Autophagy Function and Regulation in Kidney Disease


Abstract
1. Introduction
2. Basal Autophagy in the Kidney and Other Organs
3. Autophagy in Response to Stress and Kidney Injury
3.1. Autophagy in AKI
3.2. Autophagy in Renal Interstitial Fibrosis and Progressive Kidney Disease
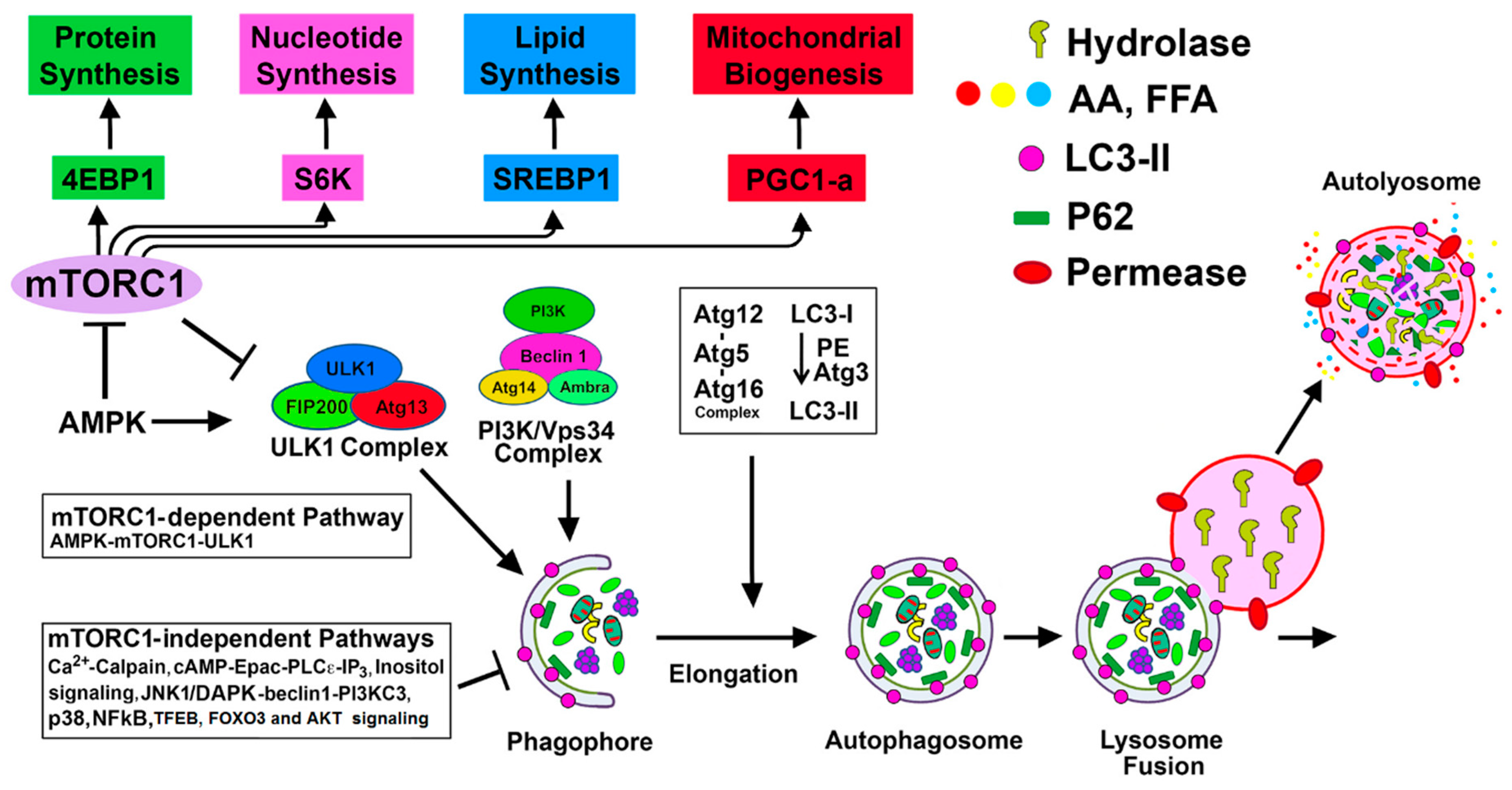
4. Regulation of Autophagy by mTORC1-Dependent and Alternative mTORC1-Independent Pathways in Renal Injury
4.1. mTORC1-Dependent Autophagy Regulation
4.1.1. mTORC1-ULK1 Pathway
4.1.2. mTORC1-ULK1-Mediated Autophagy in Renal Injury

{kind=link}
{kind=link}
{kind=link}
| mTORC1-ULK1 Pathway in Kidney | |||
|---|---|---|---|
| Model/ Kidney Disease | Agent/Drug | Effect on Autophagy | Reference |
| Murine model of sepsis | Temsirolimus | ↑ autophagy via inhibition of mTORC1 | [85] |
| AMPK activator AICAR | |||
| Adriamycin-induced podocyte injury | Rapamycin and podocyte-specific Atg7 KO | ↑ autophagy | [86] |
| G1 and G2 generic variants in APOL1 gene in podocytes | ↓ autophagic flux by miR-193a mediated mTOR inhibition | [87] | |
| High glucose-induced podocyte injury | Ursolic acid | ↑ autophagy and improves podocyte injury | [88] |
| db/db mouse model | AGEs | ↓ podocyte autophagy via activation of mTORC1 and inhibition of nuclear translocation of TFEB | [89] |
| Pyridoxamine | ↑ autophagic flux | ||
| kkAy mice with DN | Apelin | ↓ autophagic flux and promotes podocyte injury and progression of DN | [90] |
| High glucose-induced podocyte injury | Notoginsenoside R1 | ↑ autophagy | [91] |
| MRLlpr/lprmice model of LN | Rapamycin | ↑ autophagy | [92] |
| Renal transplant | Sirolimus | ↑ autophagy | [93] |
| AMPK-mTORC1/ULK1 pathway in Kidney | |||
| Renal IR injury | AMPK activators (AICAR, metformin) | ↑ autophagy | [94] |
| Quercetin, omega 3-PUFA and pioglitazone | ↑ autophagy | [94,95,96,97] | |
| Cisplatin nephrotoxicity | Metformin and neferine | ↑ autophagy | [98,99] |
| Sepsis by CLP | SIRT3 | ↑ autophagy | [100] |
| STZ induced DN | Saponin astragaloside and mangiferin | ↑ autophagy | [101,102] |
| db/db diabetic nephropathy | Cincalcet (type II agonist of calcium sensing receptor) | ↑ autophagy | [103] |
| HO-1, berberine and progranulin | ↑ autophagy | [104,105,106] | |
| HFD | AICAR | ↑ autophagy | [107] |
| Fenofibrate | ↑ autophagy | [108,109] | |
4.1.3. AMPK-mTORC1/ULK1 Pathway
4.1.4. AMPK-mTORC1/ULK1-mediated Autophagy in Renal Injury
4.1.5. SIRT1-mTORC1 Pathway
4.2. mTORC1-Independent Alternative Pathways of Autophagy Regulation in Renal Injury

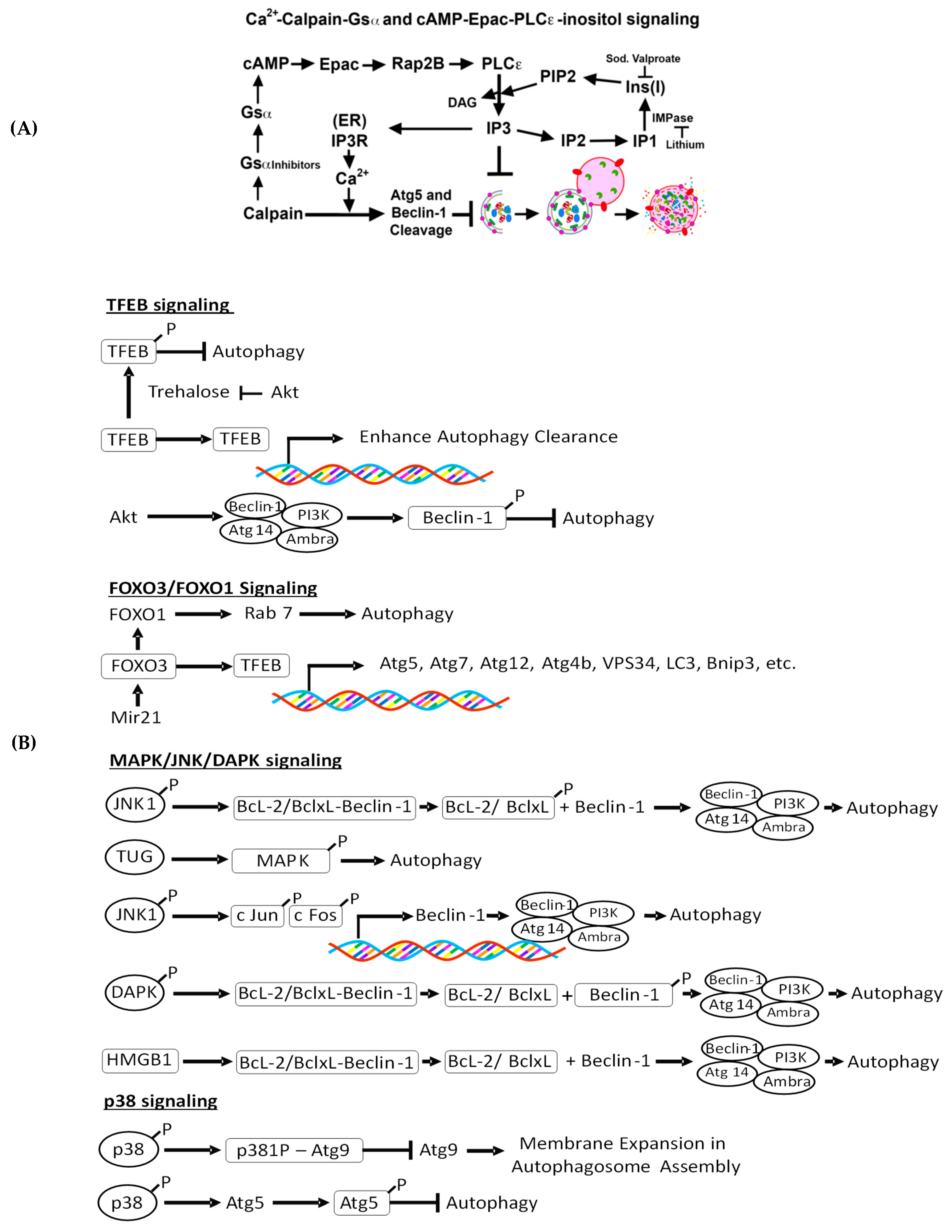
4.2.1. Intracellular Ca 2+-Mediated Autophagy Regulation and Renal Injury
4.2.2. Ca2+-Calpain-Mediated Autophagy Regulation and Renal Injury
4.2.3. cAMP-Epac-PLC-ε-IP3 Pathway in Autophagy Regulation and Renal Injury
4.2.4. c-Jun N-Terminal Kinase (JNK)-Beclin-1, p38, and Akt Signaling and Renal Injury
4.2.5. Trehalose-Mediated Autophagy Regulation and Renal Injury
4.2.6. TFEB-Mediated Autophagy Induction and Renal Injury
4.2.7. MAPK/JNK/DAPK-Mediated Autophagy Induction and Kidney Injury
4.2.8. FoxO3-Mediated Autophagy Induction and Kidney Disease
| Model/ Kidney Disease | Agent/Drug | Effect on Autophagy | Reference |
|---|---|---|---|
| Cadmium-induced nephrotoxicity | Increased calcium release from ER | ↓ autophagy due to blockage in Rab7 recruitment to autophagosome | [136,137,138,139] |
| Cisplatin-nephrotoxicity | zVAD-fmk | ↓ autophagic flux | [147] |
| Diabetic kidney disease | Increased expression of MIOX | ↓ autophagy | [152] |
| Xestospongin B | ↑ autophagy | [154] | |
| PAN-induced podocyte injury | Trehalose and analogs, known as lentztrehaloses | ↑ autophagy | [169] |
| Zebrafish model of polycystic kidney disease | carbamazepine and minoxidil | ↑ autophagy | [171] |
| JACK2-deficient podocyte mice | TFEB overexpression | ↑ autophagy | [181] |
| Renal IR injury | Urolithin-A | ↑ autophagy by enhanced TFEB expression | [183] |
| Tacrolimus-induced renal injury | Klotho | ↑ autophagy by enhanced nuclear translocation of TFEB | [184] |
| Lipopolysaccharide (LPS)-induced podocyte injury | Overexpression of TUG1 | ↑ autophagy | [190] |
| Cisplatin nephrotoxicity | ↑transforming growth factor-beta-activated kinase 1 (TAK1) | ↑ autophagy | [191] |
| Aristolochic acid (AA)-induced nephropathy | AA exposure | ↑ autophagy with ↑ ERK1/2 activity | [192] |
| UUO | FoxO3 | ↑ autophagy through nuclear expression of Atg proteins | [193] |
| Prolonged IR (AKI to CKD transition) | FoxO3 | ↑ autophagy | [194] |
| Ischemic preconditioning | Increased FoxO3 by overexpression of SGK1 | ↑ autophagy | [196] |
| Podocyte injury | Aldosterone | ↑ autophagy through FoxO1-Rab7 | [195] |
| STZ-induced DN | FoxO1 overexpression | ↑ autophagy | [196] |
| Podocyte injury | Atrasentan | ↑ autophagy by increased expression of FoxO1 by downregulating miR21 | [197] |
4.2.9. GCN2 Kinase-Mediated Autophagy Induction and Kidney Disease
5. Cell Fate in Renal Injury and Regulation by Autophagy
5.1. Cell Fate in Renal Injury
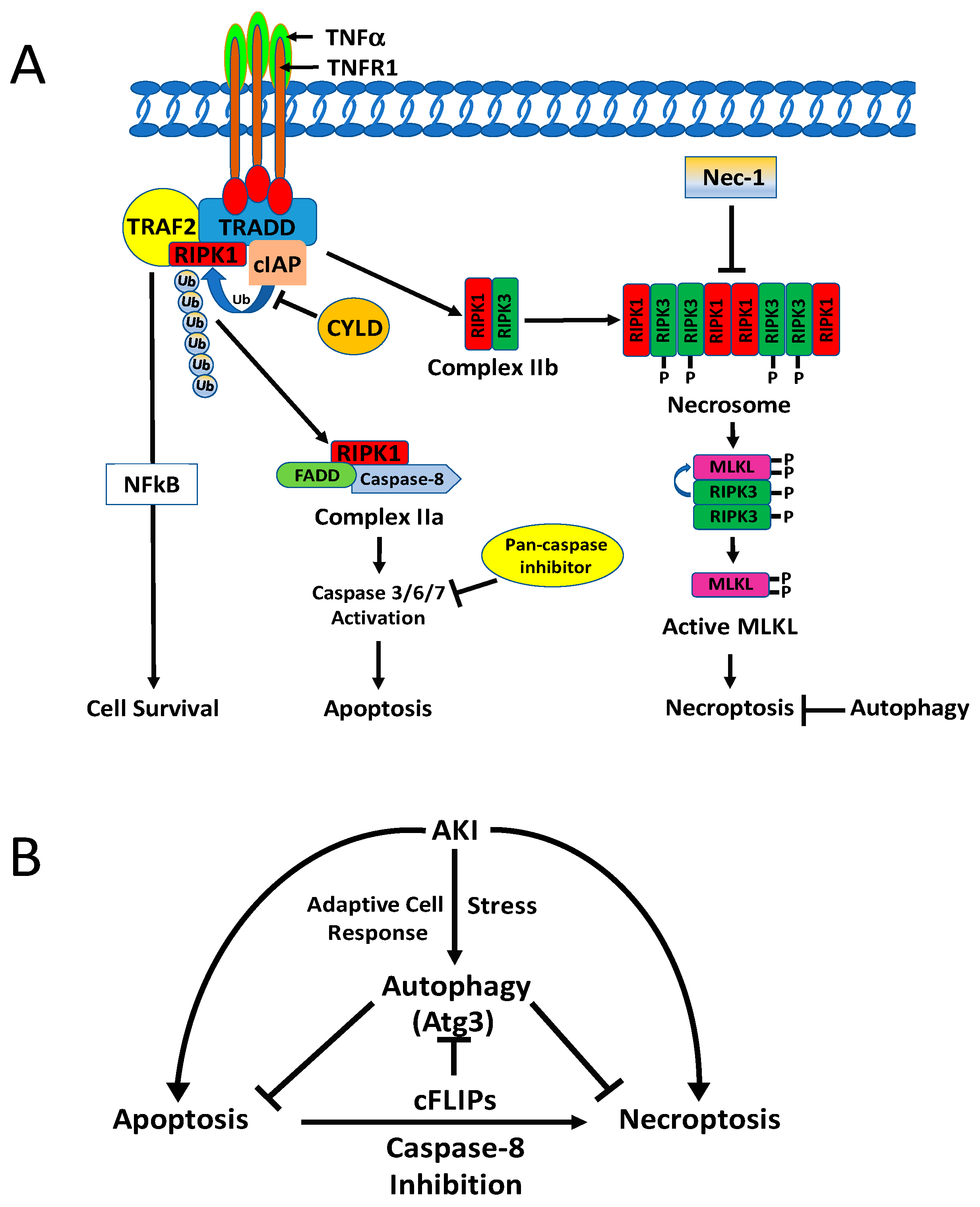
5.1.1. Apoptosis and Necrosis
5.1.2. Necroptosis
5.1.3. Ferroptosis
5.1.4. Mitochondrial Permeability Transition (MPT)-Regulated Necrosis (MPT-RN)
5.2. Molecular Interaction of Autophagy with Apoptosis and Regulated Necrosis
5.2.1. Autophagy and Apoptosis
5.2.2. Autophagy and Necroptosis
5.2.3. Autophagy and Ferroptosis
5.2.4. Autophagy and PARP-Mediated Necrosis
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell Mol. Life Sci. 2016, 73, 2405–2410. [Google Scholar] [CrossRef]
