Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement

 ,
,  , , and
, , and
Abstract
1. Introduction
2. The Cholinergic Hypothesis of Alzheimer’s Disease
2.1. Acetylcholine—The Neurotransmitter of The Cholinergic Synapse: Synthesis, Depolarization, Release, Inactivation, and Recovery
2.2. Cholinergic Receptors
3. Cholinesterase Inhibitors, A Positive Approach to Delay the Progress of Alzheimer’s Disease
4. Different Preclinical Models to Study Cholinergic Hypothesis of Alzheimer’s Disease
5. From Single Targets towards Multi-Target Directed Ligands
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| WHO | World Health Organization |
| AChE | acetylcholinesterase |
| AChEIs | acetylcholinesterase inhibitors |
| BuChE | butyrylcholinesterase |
| ChEIs | cholinesterase inhibitors |
| CYP | cytochrome system |
| USA | United States of America |
| Aβ | amyloid beta |
| APP | amyloid precursor protein |
| ACh | acetylcholine |
| CSF | cerebrospinal fluid |
| ChAT | choline acetyltransferase |
| acetyl-CoA | acetyl-coenzyme A |
| CNS | central nervous system |
| Gq | G-stimulatory protein |
| PLC | Phospholipase C |
| IP3 | inositoltrophosphate |
| DAG | diacylglycerol |
| cAMP | cyclic adenosine monophosphate |
| BBB | blood brain barrier |
| P-gp | P-glycoprotein 1 |
| MTDLs | multi-target-directed ligands |
| BACE 1 | β-secretase, β-site APP cleaving enzyme 1 |
| ABAD | amyloid-binding alcohol dehydrogenase |
References
- Ghumatkar, P.J.; Patil, S.P.; Jain, P.D.; Tambe, R.M.; Sathaye, S. Nootropic, neuroprotective and neurotrophic effects of phloretin in scopolamine induced amnesia in mice. Pharmacol. Biochem. Behav. 2015, 135, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 5, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International (ADI): London, UK, 2018. [Google Scholar]
- Maresova, P.; Mohelska, H.; Dolejs, J.; Kuca, K. Socio-economic Aspects of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer Dement. 2013, 9, 63–75. [Google Scholar] [CrossRef]
- Gustavsson, A.; Green, C.; Jones, R.W.; Förstl, H.; Simsek, D.; de Reydet de Vulpillieres, F.; Luthman, S.; Adlard, N.; Bhattacharyya, S.; Wimo, A. Current issues and future research priorities for health economic modelling across the full continuum of Alzheimer’s disease. Alzheimer Dement. 2017, 13, 312–321. [Google Scholar] [CrossRef]
- Cimler, R.; Maresova, P.; Kuhnova, J.; Kuca, K. Predictions of Alzheimer’s disease treatment and care costs in European countries. PLoS ONE 2019, 14, e0210958. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, 1–23. [Google Scholar] [CrossRef]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s disease and some therapeutic approaches targeting A β by using several synthetic and herbal compounds. Oxid. Med. Cell. Longev. 2016, 2016, 22. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.E.; Ginsberg, S.D.; Counts, S.E.; Mufson, E.J. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2009, 8, 1703–1718. [Google Scholar]
- Ferreira-Vieira, T.; Guimaraes, I.; Silva, F.; Ribeiro, F. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, S.; Morioka, M.S.; Kikuchi, M. Dysregulation of Iron Metabolism in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Adv. Pharmacol. Sci. 2011, 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.; Cotman, C.; Lynch, G.; Blass, J. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer Dement. 2017, 13, 178–182. [Google Scholar]
- Gamba, P.; Staurenghi, E.; Testa, G.; Giannelli, S.; Sottero, B.; Leonarduzzi, G. A crosstalk between brain cholesterol oxidation and glucose metabolism in Alzheimer’s disease. Front. Neurosci. 2019, 13, 1–9. [Google Scholar] [CrossRef]
- Martinez, A.; Castro, A. Novel cholinesterase inhibitors as future effective drugs for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2006, 15, 1–12. [Google Scholar] [CrossRef]
- Mehta, M.; Adem, A.; Sabbagh, M. New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Int. J. Alzheimer Dis. 2012, 2012, 728983. [Google Scholar] [CrossRef]
- Sun, Y.; Lai, M.; Lu, C.; Chen, R. How long can patients with mild or moderate Alzheimer’s dementia maintain both the cognition and the therapy of cholinesterase inhibitors: A national population-based study. Eur. J. Neurol. 2008, 15, 278–283. [Google Scholar] [CrossRef]
- Nestor, P.J.; Scheltens, P.; Hodges, J.R. Advances in the early detection of Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 10, S34. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatr. 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Mobley, W.C. Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons:Converging insights from alternative hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef] [PubMed]
