://doi.org/10.3390/biom10010062
Hamamoto R, Komatsu M, Takasawa K, Asada K, Kaneko S. Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. Biomolecules. 2020; 10(1):62. https://doi.org/10.3390/biom10010062
Chicago/Turabian StyleHamamoto, Ryuji, Masaaki Komatsu, Ken Takasawa, Ken Asada, and Syuzo Kaneko. 2020. "Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine" Biomolecules 10, no. 1: 62. https://doi.org/10.3390/biom10010062
APA StyleHamamoto, R., Komatsu, M., Takasawa, K., Asada, K., & Kaneko, S. (2020). Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. Biomolecules, 10(1), 62. https://doi.org/10.3390/biom10010062