- Ktistakis, N.T.; Tooze, S.A. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Anding, A.L.; Baehrecke, E.H. Autophagy in Cell Life and Cell Death. Curr. Top. Dev. Biol. 2015, 114, 67–91. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. Recent insights into cell death and autophagy. FEBS J. 2015, 282, 4279–4288. [Google Scholar] [CrossRef]
- Altman, B.J.; Rathmell, J.C. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a008763. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., III; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef]
- Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury. Semin. Nephrol. 2014, 34, 17–26. [Google Scholar] [CrossRef]
- Choi, M.E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 2019. [Google Scholar] [CrossRef]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sorensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy Induces Prosenescent Changes in Proximal Tubular S3 Segments. J. Am. Soc. Nephrol. 2016, 27, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Kers, J.; Leemans, J.C.; Linkermann, A. An Overview of Pathways of Regulated Necrosis in Acute Kidney Injury. Semin. Nephrol. 2016, 36, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef] [PubMed]
- Havasi, A.; Dong, Z. Autophagy and Tubular Cell Death in the Kidney. Semin. Nephrol. 2016, 36, 174–188. [Google Scholar] [CrossRef]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Bohle, A.; Strutz, F.; Muller, G.A. On the pathogenesis of chronic renal failure in primary glomerulopathies: A view from the interstitium. Exp. Nephrol. 1994, 2, 205–210. [Google Scholar]
- Nath, K.A. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis. 1992, 20, 1–17. [Google Scholar] [CrossRef]
- Boor, P.; Ostendorf, T.; Floege, J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 2010, 6, 643–656. [Google Scholar] [CrossRef]
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Nam, S.A.; Song, H.C.; Ko, J.S.; Park, S.H.; Kim, H.L.; Choi, E.J.; Kim, Y.S.; Kim, J.; Kim, Y.K. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology 2012, 17, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Na, H.J.; Ding, Y.; Wang, Z.; Lee, S.J.; Choi, M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J. Biol. Chem. 2012, 287, 11677–11688. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef]
- Nam, S.A.; Kim, W.-Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.-S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 78. [Google Scholar] [CrossRef]
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486. [Google Scholar] [CrossRef]
- Peng, X.; Wang, Y.; Li, H.; Fan, J.; Shen, J.; Yu, X.; Zhou, Y.; Mao, H. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019, 10, 253. [Google Scholar] [CrossRef]
- Bhatia, D.; Chung, K.P.; Nakahira, K.; Patino, E.; Rice, M.C.; Torres, L.K.; Muthukumar, T.; Choi, A.M.; Akchurin, O.M.; Choi, M.E. Mitophagy dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 2019. [Google Scholar] [CrossRef]
- Kawaoka, K.; Doi, S.; Nakashima, A.; Yamada, K.; Ueno, T.; Doi, T.; Masaki, T. Valproic acid attenuates renal fibrosis through the induction of autophagy. Clin. Exp. Nephrol. 2017, 21, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, S.; Wu, D.; Liu, X.; Shi, M.; Wang, Y.; Zhang, F.; Ding, J.; Xiao, Y.; Guo, B. MicroRNA-22 Promotes Renal Tubulointerstitial Fibrosis by Targeting PTEN and Suppressing Autophagy in Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 4728645. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Wang, S.-S.; Han, Z.; Han, F.; Chang, Y.-P.; Yang, Y.; Xue, M.; Sun, B.; Chen, L.-M. Triptolide Restores Autophagy to Alleviate Diabetic Renal Fibrosis through the miR-141-3p/PTEN/Akt/mTOR Pathway. Mol. Ther. Nucleic Acids 2017, 9, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Bi, X.; Ling, L.; Ding, W. 1,25-Dihydroxyvitamin D3 Attenuates Angiotensin II-Induced Renal Injury by Inhibiting Mitochondrial Dysfunction and Autophagy. Cell. Physiol. Biochem. 2018, 51, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Chang, F.P.; Li, T.H.; Liu, C.W.; Huang, C.C.; Huang, S.F.; Yang, Y.Y.; Lee, K.C.; Hsieh, Y.C.; Wang, Y.W.; et al. Elafibranor Inhibits Chronic Kidney Disease Progression in NASH Mice. BioMed Res. Int. 2019, 2019, 6740616. [Google Scholar] [CrossRef]
- Song, Y.; Tao, Q.; Yu, L.; Li, L.; Bai, T.; Song, X.; Hu, H.; Li, Y.; Tan, X. Activation of autophagy contributes to the renoprotective effect of postconditioning on acute kidney injury and renal fibrosis. Biochem. Biophys. Res. Commun. 2018, 504, 641–646. [Google Scholar] [CrossRef]
- Kudo, A.; Kii, I. Periostin function in communication with extracellular matrices. J. Cell Commun. Signal. 2018, 12, 301–308. [Google Scholar] [CrossRef]
- Bian, X.; Bai, Y.; Su, X.; Zhao, G.; Sun, G.; Li, D. Knockdown of periostin attenuates 5/6 nephrectomy-induced intrarenal renin-angiotensin system activation, fibrosis, and inflammation in rats. J. Cell Physiol. 2019, 234, 22857–22873. [Google Scholar] [CrossRef]
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef]
- Khan, I.A.; Nasiruddin, M.; Haque, S.F.; Khan, R.A. Evaluation of Rhubarb Supplementation in Stages 3 and 4 of Chronic Kidney Disease: A Randomized Clinical Trial. Int. J. Chronic Dis. 2014, 2014, 789340. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Ren, J.; Sun, X.; Gui, Y.; Feng, Y.; Shu, B.; Wei, W.; Lu, Q.; Liang, Y.; He, W.; et al. Protein kinase Cα drives fibroblast activation and kidney fibrosis by stimulating autophagic flux. J. Biol. Chem. 2018, 293, 11119–11130. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Gu, L.; Chen, D.; Wu, W.; Liu, H.; Hu, H.; Wan, Y.; Sun, W. Rhein Inhibits Autophagy in Rat Renal Tubular Cells by Regulation of AMPK/mTOR Signaling. Sci. Rep. 2017, 7, 43790. [Google Scholar] [CrossRef]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2011, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Su, K.H.; Dai, C. mTORC1 senses stresses: Coupling stress to proteostasis. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Liko, D.; Hall, M.N. mTOR in health and in sickness. J. Mol. Med. 2015, 93, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef]