- Wurtman, R.J. American Society for Clinical Investigation. J. Clin. Investig. 1994, 94, 470. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Stevens, J.C.; Beck, C.; Dubinsky, R.M.; Kaye, J.A.; Gwyther, L.; Mohs, R.C.; Thal, L.J.; Whitehouse, P.J.; DeKosky, S.T.; et al. Practice parameter: Management of dementia (an evidence-based review): Report of the quality standards subcommittee of the American Academy of Neurology. Neurology 2001, 56, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Richter, N.; Nellessen, N.; Dronse, J.; Dillen, K.; Jacobs, H.I.L.; Langen, K.J.; Dietlein, M.; Kracht, L.; Neumaier, B.; Fink, G.R.; et al. Spatial distributions of cholinergic impairment and neuronal hypometabolism differ in MCI due to AD. NeuroImage Clin. 2019, 24, 101978. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef]
- Talesa, V.N. Acetylcholinesterase in Alzheimer’s disease. Mech. Ageing Dev. 2001, 122, 1961–1969. [Google Scholar] [CrossRef]
- Rees, T.M.; Brimijoin, S. The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today 2003, 39, 75–83. [Google Scholar] [CrossRef]
- Francis, P.T. The Interplay of Neurotransmitters in Alzheimer’s Disease. CNS Spectr. 2005, 10, 6–9. [Google Scholar] [CrossRef]
- Tabet, N. Acetylcholinesterase inhibitors for Alzheimer’s disease: Anti-inflammatories in acetylcholine clothing! Age Ageing 2006, 35, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, Y.; Zhang, C.; Zhao, Y.; Bu, G.; Xu, H.; Zhang, Y.W. M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Pavía, J.; De Ceballos, M.; Sanchez De La Cuesta, F. Alzheimer’s disease: Relationship between muscarinic cholinergic receptors, β-amyloid and tau proteins. Fundam. Clin. Pharmacol. 1998, 12, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; LaFerla, F.M. The role of nicotinic acetylcholine receptors in Alzheimer’s disease. J. Physiol. Paris 2006, 99, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.D.; Jones, A.K.; Brown, L.A.; Sattelle, D.B. Nicotinic acetylcholine receptor signalling: Roles in alzheimer’s disease and amyloid neuroprotection. Pharmacol. Rev. 2009, 61, 39–61. [Google Scholar] [CrossRef]
- Georgi, S. Nicotinic Acetylcholine Receptors and Alzheimer’s Disease Therapeutics: A Review of Current Literature. J. Young Investig. 2005, 4, 7–14. [Google Scholar]
- Cao, J.; Hou, J.; Ping, J.; Cai, D. Advances in developing novel therapeutic strategies for Alzheimer’s disease 11 Medical and Health Sciences 1109 Neurosciences. Mol. Neurodegener. 2018, 13, 64. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sánchez-López, E.; García, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Lourenco, M.V.; de Felice, F.G. Challenges for Alzheimer’s disease therapy: Insights from novel mechanisms beyond memory defects. Front. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinesterases: New Roles in Brain Function and in Alzheimer’s Disease. Neurochem. Res. 2003, 28, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Schalkt, I.; Ehret-Sabatiert, L.; Bouett, F.; Goeldnert, M.; Hirtht, C.; Axelsen, P.H.; Silmanii, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.; Gracon, S.I.; Hershey, L.A.; Lewis, K.W.; Sadowsky, C.H.; Dolan Ureno, J. A Controlled Trial of Tacrine in Alzheimer’s Disease. JAMA J. Am. Med. Assoc. 1992, 268, 2523–2529. [Google Scholar] [CrossRef]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic Effects of Tacrine Administration in Patients With Alzheimer’s Disease. JAMA J. Am. Med. Assoc. 1994, 271, 992–998. [Google Scholar] [CrossRef]
- Grossberg, G.T. Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease: Getting On and Staying On. Curr. Ther. Res. 2003, 64, 216–235. [Google Scholar] [CrossRef]
- Jann, M.W.; Shirley, K.L.; Small, G.W. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin. Pharmacokinet. 2002, 41, 719–739. [Google Scholar] [CrossRef]
- Inglis, F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int. J. Clin. Pract. Suppl. 2002, 127, 45–63. [Google Scholar]
- Onor, M.L.; Trevisiol, M.; Aguglia, E. Rivastigmine in the treatment of Alzheimer’s disease: An update. Clin. Interv. Aging 2007, 2, 17–32. [Google Scholar] [CrossRef]
- Bentham, P.; Gray, R.; Sellwood, E.; Raftery, J.; Rösler, M.; Selai, C.E.; Trimble, M.R.; Rossor, M.N.; Harvey, R.J.; Storosum, J.G.; et al. Effectiveness of rivastigmine in Alzheimer’s disease. BMJ 1999, 319, 640. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.D.; Davies, J.R.T.; Chang, X. New Gold in Them Thar Hills: Testing a Novel Supply Route for Plant-Derived Galanthamine. J. Alzheimers. Dis. 2017, 55, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- De Souza, F.M.S.; Busquet, N.; Blatner, M.; Maclean, K.N.; Restrepo, D. Galantamine improves olfactory learning in the Ts65Dn mouse model of Down syndrome. Sci. Rep. 2011, 1, 137. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Solomon, P.R.; Morris, J.C.; Kershaw, P.; Lilienfeld, S.; Ding, C. A 5-month, randomized, placebo-controlled trial of galantamine in AD. The Galantamine USA-10 Study Group. Neurology 2000, 54, 2269–2276. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cyrus, P.A.; Bieber, F.; Mas, J.; Orazem, J.; Gulanski, B. Metrifonate treatment of the cognitive deficits of Alzheimer’s disease. Metrifonate Study Group. Neurology 1998, 50, 1214–1221. [Google Scholar] [CrossRef]
