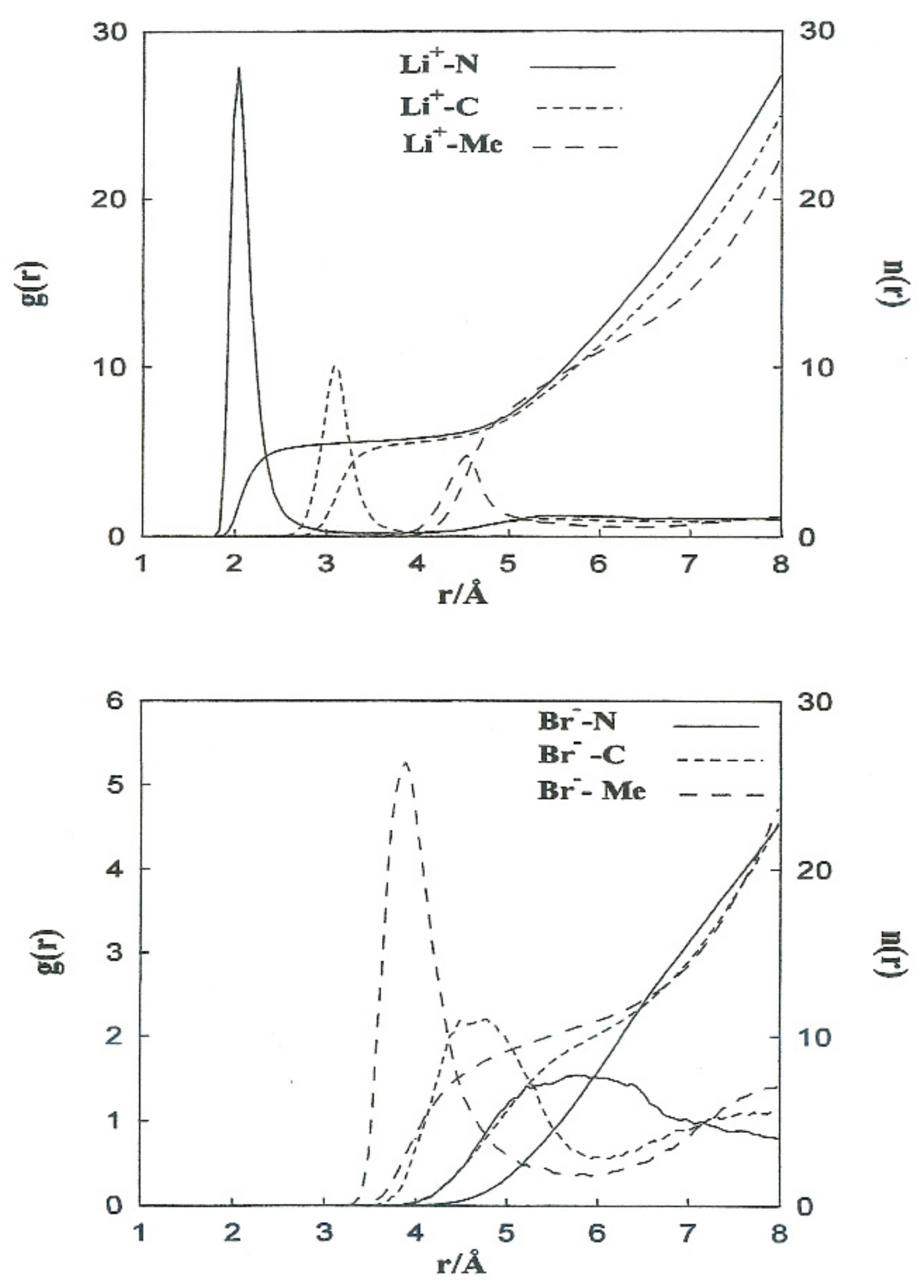
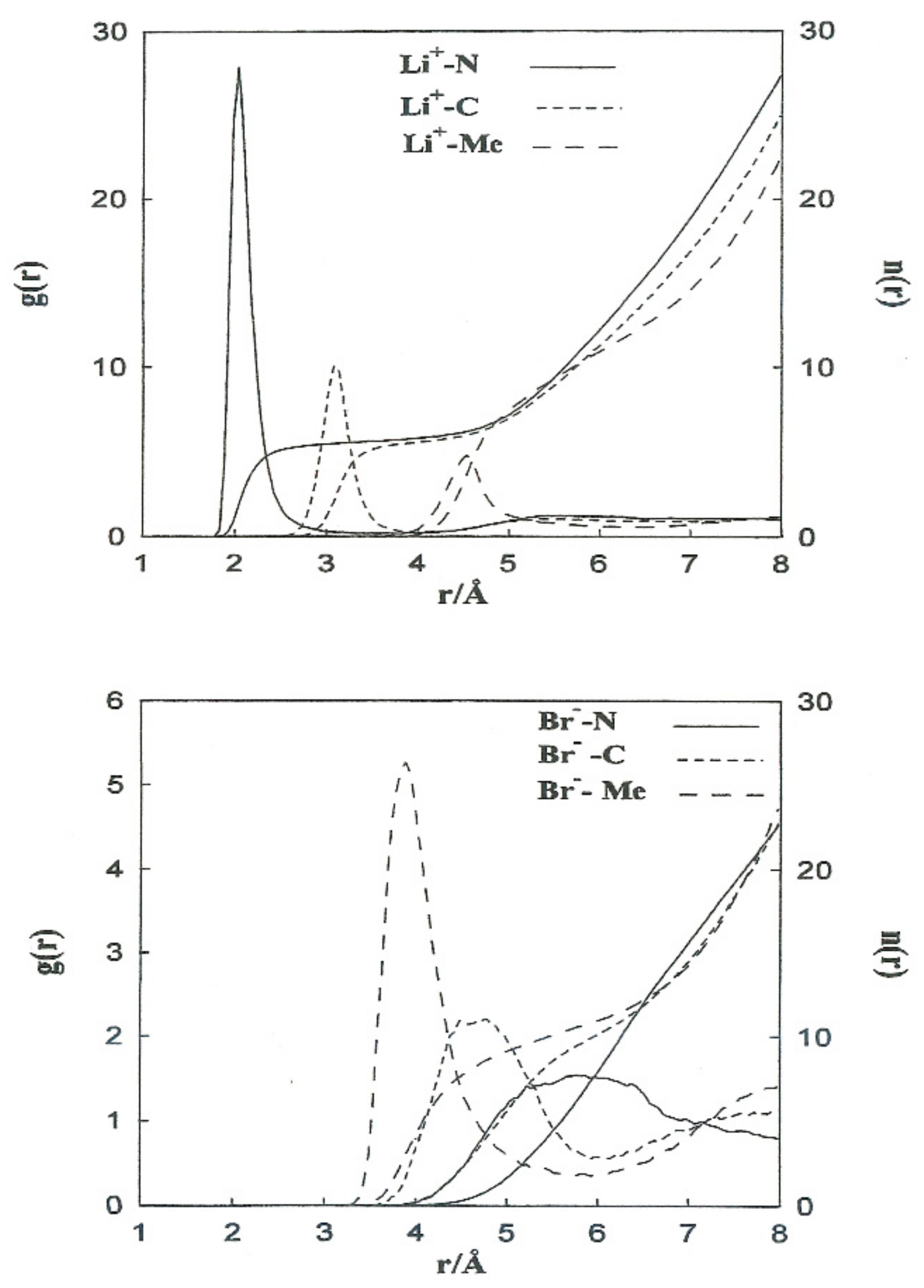
3.1. Ion–Acetonitrile Structural Properties
The structural properties obtained from the non-CMD simulations of Li
+ and Br
− ions in acetonitrile are shown in
Figure 2 (left axis) in the form of ion-acetonitrile radial distribution functions
. We similarly calculated the running coordination numbers (
) which is giving by
where
is the numeric solvent density. The function
gives the mean number of molecules within a sphere of radius r centered on the ion. The coordination number is defined as the plateau value of
at distances close to the first g(r) minimum (
Figure 2, right axis).
Table 4 summarizes the positions of the first peaks of
, and the coordination numbers obtained from our simulations and neutron diffraction experiments. The most probable distances, Li
+-N and Li
+-C, are well-reproduced. For Li
+ in acetonitrile, we obtain a coordination number slightly higher than the experimental one (5.5 instead of 3).