- Grahammer, F.; Haenisch, N.; Steinhardt, F.; Sandner, L.; Roerden, M.; Arnold, F.; Cordts, T.; Wanner, N.; Reichardt, W.; Kerjaschki, D.; et al. mTORC1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc. Natl. Acad. Sci. USA 2014, 111, E2817–E2826. [Google Scholar] [CrossRef]
- Lieberthal, W.; Fuhro, R.; Andry, C.C.; Rennke, H.; Abernathy, V.E.; Koh, J.S.; Valeri, R.; Levine, J.S. Rapamycin impairs recovery from acute renal failure: Role of cell-cycle arrest and apoptosis of tubular cells. Am. J. Physiol. Ren. Physiol. 2001, 281, F693–F706. [Google Scholar] [CrossRef]
- Nakagawa, S.; Nishihara, K.; Inui, K.; Masuda, S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur. J. Pharmacol. 2012, 696, 143–154. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Ruegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef]
- Godel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mor, A.; Lindenmeyer, M.T.; Rastaldi, M.P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Distefano, G.; Boca, M.; Rowe, I.; Wodarczyk, C.; Ma, L.; Piontek, K.B.; Germino, G.G.; Pandolfi, P.P.; Boletta, A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell Biol. 2009, 29, 2359–2371. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Fuhro, R.; Andry, C.; Patel, V.; Levine, J.S. Rapamycin delays but does not prevent recovery from acute renal failure: Role of acquired tubular resistance. Transplantation 2006, 82, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, G.M.; Cenedeze, M.A.; Feitoza, C.Q.; de Paula, C.B.; Marques, G.D.; Pinheiro, H.S.; de Paula Antunes Teixeira, V.; Antonia dos Reis, M.; Pacheco-Silva, A.; Camara, N.O. The role of immunosuppressive drugs in aggravating renal ischemia and reperfusion injury. Transplant. Proc. 2007, 39, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.M.; Gomez, H.; Collage, R.D.; Loughran, P.; Zhang, X.; Escobar, D.A.; Billiar, T.R.; Zuckerbraun, B.S.; Rosengart, M.R. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS ONE 2013, 8, e69520. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zhang, L.; Liu, Y.; Livingston, M.J.; Chen, J.-K.; Nahman, N.S., Jr.; Liu, F.; Dong, Z. Autophagy is activated to protect against podocyte injury in adriamycin-induced nephropathy. Am. J. Physiol. Ren. Physiol. 2017, 313, F74–F84. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ayasolla, K.; Jha, A.; Mishra, A.; Vashistha, H.; Lan, X.; Qayyum, M.; Chinnapaka, S.; Purohit, R.; Mikulak, J.; et al. Disrupted apolipoprotein L1-miR193a axis dedifferentiates podocytes through autophagy blockade in an APOL1 risk milieu. Am. J. Physiol. -Cell Physiol. 2019, 317, C209–C225. [Google Scholar] [CrossRef]
- Xu, L.; Fan, Q.; Wang, X.; Li, L.; Lu, X.; Yue, Y.; Cao, X.; Liu, J.; Zhao, X.; Wang, L. Ursolic acid improves podocyte injury caused by high glucose. Nephrol. Dial. Transplant. 2017, 32, 1285–1293. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, Y.; Tan, X.; Zhang, L.; Zhang, H.; Li, Z.; Liu, S.; Li, R.; Lin, T.; Liao, R.; et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J. Pathol. 2018, 245, 235–248. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Wang, Y.; Zeng, X. Apelin involved in progression of diabetic nephropathy by inhibiting autophagy in podocytes. Cell Death Dis. 2017, 8, e3006. [Google Scholar] [CrossRef]
- Huang, G.; Zou, B.; Lv, J.; Li, T.; Huai, G.; Xiang, S.; Lu, S.; Luo, H.; Zhang, Y.; Jin, Y.; et al. Notoginsenoside R1 attenuates glucose-induced podocyte injury via the inhibition of apoptosis and the activation of autophagy through the PI3K/Akt/mTOR signaling pathway. Int. J. Mol. Med. 2017, 39, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.Y.; Zhou, X.J.; Cheng, F.J.; Hou, P.; Ren, Y.L.; Wang, S.X.; Zhao, M.H.; Yang, L.; Martinez, J.; Zhang, H. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Ann. Rheum. Dis. 2018, 77, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Bhayana, S.; Baisantry, A.; Kraemer, T.D.; Wrede, C.; Hegermann, J.; Brasen, J.H.; Bockmeyer, C.; Ulrich Becker, J.; Ochs, M.; Gwinner, W.; et al. Autophagy in kidney transplants of sirolimus treated recipients. J. Nephropathol. 2017, 6, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, J.; Finckenberg, P.; Levijoki, J.; Mervaala, E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br. J. Pharmacol. 2012, 166, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-L.; Wang, L.-T.; Huang, K.-H.; Wang, C.-C.; Chiang, C.-K.; Liu, S.-H. Quercetin attenuates renal ischemia/reperfusion injury via an activation of AMP-activated protein kinase-regulated autophagy pathway. J. Nutr. Biochem. 2014, 25, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Gwon, D.H.; Hwang, T.W.; Ro, J.Y.; Kang, Y.J.; Jeong, J.Y.; Kim, D.K.; Lim, K.; Kim, D.W.; Choi, D.E.; Kim, J.J. High Endogenous Accumulation of omega-3 Polyunsaturated Fatty Acids Protect against Ischemia-Reperfusion Renal Injury through AMPK-Mediated Autophagy in Fat-1 Mice. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Chen, W.; Xi, X.; Zhang, S.; Zou, C.; Kuang, R.; Ye, Z.; Huang, Y.; Hu, H. Pioglitazone Protects Against Renal Ischemia-Reperfusion Injury via the AMP-Activated Protein Kinase-Regulated Autophagy Pathway. Front. Pharmacol. 2018, 9, 851. [Google Scholar] [CrossRef]