- Schneider, L.S.; Giacobini, E. Metrifonate: A cholinesterase inhibitor for Alzheimer’s disease therapy. CNS Drug Rev. 1999, 5, 13–26. [Google Scholar] [CrossRef]
- Lilja, A.M.; Luo, Y.; Yu, Q.S.; Röjdner, J.; Li, Y.; Marini, A.M.; Marutle, A.; Nordberg, A.; Greig, N.H. Neurotrophic and Neuroprotective Actions of (-)- and (+)-Phenserine, Candidate Drugs for Alzheimer’s Disease. PLoS ONE 2013, 8, e54887. [Google Scholar] [CrossRef]
- Klein, J. Phenserine. Phenserine. Expert Opin. Investig. Drugs 2007, 16, 1087–1097. [Google Scholar] [CrossRef]
- Nordberg, A.; Kadir, A.; Andreasen, N.; Almkvist, O.; Wall, A.; Blennow, K.; Langstrom, B.; Zetterberg, H. Correlations between Alzheimer’s Disease Cerebrospinal Fluid Biomarkers and Cerebral Glucose Metabolism after 12 Months of Phenserine Treatment. J. Alzheimer Dis. 2015, 47, 691–704. [Google Scholar]
- Kamal, M.A.; Greig, N.H.; Alhomida, A.S.; Al-Jafari, A.A. Kinetics of human acetylcholinesterase inhibition by the novel experimental alzheimer therapeutic agent, tolserine. Biochem. Pharmacol. 2000, 60, 561–570. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Q.; Hu, X.; Wang, W.; Zhu, X.; Yuan, Z. Pharmacodynamics in Alzheimer’s disease model rats of a bifunctional peptide with the potential to accelerate the degradation and reduce the toxicity of amyloid β-Cu fibrils. Acta Biomater. 2018, 65, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.J.; Bian, H.L.; Wang, J.W.; Shan, W.G. Synthesis of physostigmine analogues and evaluation of their anticholinesterase activities. Bioorganic Med. Chem. Lett. 2010, 20, 1532–1534. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yan, H.; Tang, X.-C. Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine 1. Acta Pharmacol. Sin. 2006, 27, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Braga de Andrade Teles, R.; Coimbra Diniz, T.; Coimbra Costa Pinto, T.; Gonçalves de Oliveira Júnior, R.; Gama Silva, M.; Martins de Lavor, É.; Wilton Cavalcante Fernandes, A.; Paula de Oliveira, A.; Pires Rodrigues de Almeida Ribeiro, F.; Alves Marcelino da Silva, A.; et al. Flavonoids as Therapeutic Agents in Alzheimer’s and Parkinson’s Diseases: A Systematic Review of Preclinical Evidences. Oxidative Med. Cell. Longevity 2018, 3, 1–21. [Google Scholar] [CrossRef] [PubMed]
- De Paula, A.A.N.; Martins, J.B.L.; dos Santos, M.L.; Nascente, L.D.C.; Romeiro, L.A.S.; Areas, T.F.M.A.; Vieira, K.S.T.; Gambôa, N.F.; Castro, N.G.; Gargano, R. New potential AChE inhibitor candidates. Eur. J. Med. Chem. 2009, 44, 3754–3759. [Google Scholar] [CrossRef] [PubMed]
- Taiwo, E.A. Cashew Nut Shell Oil—A Renewable and Reliable Petrochemical Feedstock. In Advances in Petrochemicals; IntechOpen: London, UK, 2015. [Google Scholar]
- Lemes, L.F.N.; De Andrade Ramos, G.; De Oliveira, A.S.; Da Silva, F.M.R.; De Castro Couto, G.; Da Silva Boni, M.; Guimarães, M.J.R.; Souza, I.N.O.; Bartolini, M.; Andrisano, V.; et al. Cardanol-derived AChE inhibitors: Towards the development of dual binding derivatives for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 108, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Bartolini, M.; Ceccarini, L.; Piazzi, L.; Gobbi, S.; Cavalli, A.; Recanatini, M.; Andrisano, V.; Rampa, A. Targeting Alzheimer’s disease: Novel indanone hybrids bearing a pharmacophoric fragment of AP2238. Bioorganic Med. Chem. 2010, 18, 1749–1760. [Google Scholar] [CrossRef]
- Piazzi, L.; Rampa, A.; Bisi, A.; Gobbi, S.; Belluti, F.; Cavalli, A.; Bartolini, M.; Andrisano, V.; Valenti, P.; Recanatini, M. 3-(4-{[benzyl(methyl)amino]methyl}-phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: A dual function lead for Alzheimer’s disease therapy. J. Med. Chem. 2003, 46, 2279–2282. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef]
- Camps, P.; Formosa, X.; Galdeano, C.; Muñoz-Torrero, D.; Ramírez, L.; Gómez, E.; Isambert, N.; Lavilla, R.; Badia, A.; Clos, M.V.; et al. Pyrano[3,2-c]quinoline—6-chlorotacrine hybrids as a novel family of acetylcholinesterase-and β-amyloid-directed anti-Alzheimer compounds. J. Med. Chem. 2009, 52, 5365–5379. [Google Scholar] [CrossRef]