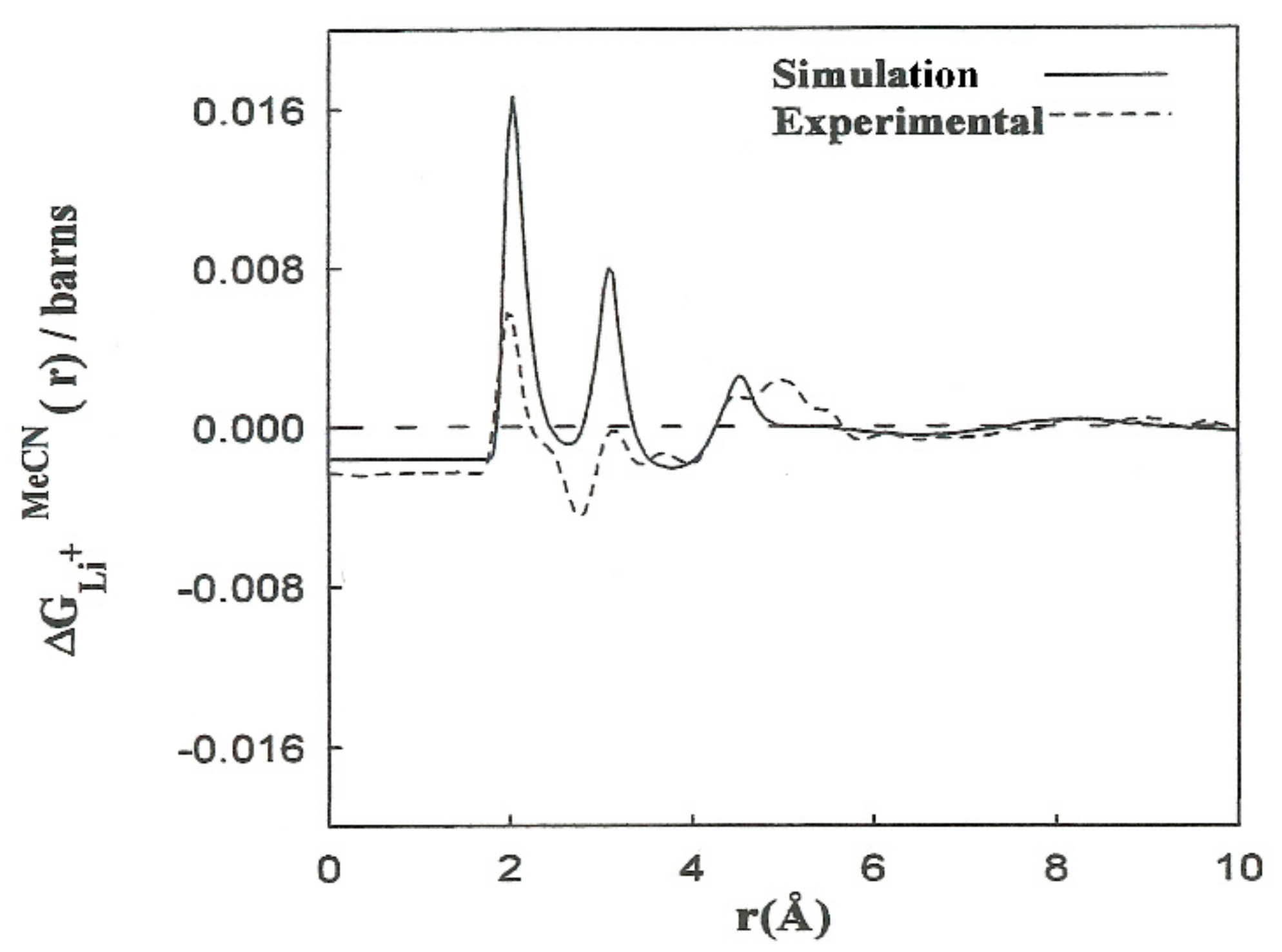
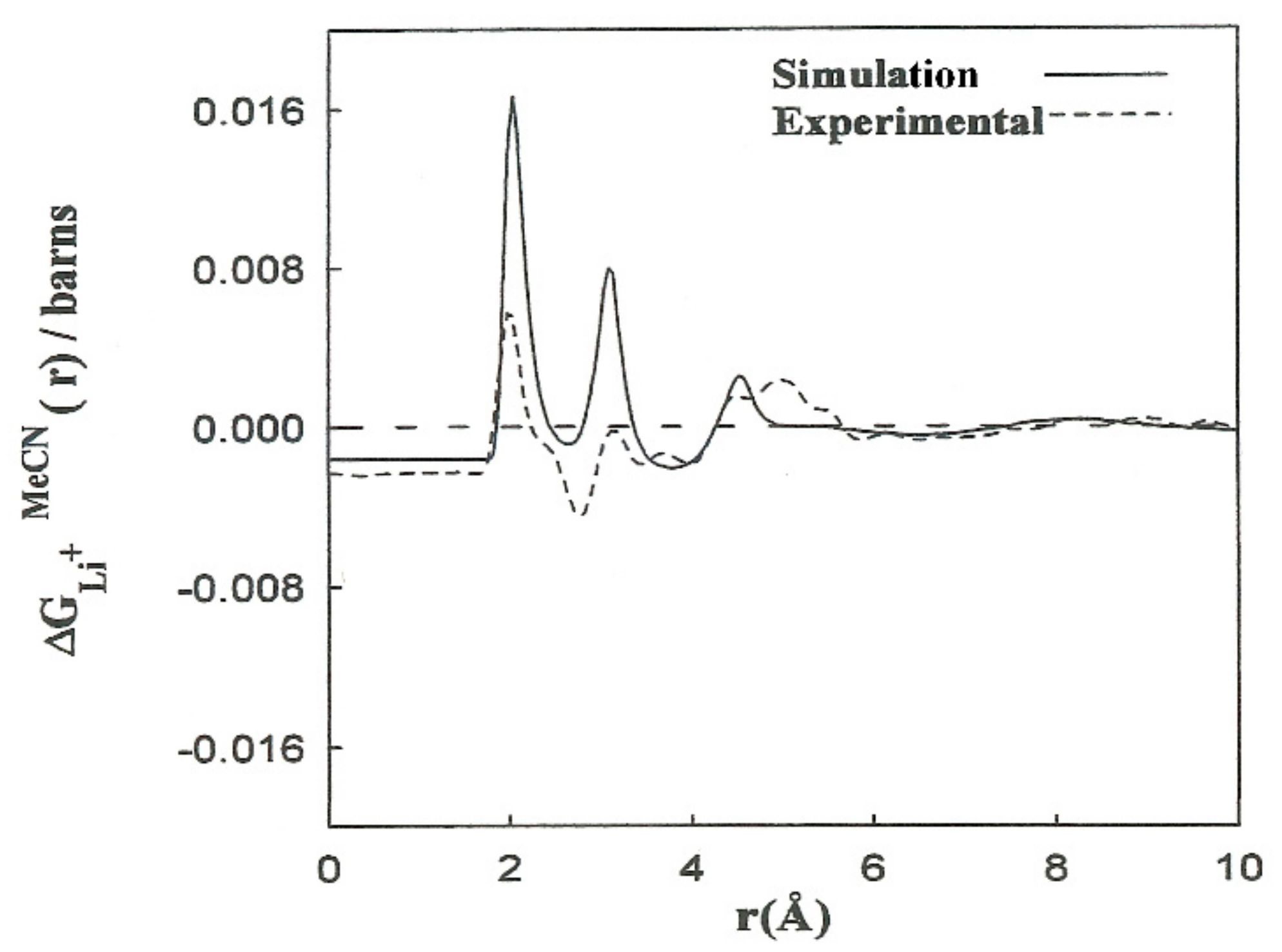
Elastic neutron scattering measurements obtained by applying the first-order difference approach [
22,
23] yield experimental information about the arrangement of solvent molecules surrounding ions. This procedure establishes a difference function
, which is a combination of the partial radial distribution functions corresponding to each solvent atom:
,
,
. The experimental solubility of LiBr in acetonitrile is 0.58 M. We calculated the
corresponding to this concentration from the
for Li
+ as follows:
where the constants A–F depend on the concentration and scattering lengths. The contribution of the first two terms of (4) pertains to distances beyond the first hydration shell,
. The shape of the
function resulting from the simulations is in agreement with the experimental results (
Figure 3). We obtained the contribution to
for the last three terms of (4). The results are consistent with the interpretation regarding the principal
peaks. The discrepancies in the height of the
peaks can be attributed to the deficiency in the interaction potentials used, as well as to experimental inaccuracies and the hydrogen atoms of the methyl group not being taken into account. Nevertheless, nearly all of the computer simulation studies described for acetonitrile and acetonitrile solutions are based on the hypothesis of rigid molecular models that do not explicitly consider the hydrogen atoms of the methyl group. With this assumption, the computing time is substantially decreased, and longer simulations of larger systems can be carried out.
Table 4 summarizes the positions of the first peaks of
, and the coordination numbers obtained from our simulations and neutron diffraction experiments. The most probable distances, Li
+-N and Li
+-C, are well-reproduced. For Li
+ in acetonitrile, we obtain a coordination number slightly higher than the experimental one (5.5 instead of 3). This failure of simulations can be attributed to the deficiency in the interaction potentials used and the hydrogen atoms of the methyl group not being considered explicit.