- Li, J.; Gui, Y.; Ren, J.; Liu, X.; Feng, Y.; Zeng, Z.; He, W.; Yang, J.; Dai, C. Metformin Protects Against Cisplatin-Induced Tubular Cell Apoptosis and Acute Kidney Injury via AMPKα-regulated Autophagy Induction. Sci. Rep. 2016, 6, 23975. [Google Scholar] [CrossRef]
- Li, H.; Tang, Y.; Wen, L.; Kong, X.; Chen, X.; Liu, P.; Zhou, Z.; Chen, W.; Xiao, C.; Xiao, P.; et al. Neferine reduces cisplatin-induced nephrotoxicity by enhancing autophagy via the AMPK/mTOR signaling pathway. Biochem. Biophys. Res. Commun. 2017, 484, 694–701. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, L.; Chen, R.; Lu, H.; Sui, M.; Zhu, Y.; Zeng, L. SIRT3 Protects Against Acute Kidney Injury via AMPK/mTOR-Regulated Autophagy. Front. Physiol. 2018, 9, 1526. [Google Scholar] [CrossRef]
- Elimam, H.; Papillon, J.; Guillemette, J.; Navarro-Betancourt, J.R.; Cybulsky, A.V. Genetic Ablation of Calcium-independent Phospholipase A2gamma Exacerbates Glomerular Injury in Adriamycin Nephrosis in Mice. Sci. Rep. 2019, 9, 16229. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zheng, H.; Huang, S.; You, N.; Xu, J.; Ye, X.; Zhu, Q.; Feng, Y.; You, Q.; Miao, H.; et al. Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp. Cell Res. 2015, 337, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, Y.; Zhang, X.; Zang, Y.; Zhang, Y.; Wang, L.; Wang, H.; Wang, Y.; Cao, A.; Peng, W. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKα-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci. Rep. 2017, 7, 6852. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, L.; Lin, H.; Song, J.; Wang, J.; Yin, Y.; Zhao, J.; Xu, X.; Li, Z.; Li, L. Mangiferin prevents diabetic nephropathy progression and protects podocyte function via autophagy in diabetic rat glomeruli. Eur. J. Pharmacol. 2018, 824, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, H.W.; Kim, M.Y.; Kim, T.W.; Kim, E.N.; Kim, Y.; Chung, S.; Kim, Y.S.; Choi, B.S.; Kim, Y.-S.; et al. Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis. 2018, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liu, S.; Ma, Q.; Xiao, D.; Chen, L. Berberine enhances the AMPK activation and autophagy and mitigates high glucose-induced apoptosis of mouse podocytes. Eur. J. Pharmacol. 2017, 794, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Zhou, Y.; et al. Progranulin alleviates podocyte injury via regulating CAMKK/AMPK-mediated autophagy under diabetic conditions. J. Mol. Med. 2019, 97, 1507–1520. [Google Scholar] [CrossRef]
- Decleves, A.E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef]
- Sohn, M.; Kim, K.; Uddin, M.J.; Lee, G.; Hwang, I.; Kang, H.; Kim, H.; Lee, J.H.; Ha, H. Delayed treatment with fenofibrate protects against high-fat diet-induced kidney injury in mice: The possible role of AMPK autophagy. Am. J. Physiol. Ren. Physiol. 2017, 312, F323–F334. [Google Scholar] [CrossRef]
- Li, W.; Zhang, P.L. Up-regulated mTOR pathway indicates active disease in both human native and transplant kidneys. Ann. Clin. Lab. Sci. 2013, 43, 378–388. [Google Scholar]
- Beckerman, P.; Bi-Karchin, J.; Park, A.S.; Qiu, C.; Dummer, P.D.; Soomro, I.; Boustany-Kari, C.M.; Pullen, S.S.; Miner, J.H.; Hu, C.A.; et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017, 23, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Cina, D.P.; Onay, T.; Paltoo, A.; Li, C.; Maezawa, Y.; De Arteaga, J.; Jurisicova, A.; Quaggin, S.E. Inhibition of MTOR disrupts autophagic flux in podocytes. J. Am. Soc. Nephrol. 2012, 23, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Mack, H.I.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1α in db/db mice. PLoS ONE 2014, 9, e96147. [Google Scholar] [CrossRef] [PubMed]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, S.; Hosojima, M.; Kaneko, R.; Aoki, H.; Nakano, D.; Sasagawa, T.; Kabasawa, H.; Kaseda, R.; Yasukawa, R.; Ishikawa, T.; et al. Megalin-Mediated Tubuloglomerular Alterations in High-Fat Diet-Induced Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1996–2008. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wang, Z.; Tao, L.; Wang, Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010, 6, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Banhegyi, G.; Kapuy, O. GADD34 Keeps the mTOR Pathway Inactivated in Endoplasmic Reticulum Stress Related Autophagy. PLoS ONE 2016, 11, e0168359. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Inagi, R.; Takano, H.; Sato, S.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol. Dial. Transplant. 2009, 24, 2665–2672. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, B.B.; Yang, C.; Ou, Y.; Feng, X.; Muhoza, D.; Holmes, A.F.; Theus, S.; Deshmukh, S.; Haun, R.S.; Kaushal, G.P. Endoplasmic Reticulum Stress-Induced Autophagy Provides Cytoprotection from Chemical Hypoxia and Oxidant Injury and Ameliorates Renal Ischemia-Reperfusion Injury. PLoS ONE 2015, 10, e0140025. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Liu, F.; Li, Z.F.; Wang, Z.Y.; Wang, L. Role of subcellular calcium redistribution in regulating apoptosis and autophagy in cadmium-exposed primary rat proximal tubular cells. J. Inorg. Biochem. 2016, 164, 99–109. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.Y.; Zhou, X.P.; Liu, Z.P.; Song, X.B.; Wang, Z.Y.; Wang, L. Cadmium disrupts autophagic flux by inhibiting cytosolic Ca2+-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 2017, 383, 13–23. [Google Scholar] [CrossRef]
- Gu, J.; Dai, S.; Liu, Y.; Liu, H.; Zhang, Y.; Ji, X.; Yu, F.; Zhou, Y.; Chen, L.; Tse, W.K.F.; et al. Activation of Ca2+-sensing receptor as a protective pathway to reduce Cadmium-induced cytotoxicity in renal proximal tubular cells. Sci. Rep. 2018, 8, 1092. [Google Scholar] [CrossRef]
- Ono, Y.; Sorimachi, H. Calpains: An elaborate proteolytic system. Biochim. Biophys. Acta 2012, 1824, 224–236. [Google Scholar] [CrossRef]