- Girek, M.; Szymański, P. Tacrine hybrids as multi-target-directed ligands in Alzheimer’s disease: Influence of chemical structures on biological activities. Chem. Pap. 2019, 73, 269–289. [Google Scholar] [CrossRef]
- Pi, R.; Mao, X.; Chao, X.; Cheng, Z.; Liu, M.; Duan, X.; Ye, M.; Chen, X.; Mei, Z.; Liu, P.; et al. Tacrine-6-ferulic acid, a novel multifunctional dimer, inhibits amyloid-β-mediated Alzheimer’s disease-associated pathogenesis in vitro and in vivo. PLoS ONE 2012, 7, e31921. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Muñoz, G.C.; Conde, S.; Lόpez, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel Tacrine−8-Hydroxyquinoline Hybrids as Multifunctional Agents for the Treatment of Alzheimer’s Disease, with Neuroprotective, Cholinergic, Antioxidant, and Copper-Complexing Properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, P.; Saavedra, L.A.; Caballero, J.; Quiroga, J.; Alzate-Morales, J.H.; Cabrera, M.G.; Trilleras, J. A novel class of selective acetylcholinesterase inhibitors: Synthesis and evaluation of (E)-2-(benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules 2012, 17, 12072–12085. [Google Scholar] [CrossRef] [PubMed]
- El-Malah, A.; Gedawy, E.M.; Kassab, A.E.; Salam, R.M.A. Novel tacrine analogs as potential cholinesterase inhibitors in Alzheimer’s disease. Arch. Pharm. 2014, 347, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Holas, O.; Binder, J.; Zemek, F.; Marek, J.; Pohanka, M.; Opletalova, V.; Dohnal, V.; Kuca, K. Synthesis and in vitro evaluation of N-alkyl-7-methoxytacrine hydrochlorides as potential cholinesterase inhibitors in Alzheimer disease. Bioorganic Med. Chem. Lett. 2010, 20, 6093–6095. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. A Novel Anti-Alzheimer Disease Drug, Ladostigil. Neuroprotective, Multimodal Brain-Selective Monoamine Oxidase and Cholinesterase Inhibitor, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 100, ISBN 9-78012-386-4673. [Google Scholar]
- Birks, J.S. Cholinesterase inhibitors for Alzheimer’s disease. In Cochrane Database of Systematic Reviews; Birks, J.S., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Dou, K.-X.; Tan, M.-S.; Tan, C.-C.; Cao, X.-P.; Hou, X.-H.; Guo, Q.-H.; Tan, L.; Mok, V.; Yu, J.-T. Comparative safety and effectiveness of cholinesterase inhibitors and memantine for Alzheimer’s disease: A network meta-analysis of 41 randomized controlled trials. Alzheimer Res. Ther. 2018, 10, 126. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, nyz310_208. [Google Scholar] [CrossRef]
- Cai, Z.; Qiao, P.F.; Wan, C.Q.; Cai, M.; Zhou, N.K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimer Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.; Sousa, J.; Sousa, N.; Palha, J. Blood–brain-barriers in aging and in Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. Blood Brain Barrier Dysfunction and Cerebrovascular Degeneration in Alzheimer’s Disease. In Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Related Disorders; Springer: Dordrecht, The Netherlands, 2000; pp. 189–206. [Google Scholar]
- Banks, W.A. Drug delivery to the brain in Alzheimer’s disease: Consideration of the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 629–639. [Google Scholar] [CrossRef]
- Matsui, K.; Taniguchi, S.; Yoshimura, T. Correlation of the intrinsic clearance of donepezil (Aricept®) between in vivo and in vitro studies in rat, dog and human. Xenobiotica 1999, 29, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Geerts, H.; Guillaumat, P.O.; Grantham, C.; Bode, W.; Anciaux, K.; Sachak, S. Brain levels and acetylcholinesterase inhibition with galantamine and donepezil in rats, mice, and rabbits. Brain Res. 2005, 1033, 186–193. [Google Scholar] [CrossRef]
- Chakraborty, A.; de Wit, N.M.; van der Flier, W.M.; de Vries, H.E. The blood brain barrier in Alzheimer’s disease. Vascul. Pharmacol. 2017, 89, 12–18. [Google Scholar] [CrossRef]
- Zdarova Karasova, J.; Sestak, V.; Korabecny, J.; Mezeiova, E.; Palicka, V.; Kuca, K.; Mzik, M. 1-Benzyl-4-methylpiperidinyl moiety in donepezil: The priority ticket across the blood-brain-barrier in rats. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1092, 350–358. [Google Scholar] [CrossRef]
- Nordberg, A.; Svensson, A.L. Cholinesterase inhibitors in the treatment of Alzheimer’s disease. A comparison of tolerability and pharmacology. Drug Saf. 1998, 19, 465–480. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; McTavish, D. Tacrine: A Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy in Alzheimer’s Disease. Drugs Aging 1994, 4, 510–540. [Google Scholar] [CrossRef]
- Mishra, P.; Kumar, A.; Panda, G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998–2018). Bioorganic Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef] [PubMed]