3.2. Mean Force Potentials
The force (∆
F(
t;
r)) applied by N solvent molecules along the intermolecular axis of two A and B ions is indicated by:
the resultant forces
FAS(
t;
r) and
FBS(
t;
r) acting on the solute caused by the molecular liquid,
mA and
mB are the masses of the solute,
is the unit vector alongside the AB path and
is the reduced mass. This expression is calculated at every single time interval and averaged throughout the entire simulation. Where
Fd(r) is the direct solute force, and the resultant MF between ions is:
where
.
W(r) was calculated by integrating the mean force:
W(r0) was calculated so the PMF values matched the macroscopic Coulomb potential at large distances (
and used the experimental acetonitrile relative permittivity (
).
Table 2 summarizes the range of interionic separation for each pair and the total number of simulations performed.
The statistical errors for
W(r) and Δ
F were analyzed as stated by the method outlined in the ref. [
24].
Table 5 shows the statistical inefficiency (
), standard deviation (
) and estimated statistical errors (
) values according to several interionic distances and simulation lengths. Simulations beyond 50 ps provided no further benefit. The estimated errors for the mean force potentials for the various ion pairs range between
and
.
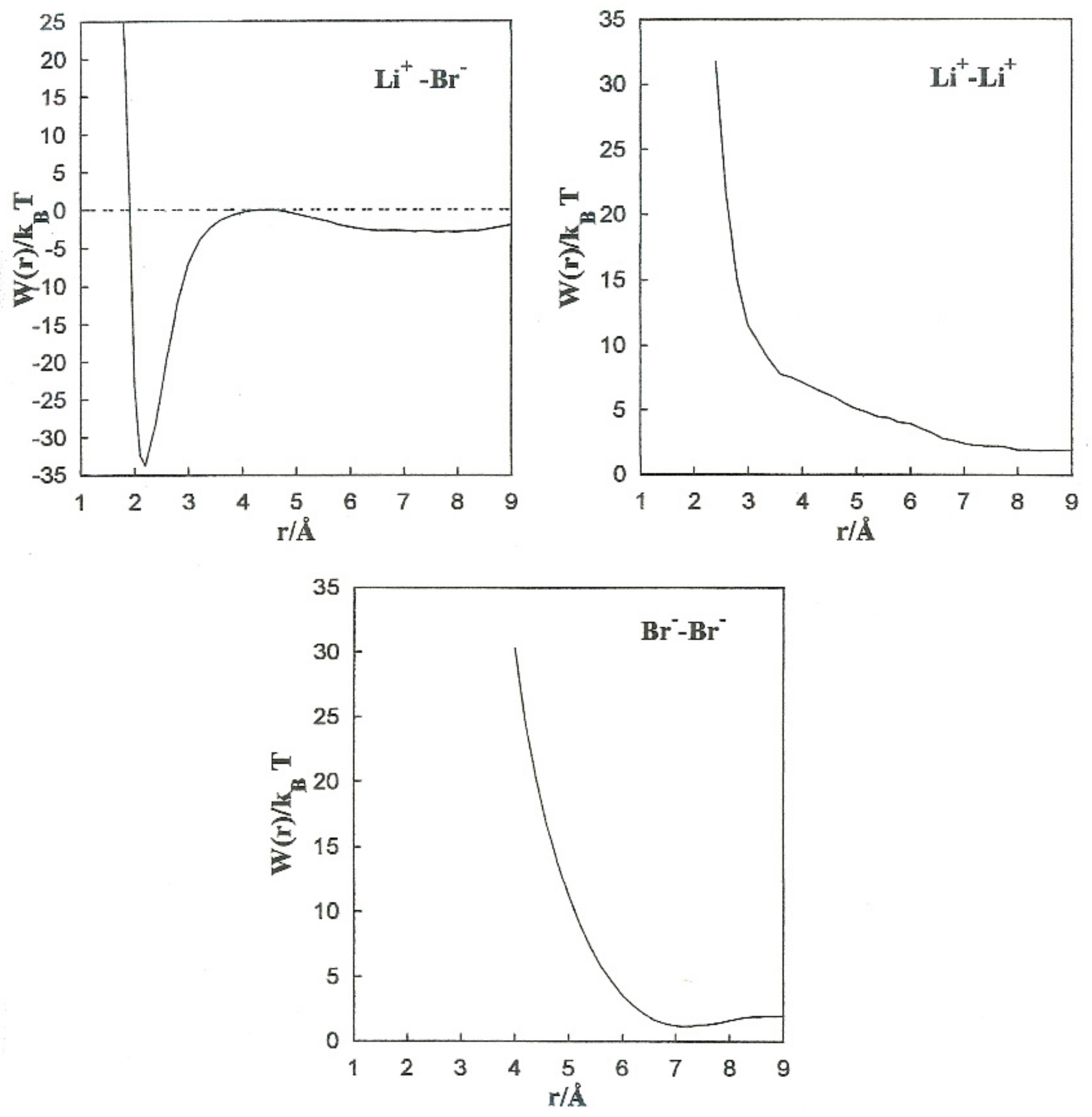
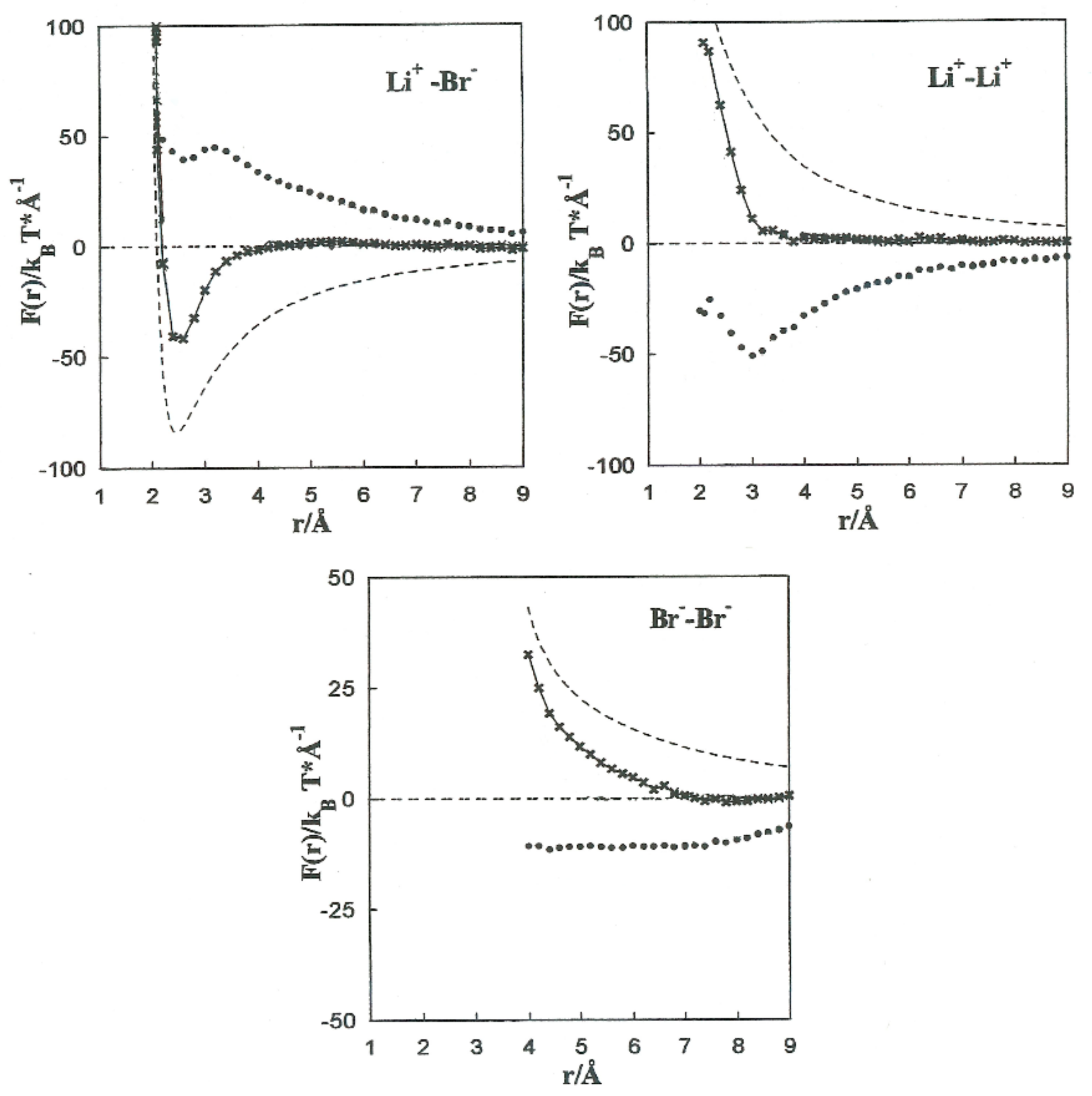
The MF acting on every single ion is shown in
Figure 4, jointly with the direct (
Fd) and solvent (∆
F(
r)) contributions to the Li
+-Br
−, Li
+-Li
+, and Br
−-Br
− pairs. The contributions of the solvent (∆
F(
r)) are opposite to the direct ion–ion force in all the systems.