- Menzies, F.M.; Garcia-Arencibia, M.; Imarisio, S.; O’Sullivan, N.C.; Ricketts, T.; Kent, B.A.; Rao, M.V.; Lam, W.; Green-Thompson, Z.W.; Nixon, R.A.; et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 2015, 22, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.G.; Zhang, L.; Chen, G.; Zhang, T.; Liu, J.; Jin, M.; Ma, X.; Ma, D.; Yuan, J. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 2010, 6, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [PubMed]
- Marcassa, E.; Raimondi, M.; Anwar, T.; Eskelinen, E.L.; Myers, M.P.; Triolo, G.; Schneider, C.; Demarchi, F. Calpain mobilizes Atg9/Bif-1 vesicles from Golgi stacks upon autophagy induction by thapsigargin. Biol. Open 2017, 6, 551–562. [Google Scholar] [CrossRef]
- Herzog, C.; Yang, C.; Holmes, A.; Kaushal, G.P. zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am. J. Physiol. Ren. Physiol. 2012, 303, F1239–F1250. [Google Scholar] [CrossRef]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef]
- Rose, C.; Menzies, F.M.; Renna, M.; Acevedo-Arozena, A.; Corrochano, S.; Sadiq, O.; Brown, S.D.; Rubinsztein, D.C. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 2144–2153. [Google Scholar] [CrossRef]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef]
- Nayak, B.; Xie, P.; Akagi, S.; Yang, Q.; Sun, L.; Wada, J.; Thakur, A.; Danesh, F.R.; Chugh, S.S.; Kanwar, Y.S. Modulation of renal-specific oxidoreductase/myo-inositol oxygenase by high-glucose ambience. Proc. Natl. Acad. Sci. USA 2005, 102, 17952–17957. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgo, J.; Agostinis, P.; Missiaen, L.; De Smedt, H.; Parys, J.B.; et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; Fiebig, A.A.; Andrews, D.; Molgo, J.; Diaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.-P.; Kindt, D.; Luyten, T.; Welkenhuyzen, K.; Missiaen, L.; De Smedt, H.; Bultynck, G.; Parys, J.B. mTOR-Controlled Autophagy Requires Intracellular Ca2+ Signaling. PLoS ONE 2013, 8, e61020. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., III; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Wang, L.L.; Deng, R.; Tang, J.; Shen, Y.; Guo, J.F.; Wang, Y.; Xia, L.P.; Feng, G.K.; Liu, Q.Q.; et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009, 28, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.L.; Tooze, S.A. Coordinated regulation of autophagy by p38α MAPK through mAtg9 and p38IP. EMBO J. 2010, 29, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Keil, E.; Hocker, R.; Schuster, M.; Essmann, F.; Ueffing, N.; Hoffman, B.; Liebermann, D.A.; Pfeffer, K.; Schulze-Osthoff, K.; Schmitz, I. Phosphorylation of Atg5 by the Gadd45β-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 2013, 20, 321–332. [Google Scholar] [CrossRef]
- Copetti, T.; Demarchi, F.; Schneider, C. p65/RelA binds and activates the beclin 1 promoter. Autophagy 2009, 5, 858–859. [Google Scholar] [CrossRef]
- Kang, Y.L.; Saleem, M.A.; Chan, K.W.; Yung, B.Y.; Law, H.K. Trehalose, an mTOR independent autophagy inducer, alleviates human podocyte injury after puromycin aminonucleoside treatment. PLoS ONE 2014, 9, e113520. [Google Scholar] [CrossRef]
- O’Neill, M.K.; Piligian, B.F.; Olson, C.D.; Woodruff, P.J.; Swarts, B.M. Tailoring Trehalose for Biomedical and Biotechnological Applications. Pure Appl. Chem. 2017, 89, 1223–1249. [Google Scholar] [CrossRef]
- Zhu, P.; Sieben, C.J.; Xu, X.; Harris, P.C.; Lin, X. Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum. Mol. Genet. 2017, 26, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zha, W.; Peng, H.; Wang, Q.; Zhang, S.; Ren, J. Trehalose Protects against Insulin Resistance-Induced Tissue Injury and Excessive Autophagy in Skeletal Muscles and Kidney. Curr. Pharm. Des. 2019, 25, 2077–2085. [Google Scholar] [CrossRef] [PubMed]
- Talab, S.S.; Emami, H.; Elmi, A.; Nezami, B.G.; Assa, S.; Deroee, A.F.; Daneshmand, A.; Tavangar, S.M.; Dehpour, A.R. Chronic lithium treatment protects the rat kidney against ischemia/reperfusion injury: The role of nitric oxide and cyclooxygenase pathways. Eur. J. Pharmacol. 2010, 647, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Ge, Y.; Wang, Z.; Zhuang, S.; Dworkin, L.; Peng, A.; Gong, R. Delayed administration of a single dose of lithium promotes recovery from AKI. J. Am. Soc. Nephrol. 2014, 25, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Liu, A.; Fang, H.; Dirsch, O.; Dahmen, U.; Boettcher, M. Corrigendum to Induction of Autophagy Reduces Ischemia/Reperfusion Injury in Steatotic Rat Livers [Journal of Surgical Research 216 (2017) 207-218]. J. Surg. Res. 2019, 237, 138. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Yang, X.J.; Fu, Q.; Han, B.; Ling, L.; Bai, J.; Zong, B.; Jiang, C.Y. Intermedin attenuates myocardial infarction through activation of autophagy in a rat model of ischemic heart failure via both cAMP and MAPK/ERK1/2 pathways. Int. J. Clin. Exp. Pathol. 2015, 8, 9836–9844. [Google Scholar]
- Yang, J.; Chen, Q.; Tian, S.; Song, S.; Liu, F.; Wang, Q.; Fu, Z. The role of 1, 25-dyhydroxyvitamin D3 in mouse liver ischemia reperfusion injury: Regulation of autophagy through activation of MEK/ERK signaling and PTEN/PI3K/Akt/mTORC1 signaling. Am. J. Transl. Res. 2015, 7, 2630–2645. [Google Scholar]
- Pal, R.; Bajaj, L.; Sharma, J.; Palmieri, M.; Di Ronza, A.; Lotfi, P.; Chaudhury, A.; Neilson, J.; Sardiello, M.; Rodney, G.G. NADPH oxidase promotes Parkinsonian phenotypes by impairing autophagic flux in an mTORC1-independent fashion in a cellular model of Parkinson’s disease. Sci. Rep. 2016, 6, 22866. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, T.A.; Majumder, S.; Thieme, K.; Batchu, S.N.; White, K.E.; Liu, Y.; Brijmohan, A.S.; Bowskill, B.B.; Advani, S.L.; Woo, M.; et al. Janus Kinase 2 Regulates Transcription Factor EB Expression and Autophagy Completion in Glomerular Podocytes. J. Am. Soc. Nephrol. 2017, 28, 2641–2653. [Google Scholar] [CrossRef] [PubMed]