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer’s disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Pepeu, G. Overview and perspective on the therapy of Alzheimer’s disease from a preclinical viewpoint. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2001, 25, 193–209. [Google Scholar] [CrossRef]
- Kurz, A.; Perneczky, R. Novel insights for the treatment of Alzheimer’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2011, 35, 373–379. [Google Scholar] [CrossRef]
- Pepeu, G. Mild cognitive impairment: Animal models. Dialogues Clin. Neurosci. 2004, 6, 369–377. [Google Scholar]
- Zhu, H.; Yan, H.; Tang, N.; Li, X.; Pang, P.; Li, H.; Chen, W.; Guo, Y.; Shu, S.; Cai, Y.; et al. Impairments of spatial memory in an Alzheimer’s disease model via degeneration of hippocampal cholinergic synapses. Nat. Commun. 2017, 8, 1676. [Google Scholar] [CrossRef]
- Zilony-Hanin, N.; Rosenberg, M.; Richman, M.; Yehuda, R.; Schori, H.; Motiei, M.; Rahimipour, S.; Groisman, A.; Segal, E.; Shefi, O. Neuroprotective Effect of Nerve Growth Factor Loaded in Porous Silicon Nanostructures in an Alzheimer’s Disease Model and Potential Delivery to the Brain. Small 2019, 15. [Google Scholar] [CrossRef]
- Zhen, J.; Qian, Y.; Fu, J.; Su, R.; An, H.; Wang, W.; Zheng, Y.; Wang, X. Deep brain magnetic stimulation promotes neurogenesis and restores cholinergic activity in a transgenic mouse model of Alzheimer’s disease. Front. Neural Circuits 2017, 11, 48. [Google Scholar] [CrossRef]
- Xia, E.; Xu, F.; Hu, C.; Kumal, J.P.P.; Tang, X.; Mao, D.; Li, Y.; Wu, D.; Zhang, R.; Wu, S.; et al. Young Blood Rescues the Cognition of Alzheimer’s Model Mice by Restoring the Hippocampal Cholinergic Circuit. Neuroscience 2019, 417, 57–69. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, X.; Chen, X.; Cai, Y.; Ma, Y.; Ma, J.; Zhang, Q.; Dai, L.; Fan, X.; Bai, Y. Choline Supplementation Ameliorates Behavioral Deficits and Alzheimer’s Disease-Like Pathology in Transgenic APP/PS1 Mice. Mol. Nutr. Food Res. 2019, 63, 2–36. [Google Scholar] [CrossRef]
- Park, T.Y.; Kim, S.H.; Shin, Y.C.; Lee, N.H.; Lee, R.K.C.; Shim, J.H.; Glimcher, L.H.; Mook-Jung, I.; Cheong, E.; Kim, W.K.; et al. Amelioration of neurodegenerative diseases by cell death-induced cytoplasmic delivery of humanin. J. Control. Release 2013, 166, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Park, S.Y.; Lee, S.H.; Kim, Y.; Kim, Y.J.; Jun, W.; Yoon, H.G. Ameliorative Effects of Dendropanax morbifera on Cognitive Impairment Via Enhancing Cholinergic Functions and Brain-Derived Neurotrophic Factor Expression in β-Amyloid-Induced Mice. J. Med. Food 2019, 22, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Karthivashan, G.; Park, S.Y.; Kweon, M.H.; Kim, J.; Haque, M.E.; Cho, D.Y.; Kim, I.S.; Cho, E.A.; Ganesan, P.; Choi, D.K. Ameliorative potential of desalted Salicornia europaea L. extract in multifaceted Alzheimer’s-like scopolamine-induced amnesic mice model. Sci. Rep. 2018, 8, 7174. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ohi, Y.; Kato, D.; Mizuno, M.; Takase, H.; Kanamori, T.; Borlongan, C.V.; Haji, A.; Matsukawa, N. Hippocampal Cholinergic Neurostimulating Peptide as a Possible Modulating Factor against Glutamatergic Neuronal Disability by Amyloid Oligomers. Cell Transplant. 2017, 26, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Kim, S.Y.; Lee, S.Y.; Jang, C.G. 6,7,4′-Trihydroxyisoflavone, a major metabolite of daidzein, improves learning and memory via the cholinergic system and the p-CREB/BDNF signaling pathway in mice. Eur. J. Pharmacol. 2018, 826, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; D’Angelo, C.; Costantini, E.; Di Nicola, M.; Yarla, N.S.; Kamal, M.A.; Salvador, N.; Perry, G. Expression Profiling of Cytokine, Cholinergic Markers, and Amyloid-β Deposition in the APP SWE /PS1dE9 Mouse Model of Alzheimer’s Disease Pathology. J. Alzheimer Dis. 2018, 62, 467–476. [Google Scholar] [CrossRef]
- Karami, A.; Eriksdotter, M.; Kadir, A.; Almkvist, O.; Nordberg, A.; Darreh-Shori, T. CSF Cholinergic Index, a New Biomeasure of Treatment Effect in Patients With Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 12, 239. [Google Scholar] [CrossRef]
- Hall, H.; Iulita, M.F.; Gubert, P.; Flores Aguilar, L.; Ducatenzeiler, A.; Fisher, A.; Cuello, A.C. AF710B, an M1/sigma-1 receptor agonist with long-lasting disease-modifying properties in a transgenic rat model of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 811–823. [Google Scholar] [CrossRef]
- McKeever, P.M.; Kim, T.H.; Hesketh, A.R.; MacNair, L.; Miletic, D.; Favrin, G.; Oliver, S.G.; Zhang, Z.; St George-Hyslop, P.; Robertson, J. Cholinergic neuron gene expression differences captured by translational profiling in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 57, 104–119. [Google Scholar] [CrossRef]