- Rega, L.R.; Polishchuk, E.; Montefusco, S.; Napolitano, G.; Tozzi, G.; Zhang, J.; Bellomo, F.; Taranta, A.; Pastore, A.; Polishchuk, R.; et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016, 89, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, H.; Jin, Y.; Shen, K.; Chen, X.; Xu, Z.; Jin, B.; Pan, H. Role of TFEB in autophagic modulation of ischemia reperfusion injury in mice kidney and protection by urolithin A. Food Chem. Toxicol. 2019, 131, 110591. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.W.; Shin, Y.J.; Luo, K.; Quan, Y.; Ko, E.J.; Chung, B.H.; Yang, C.W. Effect of Klotho on autophagy clearance in tacrolimus-induced renal injury. FASEB J. 2019, 33, 2694–2706. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Liu, Z.; Zhang, H. The protective effect of the TUG1/miR197/MAPK1 axis on lipopolysaccharideinduced podocyte injury. Mol. Med. Rep. 2019, 20, 49–56. [Google Scholar] [CrossRef]
- Zhou, J.; Fan, Y.; Zhong, J.; Huang, Z.; Huang, T.; Lin, S.; Chen, H. TAK1 mediates excessive autophagy via p38 and ERK in cisplatin-induced acute kidney injury. J. Cell Mol. Med. 2018, 22, 2908–2921. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, X.; Wang, J.; Fan, J.; Kong, Q.; Yu, X. Aristolochic acid I induced autophagy extenuates cell apoptosis via ERK 1/2 pathway in renal tubular epithelial cells. PLoS ONE 2012, 7, e30312. [Google Scholar] [CrossRef]
- Li, L.; Zviti, R.; Ha, C.; Wang, Z.V.; Hill, J.A.; Lin, F. Forkhead box O3 (FoxO3) regulates kidney tubular autophagy following urinary tract obstruction. J. Biol. Chem. 2017, 292, 13774–13783. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Investig. 2019, 129, 2374–2389. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, X.; Zhang, Y.; Ding, G.; Zhu, C.; Huang, S.; Jia, Z.; Zhang, A. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Jiang, D.; Xiao, J.; Fu, C.; Zhang, Z.; Ye, Z.; Zhang, X. Ischemic preconditioning attenuates ischemia/reperfusion-induced kidney injury by activating autophagy via the SGK1 signaling pathway. Cell Death Dis. 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liao, W.; Yang, J.; Ma, K.; Li, X.; Wang, Y.; Wang, D.; Wang, L.; Zhang, Y.; Yin, Y.; et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 2012, 8, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ding, W.; Zhang, M.; Li, H.; Guo, H.; Lin, L.; Chen, J.; Gu, Y. Role of FOXO1 in aldosterone-induced autophagy: A compensatory protective mechanism related to podocyte injury. Oncotarget 2016, 7, 45331–45351. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Du, M.; Wang, Q.; Ma, X.; Wu, L.; Guo, F.; Ji, H.; Huang, F.; Qin, G. FoxO1 Promotes Mitophagy in the Podocytes of Diabetic Male Mice via the PINK1/Parkin Pathway. Endocrinology 2017, 158, 2155–2167. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, L.; Hong, H.; Li, J.; Wang, H.; Li, X. Atrasentan alleviates high glucose-induced podocyte injury by the microRNA-21/forkhead box O1 axis. Eur. J. Pharmacol. 2019, 852, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.K.; Menshikova, E.V.; Mi, Z.; Verrier, J.D.; Bansal, R.; Janesko-Feldman, K.; Jackson, T.C.; Kochanek, P.M. Renal 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase Is an Important Determinant of AKI Severity after Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2016, 27, 2069–2081. [Google Scholar] [CrossRef]
- Fougeray, S.; Mami, I.; Bertho, G.; Beaune, P.; Thervet, E.; Pallet, N. Tryptophan depletion and the kinase GCN2 mediate IFN-γ-induced autophagy. J. Immunol. 2012, 189, 2954–2964. [Google Scholar] [CrossRef]
- Chaudhary, K.; Shinde, R.; Liu, H.; Gnana-Prakasam, J.P.; Veeranan-Karmegam, R.; Huang, L.; Ravishankar, B.; Bradley, J.; Kvirkvelia, N.; McMenamin, M.; et al. Amino acid metabolism inhibits antibody-driven kidney injury by inducing autophagy. J. Immunol. 2015, 194, 5713–5724. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Sounidaki, M.; Antoniadis, N.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Preconditioning of primary human renal proximal tubular epithelial cells without tryptophan increases survival under hypoxia by inducing autophagy. Int. Urol. Nephrol. 2017, 49, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed]
- Schumer, M.; Colombel, M.C.; Sawczuk, I.S.; Gobe, G.; Connor, J.; O’Toole, K.M.; Olsson, C.A.; Wise, G.J.; Buttyan, R. Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am. J. Pathol. 1992, 140, 831–838. [Google Scholar] [PubMed]
- Havasi, A.; Borkan, S.C. Apoptosis and acute kidney injury. Kidney Int. 2011, 80, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Chen, J.K.; Ramesh, G.; Dong, Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013, 84, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Shimizu, A.; Masuda, Y.; Mii, A.; Fujita, E.; Yoshizaki, K.; Higo, S.; Kanzaki, G.; Kajimoto, Y.; Takano, H.; et al. Caspase-3-independent internucleosomal DNA fragmentation in ischemic acute kidney injury. Nephron Exp. Nephrol. 2012, 120, e103–e113. [Google Scholar] [CrossRef]
- Linkermann, A.; Brasen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012, 81, 751–761. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Shah, S.V. Non-apoptotic effects of antiapoptotic agent zVAD-fmk in renal injury. Kidney Int. 2013, 83, 531. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Lau, A.; Wang, S.; Jiang, J.; Haig, A.; Pavlosky, A.; Linkermann, A.; Zhang, Z.X.; Jevnikar, A.M. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant. 2013, 13, 2805–2818. [Google Scholar] [CrossRef] [PubMed]