- Hollnagel, J.O.; Elzoheiry, S.; Gorgas, K.; Kins, S.; Beretta, C.A.; Kirsch, J.; Kuhse, J.; Kann, O.; Kiss, E. Early alterations in hippocampal perisomatic GABAergic synapses and network oscillations in a mouse model of Alzheimer’s disease amyloidosis. PLoS ONE 2019, 14, e0209228. [Google Scholar] [CrossRef]
- Llorente-Ovejero, A.; Manuel, I.; Lombardero, L.; Giralt, M.T.; Ledent, C.; Giménez-Llort, L.; Rodríguez-Puertas, R. Endocannabinoid and Muscarinic Signaling Crosstalk in the 3xTg-AD Mouse Model of Alzheimer’s Disease. J. Alzheimer Dis. 2018, 64, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Coquelle, N.; Pišlar, A.; Žakelj, S.; Jukič, M.; Sova, M.; Mravljak, J.; Nachon, F.; Brazzolotto, X.; Kos, J.; et al. Multi-target-directed ligands for treating Alzheimer’s disease: Butyrylcholinesterase inhibitors displaying antioxidant and neuroprotective activities. Eur. J. Med. Chem. 2018, 156, 598–617. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tiwari, A.; Sharma, A. Changing Paradigm from one Target one Ligand Towards Multi-target Directed Ligand Design for Key Drug Targets of Alzheimer Disease: An Important Role of In Silico Methods in Multi-target Directed Ligands Design. Curr. Neuropharmacol. 2018, 16, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.; Minarini, A.; Rosini, M.; Tumiatti, V.; Melchiorre, C. From Dual Binding Site Acetylcholinesterase Inhibitors to Multi-Target-Directed Ligands (MTDLs): A Step Forward in the Treatment of Alzheimers Disease. Mini Rev. Med. Chem. 2008, 8, 960–967. [Google Scholar] [CrossRef]
- Mohamed, T.; Shakeri, A.; Rao, P.P.N. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef]
- Bajda, M.; Guzior, N.; Ignasik, M.; Malawska, B. Multi-Target-Directed Ligands in Alzheimer’s Disease Treatment. Curr. Med. Chem. 2011, 18, 4949–4975. [Google Scholar] [CrossRef]
- Oliveira, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Sequeira, L.; Mesiti, F.; Silva, T.; Garrido, J.; Remião, F.; Vilar, S.; et al. Hydroxybenzoic Acid Derivatives as Dual-Target Ligands: Mitochondriotropic Antioxidants and Cholinesterase Inhibitors. Front. Chem. 2018, 6, 126. [Google Scholar] [CrossRef]
- Yu, Q.S.; Holloway, H.W.; Utsuki, T.; Brossi, A.; Greig, N.H. Synthesis of novel phenserine-based-selective inhibitors of butyrylcholinesterase for Alzheimer’s disease. J. Med. Chem. 1999, 42, 1855–1861. [Google Scholar] [CrossRef]
- Unzeta, M.; Esteban, G.; Bolea, I.; Fogel, W.A.; Ramsay, R.R.; Youdim, M.B.H.; Tipton, K.F.; Marco-Contelles, J. Multi-target directed donepezil-like ligands for Alzheimer’s disease. Front. Neurosci. 2016, 10, 1–24. [Google Scholar] [CrossRef]
- Lu, C.; Guo, Y.; Yan, J.; Luo, Z.; Luo, H.B.; Yan, M.; Huang, L.; Li, X. Design, synthesis, and evaluation of multitarget-directed resveratrol derivatives for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 5843–5859. [Google Scholar] [CrossRef]
- Lee, H.Y.; Fan, S.J.; Huang, F.I.; Chao, H.Y.; Hsu, K.C.; Lin, T.E.; Yeh, T.K.; Lai, M.J.; Li, Y.H.; Huang, H.L.; et al. 5-Aroylindoles Act as Selective Histone Deacetylase 6 Inhibitors Ameliorating Alzheimer’s Disease Phenotypes. J. Med. Chem. 2018, 61, 7087–7102. [Google Scholar] [CrossRef] [PubMed]
- Vohora, D.; Bhowmik, M. Histamine H3 receptor antagonists/inverse agonists on cognitive and motor processes: Relevance to Alzheimer’s disease, ADHD, schizophrenia, and drug abuse. Front. Syst. Neurosci. 2012, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Gemkow, M.J.; Davenport, A.J.; Harich, S.; Ellenbroek, B.A.; Cesura, A.; Hallett, D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov. Today 2009, 14, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Berlin, M.; Boyce, C.W.; De Lera Ruiz, M. Histamine H3 receptor as a drug discovery target. J. Med. Chem. 2011, 54, 26–53. [Google Scholar] [CrossRef]
- Muñoz-Ruiz, P.; Rubio, L.; García-Palomero, E.; Dorronsoro, I.; Del Monte-Millán, M.; Valenzuela, R.; Usán, P.; De Austria, C.; Bartolini, M.; Andrisano, V.; et al. Design, synthesis, and biological evaluation of dual binding site acetylcholinesterase inhibitors: New disease-modifying agents for Alzheimer’s disease. J. Med. Chem. 2005, 48, 7223–7233. [Google Scholar] [CrossRef]
- Lange, J.H.M.; Coolen, H.K.A.C.; Van Der Neut, M.A.W.; Borst, A.J.M.; Stork, B.; Verveer, P.C.; Kruse, C.G. Design, synthesis, biological properties, and molecular modeling investigations of novel tacrine derivatives with a combination of acetylcholinesterase inhibition and cannabinoid CB1 receptor antagonism. J. Med. Chem. 2010, 53, 1338–1346. [Google Scholar] [CrossRef]
- Smith, T.H.; Sim-Selley, L.J.; Selley, D.E. Cannabinoid CB 1 receptor-interacting proteins: Novel targets for central nervous system drug discovery? Br. J. Pharmacol. 2010, 160, 454–466. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef]
- Prati, F.; Bottegoni, G.; Bolognesi, M.L.; Cavalli, A. BACE-1 Inhibitors: From Recent Single-Target Molecules to Multitarget Compounds for Alzheimer’s Disease. J. Med. Chem. 2018, 61, 619–637. [Google Scholar] [CrossRef]
- Wu, H.; Li, X.D.; Peng, D.T.; Jiao, J.S.; Li, Y.F.; Yu, P.L.; Ji, Y. The role of butyrylcholinesterase in the pathogenesis of Alzheimer’s disease. Chin. J. Contemp. Neurol. Neurosurg. 2017, 17, 933–936. [Google Scholar]
- Wu, Y.; Li, Z.; Huang, Y.Y.; Wu, D.; Luo, H. Bin Novel Phosphodiesterase Inhibitors for Cognitive Improvement in Alzheimer’s Disease. J. Med. Chem. 2018, 61, 5467–5483. [Google Scholar] [CrossRef] [PubMed]
- Vila-Real, H.; Coelho, H.; Rocha, J.; Fernandes, A.; Ventura, M.R.; Maycock, C.D.; Iranzo, O.; Simplício, A.L. Peptidomimetic β-Secretase Inhibitors Comprising a Sequence of Amyloid-β Peptide for Alzheimer’s Disease. J. Med. Chem. 2015, 58, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Hroch, L.; Aitken, L.; Gunn-Moore, F.; Vinklarova, L.; Kuca, K.; Perez, D.I.; Perez, C.; Martinez, A.; Fisar, Z.; et al. 1-(Benzo[d]thiazol-2-yl)-3-phenylureas as dual inhibitors of casein kinase 1 and ABAD enzymes for treatment of neurodegenerative disorders. J. Enzyme Inhib. Med. Chem. 2018, 33, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Aitken, L.; Benek, O.; McKelvie, B.E.; Hughes, R.E.; Hroch, L.; Schmidt, M.; Major, L.L.; Vinklarova, L.; Kuca, K.; Smith, T.K.; et al. Novel Benzothiazole-based Ureas as 17β-HSD10 Inhibitors, A Potential Alzheimer’s Disease Treatment. Molecules 2019, 24, 182–191. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
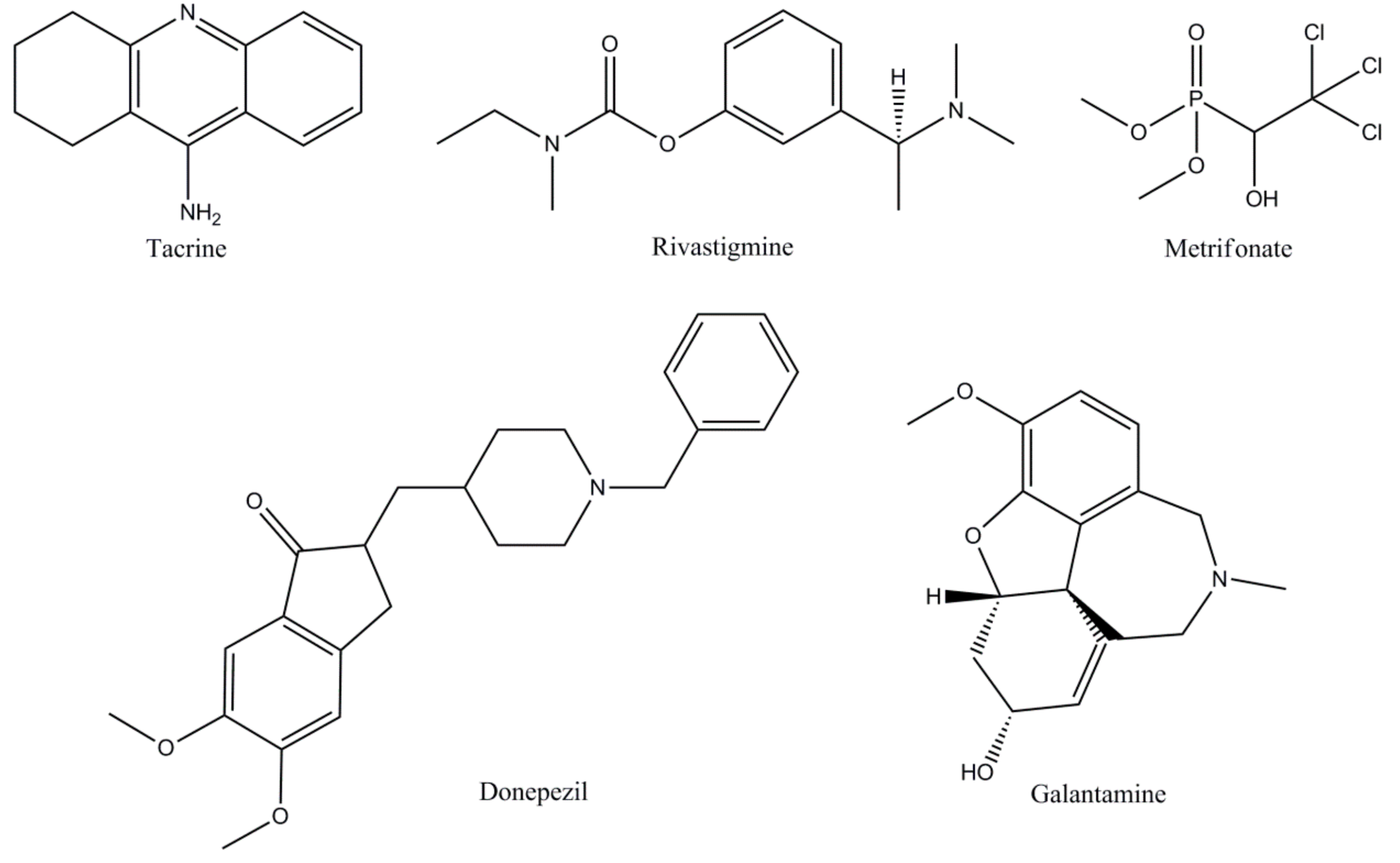
| Drug | Pharmacological Aspects | |
|---|---|---|
| Traditional ChEIs | Tacrine [25,45,46,47] | The first compound approved in 1993 for the Alzheimer’s patient’s therapy, tacrine is a non-competitive, rapidly reversible inhibitor of both AChE and BuChE. The bioavailability of tacrine varieties from 17-37%, elimination half-life ranges from 1.3 to 7.0 hours and is almost 75% protein bound. Metabolism of tacrine is achieved by CYP 450 1A2 and 2D6. |
| Donepezil [20,25,48,49] | Approved in 1996 for mild to moderate AD therapy, donepezil is a non-competitive, rapidly reversible AChE inhibitor. Bioavailability of 100%, readily absorbed after oral administration, vastly protein bound - 96%, with an excretion half-life of 60–90 hours. Donepezil is mainly metabolized by CYP 450 isoenzymes 2D6 and 3A4. | |
| Rivastigmine [49,50,51,52] | Approved in 2000 for AD treatment, the compound is considered a non-competitive, pseudo-irreversible of both AChE and BuChE with equal proficiency. Bioavailability of rivastigmine is quite low at 40%, protein binding at 40%, and elimination half-life of almost 2 hours. Rivastigmine is rapidly and primarily metabolized by cholinesterase’s. | |
| Galantamine [19,53,54,55] | A competitive, rapidly reversible potent AChE inhibitor, galantamine was approved in AD therapy in 2001. It is well absorbed with an 85% to 100% bioavailability in oral delivery. Plasma protein binding of this compound is about 18%, with a half-life of 7 hours. The main way for galantamine metabolism occurs through CYP isoenzymes 2D6 and 3A4. | |
| Metrifonate [19,56,57] | Metrifonate a long-acting irreversible inhibitor of ChEI is not an approved AD drug because of risk of neuromuscular transmission dysfunction and respiratory paralysis. Even if metrifonate has proven a strong and important clinical effect it was abandoned after Phase III. | |
| ChEIs in development | Phenserine [58,59,60] | Phenserine is a pure non-competitive, selective AChE inhibitor, being a promising agent for developing new strategies in AD therapy. It has a half-life of 10 minutes and owns an action more than 8 hours, being highly penetrative to the brain with a brain: Plasma concentration ratio of 10:1. |
| Tolserine [19,25,61] | A partial non-competitive, reversible AChE inhibitor, tolserine, has a pharmacokinetics half-life of 12 minutes and a pharmacodynamics half-life greater than 8 hours. Tolserine delivers a selectivity for AChE of 200-fold versus 75-fold for phenserine. The 29-methyl substitution additionally raises the hydrophobic properties of tolserine compared to phenserine improving its blood– brain barrier permeability, ensuring high brain absorption (brain:blood ratio 24:1). | |
| Eseroline [25,62,63] | Considered un metabolite of physostigmine, eseroline has a competitive, limited, and reversible effect on AChE inhibition. Zhan et al. [63] found that a cyclic alkyl carbamate derived from eseroline is more effective against AChE with a great selectivity compared with BChE. There have been no recent studies reporting the effect of eseroline. | |