- Tristao, V.R.; Goncalves, P.F.; Dalboni, M.A.; Batista, M.C.; Durao Mde, S., Jr.; Monte, J.C. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren. Fail. 2012, 34, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Heller, J.O.; Prokai, A.; Weinberg, J.M.; De Zen, F.; Himmerkus, N.; Szabo, A.J.; Brasen, J.H.; Kunzendorf, U.; Krautwald, S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J. Am. Soc. Nephrol. 2013, 24, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Cui, H.; Gan, H.; Xia, Y.; Wang, L.; Wang, Y.; Sun, Y. Necroptosis mediated by receptor interaction protein kinase 1 and 3 aggravates chronic kidney injury of subtotal nephrectomised rats. Biochem. Biophys. Res. Commun. 2015, 461, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [PubMed]
- Tristao, V.R.; Pessoa, E.A.; Nakamichi, R.; Reis, L.A.; Batista, M.C.; Durao Junior Mde, S.; Monte, J.C. Synergistic effect of apoptosis and necroptosis inhibitors in cisplatin-induced nephrotoxicity. Apoptosis 2016, 21, 51–59. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Dewitz, C.; Schmitz, J.; Schroder, A.S.; Brasen, J.H.; Stockwell, B.R.; Murphy, J.M.; Kunzendorf, U.; Krautwald, S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol. Life Sci. 2017, 74, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- Devalaraja-Narashimha, K.; Diener, A.M.; Padanilam, B.J. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Ren. Physiol. 2009, 297, F749–F759. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.R.; Lewington, A.J.; Hammerman, M.R.; Padanilam, B.J. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1834–R1840. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Devalaraja-Narashimha, K.; Singaravelu, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Ren. Physiol. 2005, 288, F387–F398. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Long, K.E.; Tang, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int. 2012, 82, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Shevalye, H.; Stavniichuk, R.; Xu, W.; Zhang, J.; Lupachyk, S.; Maksimchyk, Y.; Drel, V.R.; Floyd, E.Z.; Slusher, B.; Obrosova, I.G. Poly(ADP-ribose) polymerase (PARP) inhibition counteracts multiple manifestations of kidney disease in long-term streptozotocin-diabetic rat model. Biochem. Pharmacol. 2010, 79, 1007–1014. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2011, 301, F450–F459. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.C.; Livingston, M.J.; Liang, X.L.; Dong, Z. Cell Apoptosis and Autophagy in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 557–584. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Klionsky, D.J.; Zhang, H. Podocytes and autophagy: A potential therapeutic target in lupus nephritis. Autophagy 2019, 15, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Livingston, M.J.; Liu, Z.; Dong, G.; Zhang, M.; Chen, J.K.; Dong, Z. Autophagy in diabetic kidney disease: Regulation, pathological role and therapeutic potential. Cell Mol. Life Sci. 2018, 75, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Oral, O.; Oz-Arslan, D.; Itah, Z.; Naghavi, A.; Deveci, R.; Karacali, S.; Gozuacik, D. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012, 17, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef]
- You, M.; Savaraj, N.; Kuo, M.T.; Wangpaichitr, M.; Varona-Santos, J.; Wu, C.; Nguyen, D.M.; Feun, L. TRAIL induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol. Cell. Biochem. 2013, 374, 181–190. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef]
- Pagliarini, V.; Wirawan, E.; Romagnoli, A.; Ciccosanti, F.; Lisi, G.; Lippens, S.; Cecconi, F.; Fimia, G.M.; Vandenabeele, P.; Corazzari, M.; et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012, 19, 1495–1504. [Google Scholar] [CrossRef]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Wu, Y.T.; Tan, H.L.; Huang, Q.; Kim, Y.S.; Pan, N.; Ong, W.Y.; Liu, Z.G.; Ong, C.N.; Shen, H.M. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy 2008, 4, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.C.; Yu, L.; Wang, H.J.; Tashiro, S.; Onodera, S.; Ikejima, T. TNFα-induced necroptosis and autophagy via supression of the p38-NF-kappaB survival pathway in L929 cells. J. Pharmacol. Sci. 2011, 117, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Farkas, T.; Daugaard, M.; Jaattela, M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J. Biol. Chem. 2011, 286, 38904–38912. [Google Scholar] [CrossRef] [PubMed]
- Bray, K.; Mathew, R.; Lau, A.; Kamphorst, J.J.; Fan, J.; Chen, J.; Chen, H.Y.; Ghavami, A.; Stein, M.; DiPaola, R.S.; et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS ONE 2012, 7, e41831. [Google Scholar] [CrossRef]
- Ogasawara, M.; Yano, T.; Tanno, M.; Abe, K.; Ishikawa, S.; Miki, T.; Kuno, A.; Tobisawa, T.; Muratsubaki, S.; Ohno, K.; et al. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J. Mol. Cell. Cardiol. 2017, 108, 203–213. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Safa, A.R. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy. J. Carcinog. Mutagen. 2013. [Google Scholar] [CrossRef] [PubMed]
- Radonjic-Hoesli, S.; Wang, X.; de Graauw, E.; Stoeckle, C.; Styp-Rekowska, B.; Hlushchuk, R.; Simon, D.; Spaeth, P.J.; Yousefi, S.; Simon, H.U. Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counterregulated by autophagy. J. Allergy Clin. Immunol. 2017, 140, 1632–1642. [Google Scholar] [CrossRef]
- Kharaziha, P.; Chioureas, D.; Baltatzis, G.; Fonseca, P.; Rodriguez, P.; Gogvadze, V.; Lennartsson, L.; Bjorklund, A.C.; Zhivotovsky, B.; Grander, D.; et al. Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 2015, 6, 37066–37082. [Google Scholar] [CrossRef]