| Naturally derived ChEIs | Huperzine [18,64] | A Lycopodium alkaloid extracted from the Chinese medicinal plant Huperzia serrata, huperzine is an effective, reversible, and very selective AChE inhibitor. Huperzine has an oral bioavailability of more than 96% metabolized by CYP1A2, with a possible secondary influence by CYP3A1/2. In China it is the drug selected for AD therapy. In the USA, Phase II studies have shown a modest but clinically substantial effect on the cognition of AD patients. |
| Flavonoid [19,25,65] | Flavonoids have attracted attention due to their free radical scavenging properties highlighting the ability to influence cognition and learning in humans and AD animal models. Galangin, a flavonol extracted and isolated from the rhizomes of Alpiniae officinarum, confirmed the most important inhibitory activity against AChE. Nevertheless, the toxicity of these alternative candidates for Alzheimer’s therapy has not been explored in preclinical studies and no clinical trials have been described to date. | |
| Cardanol [66,67,68] | Cardanol derivatives as new potential candidates of AChE inhibitors designed from nonisoprenoid phenolic lipids of cashew (NIPLs) of Anacardium occidentale nut-shell liquid have revealed favorable results. The development of cardanol derivatives seems to be attractive because of the abundance of the source of raw materials. However, there are no reported preclinical and toxicity studies, to date. | |
| Hybrid ChEIs | Donepezil–AP2238 hybrid [69,70,71] | The first inhibitor capable of binding to both the catalytic and peripheral sites of AChE, AP 2238 hybrid has an activity against AChE similar to that of donepezil but with a higher capacity to inhibit Aβ-mediated toxicity. At present, there are no reports of human studies, not being preclinical and clinical safety and toxicity. |
| Donepezil–tacrine hybrid [72,73] | Designed and synthetized from a unit of 6-chlorotacrine and 5,6-dimethoxy- 2-[(4-piperidinyl)methyl]-1-idanone moiety of donepezil, donepezil–tacrine hybrids have proven to be highly potent inhibitors of both AChE and BChE as well as beta-amyloid aggregation determined by AChE. | |
| Tacrine–ferulic acid (T6FA) hybrid [74] | In vitro studies have shown that T6FA can significantly inhibit auto- and AChE aggregation of Aβ (1-40), blocking Aβ-induced cell death (1-40) in PC12 cells. Moreover, in an AD rodent model, T6FA considerably enriched the cognitive capacity along with growing ChAT and superoxide dismutase activity, reducing AChE activity. | |
| Tacrine and 8-hydroxyquinoline hybrids | Designed and synthesized by Fernandez-Bachiller et al. [75], the new hybrids have been shown to be more effective than tacrine against ChEIs inhibition. The compounds revealed low cell level toxicity, antioxidant, and copper-complexing activities. | |
| Synthetic analogues | Tacrine analogues [76,77,78] | Screening results revealed that most tacrine analogues displayed important inhibition against AChE compared to tacrine. |
| (E)-2(benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles [25,76] | Developed as an inhibitor of AChE since 2012, the compound (E)-2(benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles has been shown to be more selective for AChE than galanthamine. | |
| Ladostigil [19,25,79] | Derived from the combination of two pharmacophores: The carbamate moiety of rivastigmine and the propargyl group of rasagiline, ladostigil represents a novel anti-AD compound, which combines neuroprotective proprieties with brain selective monoamine oxidase and cholinesterase inhibitory activities. The drug is currently included in a Phase IIb clinical trial for the AD therapy and comorbid dementia associated with extrapyramidal conditions and depression. | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2020, 10, 40. https://doi.org/10.3390/biom10010040
Stanciu GD, Luca A, Rusu RN, Bild V, Beschea Chiriac SI, Solcan C, Bild W, Ababei DC. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules. 2020; 10(1):40. https://doi.org/10.3390/biom10010040
Chicago/Turabian StyleStanciu, Gabriela Dumitrita, Andrei Luca, Razvan Nicolae Rusu, Veronica Bild, Sorin Ioan Beschea Chiriac, Carmen Solcan, Walther Bild, and Daniela Carmen Ababei. 2020. "Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement" Biomolecules 10, no. 1: 40. https://doi.org/10.3390/biom10010040
APA StyleStanciu, G. D., Luca, A., Rusu, R. N., Bild, V., Beschea Chiriac, S. I., Solcan, C., Bild, W., & Ababei, D. C. (2020). Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules, 10(1), 40. https://doi.org/10.3390/biom10010040