- Echeverry, N.; Ziltener, G.; Barbone, D.; Weder, W.; Stahel, R.A.; Broaddus, V.C.; Felley-Bosco, E. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual PI3K/mTOR inhibitors. Cell Death Dis. 2015, 6, e1757. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hwang, K.E.; Park, D.S.; Oh, S.H.; Jun, H.Y.; Yoon, K.H.; Jeong, E.T.; Kim, H.R.; Kim, Y.S. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J. Transl. Med. 2017, 15, 123. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Li, X.H.; Zeng, X.C.; Li, J.; Zhou, J.; Xiao, B.; Hu, K. Curcumin protects neuronal cells against status-epilepticus-induced hippocampal damage through induction of autophagy and inhibition of necroptosis. Can. J. Physiol. Pharmacol. 2017, 95, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.J.; Rizwan Alam, M.; Waldeck-Weiermair, M.; Karsten, F.; Groschner, L.; Riederer, M.; Hallstrom, S.; Rockenfeller, P.; Konya, V.; Heinemann, A.; et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J. Biol. Chem. 2012, 287, 21110–21120. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schafer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.M.; Kim, S.J.; Lee, S.M. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol. Appl. Pharmacol. 2016, 308, 1–10. [Google Scholar] [CrossRef]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef]
- Dey, A.; Mustafi, S.B.; Saha, S.; Kumar Dhar Dwivedi, S.; Mukherjee, P.; Bhattacharya, R. Inhibition of BMI1 induces autophagy-mediated necroptosis. Autophagy 2016, 12, 659–670. [Google Scholar] [CrossRef]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Padanilam, B.J. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell Mol. Life Sci. 2016, 73, 2309–2324. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vargas, J.M.; Ruiz-Magana, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodriguez, M.I.; Munoz-Gamez, J.A.; de Almodovar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Ethier, C.; Tardif, M.; Arul, L.; Poirier, G.G. PARP-1 modulation of mTOR signaling in response to a DNA alkylating agent. PLoS ONE 2012, 7, e47978. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Shen, H.M. To die or to live: The dual role of poly(ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy 2009, 5, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Chung, S.; Sundar, I.K.; Yao, H.; Arunachalam, G.; McBurney, M.W.; Rahman, I. Cigarette smoke-induced autophagy is regulated by SIRT1-PARP-1-dependent mechanism: Implication in pathogenesis of COPD. Arch. Biochem. Biophys. 2010, 500, 203–209. [Google Scholar] [CrossRef]
- Chen, Z.T.; Zhao, W.; Qu, S.; Li, L.; Lu, X.D.; Su, F.; Liang, Z.G.; Guo, S.Y.; Zhu, X.D. PARP-1 promotes autophagy via the AMPK/mTOR pathway in CNE-2 human nasopharyngeal carcinoma cells following ionizing radiation, while inhibition of autophagy contributes to the radiation sensitization of CNE-2 cells. Mol. Med. Rep. 2015, 12, 1868–1876. [Google Scholar] [CrossRef]


| Kidney Disease | Agent/Drug | Effect on Autophagy | Effect on Fibrosis | Reference |
|---|---|---|---|---|
| Autophagy suppresses fibrosis | ||||
| UUO model | 3-MA | ↓ autophagy | ↑ in interstitial fibrosis and tubular apoptosis | [42,43,44] |
| LC3 KO and beclin-1 ± | ↓ autophagy | ↑ deposition of collagen and TGF-β1 | [46] | |
| Conditional deletion of ATG7 in distal tubule | ↓ autophagy in the distal tubules | ↑ in tubulointerstitial fibrosis via the TGF-β/Smad4 and NLRP3 signaling | [47] | |
| Conditional deletion of ATG5 | ↓ autophagy | ↑ renal interstitial fibrosis and cell cycle arrest at G2/M | [48] | |
| Proximal tubule specific deletion of ATG5 | ↓ autophagy | ↑ renal fibrosis due to leukocyte infiltration and expression of pro-inflammatory cytokines | [49] | |
| Valproic acid (histone deacetylase inhibitor) | ↑ autophagy | ↓ in renal fibrosis | [51] | |
| Rubicon | ↓ autophagy | ↑ in renal fibrosis | [52] | |
| STZ- DN | miR -22 upregulation | ↓ autophagy | ↑ in renal fibrosis with increased expression of col-IV and α-SMA | [53] |
| Triptolide | ↑ autophagy via miR-141-3p/PTEN/Akt/mTOR pathway | ↓ in renal fibrosis | [54] | |
| HFD with CKD | Elafibranor (dual PPARα/δ agonist) | ↑ autophagy mediated by SIRT1 | ↓ in renal fibrosis | [56] |
| 5/6-Nephrectomy | Knockdown of periostin gene (osteoblast specific factor-2) | ↑ autophagy and upregulates periostin gene (pro-fibrotic and pro-inflammatory factor) | ↓ in renal inflammation and fibrosis | [58] |
| Proximal tubule specific deletion of ATG7 | ↑ autophagy | ↓ in renal fibrosis with pro-fibrotic FGF2 | [60] | |
| Autophagy promotes fibrosis | ||||
| Ang II- induced CKD | 1,25-dihydroxyvitamin D3 | ↓ autophagy with improved mitochondrial dysfunction | ↓ in renal fibrosis | [55] |
| Stage 3 and stage 4 CKD | Rhubarb (Rhein- bioactive component) | ↓ autophagy | ↓ in renal fibrosis | [61] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A.; Shah, S.V. Autophagy Function and Regulation in Kidney Disease. Biomolecules 2020, 10, 100. https://doi.org/10.3390/biom10010100
Kaushal GP, Chandrashekar K, Juncos LA, Shah SV. Autophagy Function and Regulation in Kidney Disease. Biomolecules. 2020; 10(1):100. https://doi.org/10.3390/biom10010100
Chicago/Turabian StyleKaushal, Gur P., Kiran Chandrashekar, Luis A. Juncos, and Sudhir V. Shah. 2020. "Autophagy Function and Regulation in Kidney Disease" Biomolecules 10, no. 1: 100. https://doi.org/10.3390/biom10010100
APA StyleKaushal, G. P., Chandrashekar, K., Juncos, L. A., & Shah, S. V. (2020). Autophagy Function and Regulation in Kidney Disease. Biomolecules, 10(1), 100. https://doi.org/10.3390/biom10010100