This is constant with the trend of polar liquids to produce stable complexes in the case of like-ion pairs, and to separate ions with opposite signs [
2,
3,
4,
5]. Equilibrium between the direct and solvent contributions results in a repulsive force acting on the Li
+-Li
+ pair at all distances. For the Li
+-Br
− pair, the resultant force is repulsive at extremely small distances and attractive at middle separation. The total force on the Br
−-Br
− pair has similar tendencies, but the attractive component is considerably less significant.
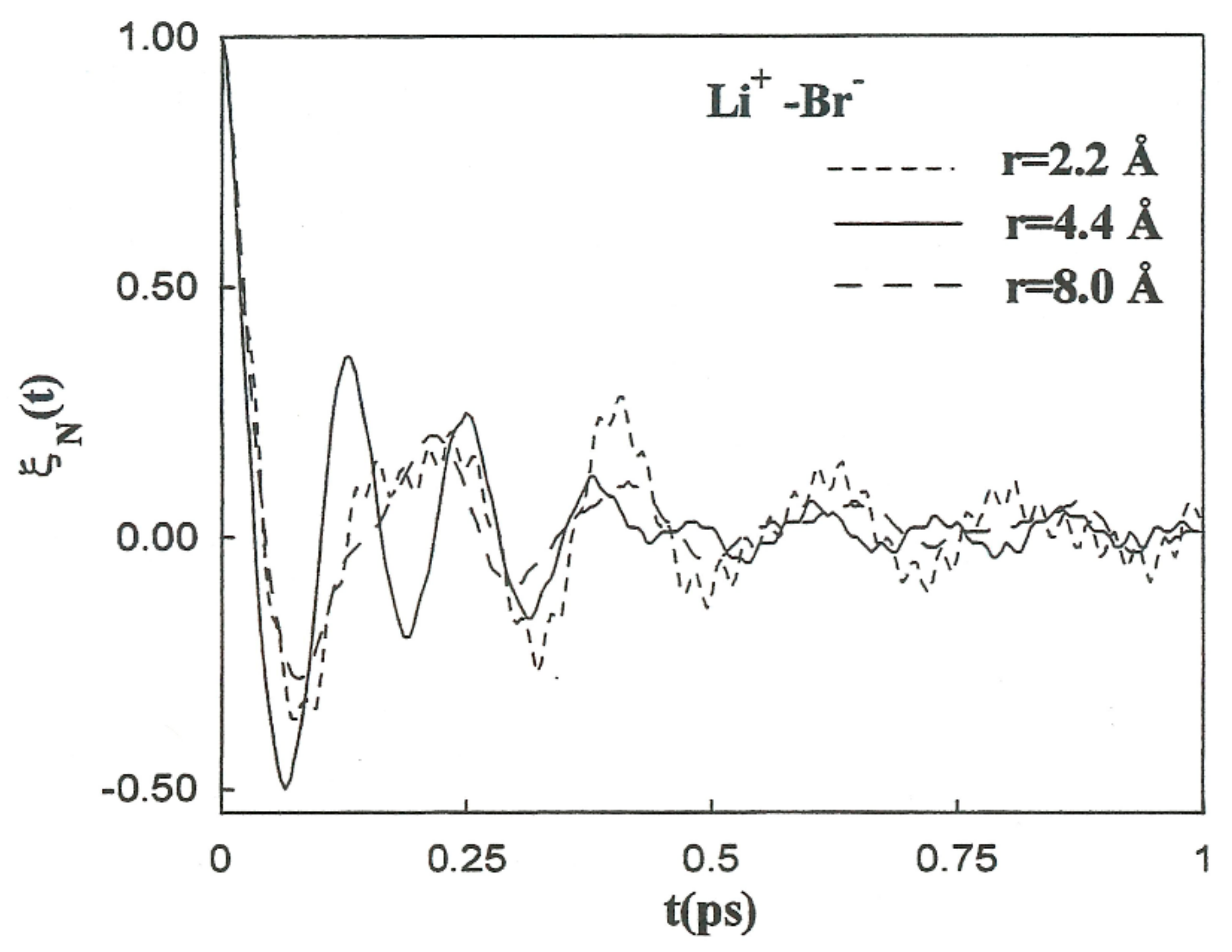
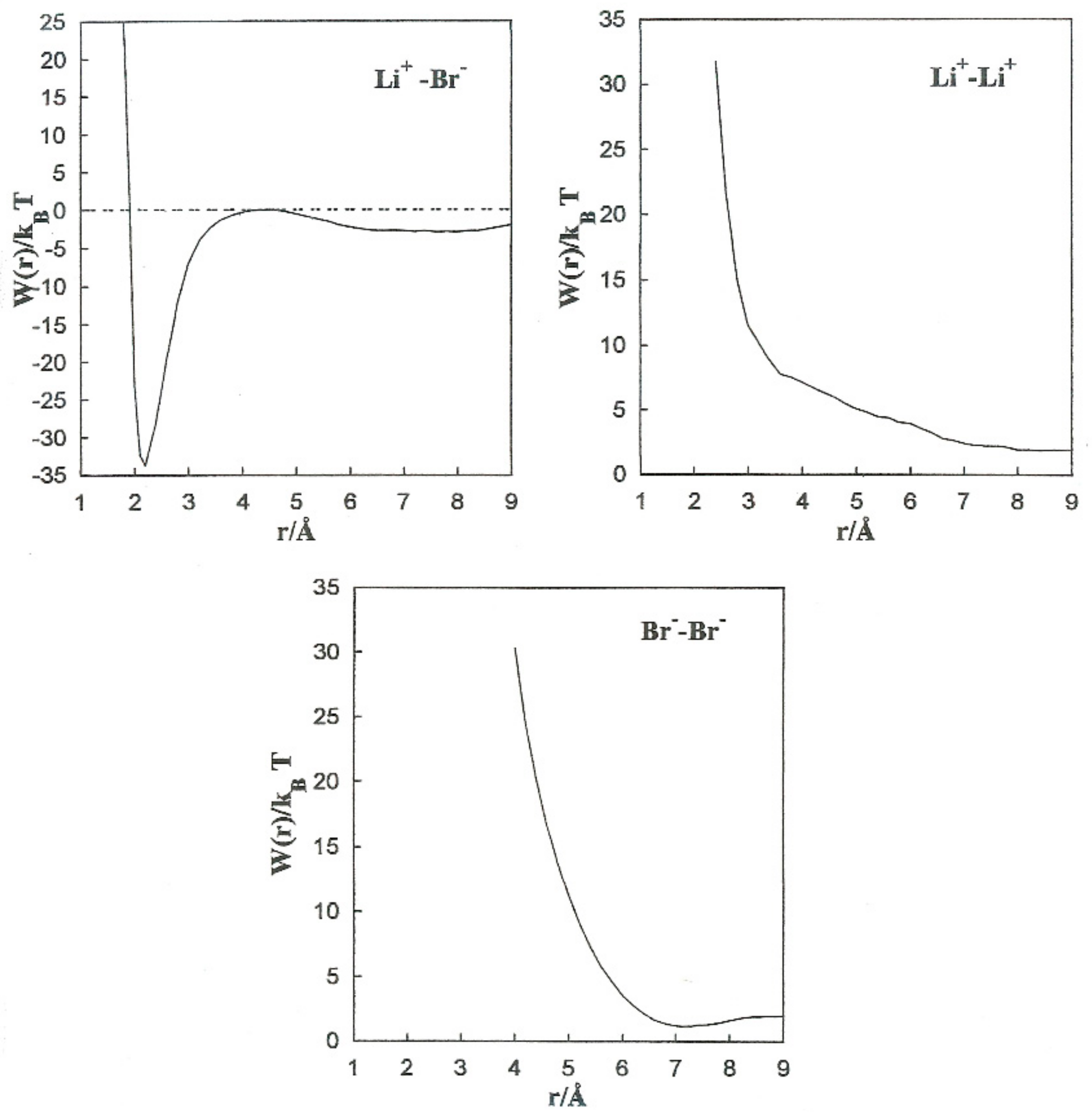
Figure 5 shows the PMF values for the various ion pairs. Oscillations in the potential corresponding to Li
+-Br
− occur in association with the structure of the solvent in the vicinity of the ions.
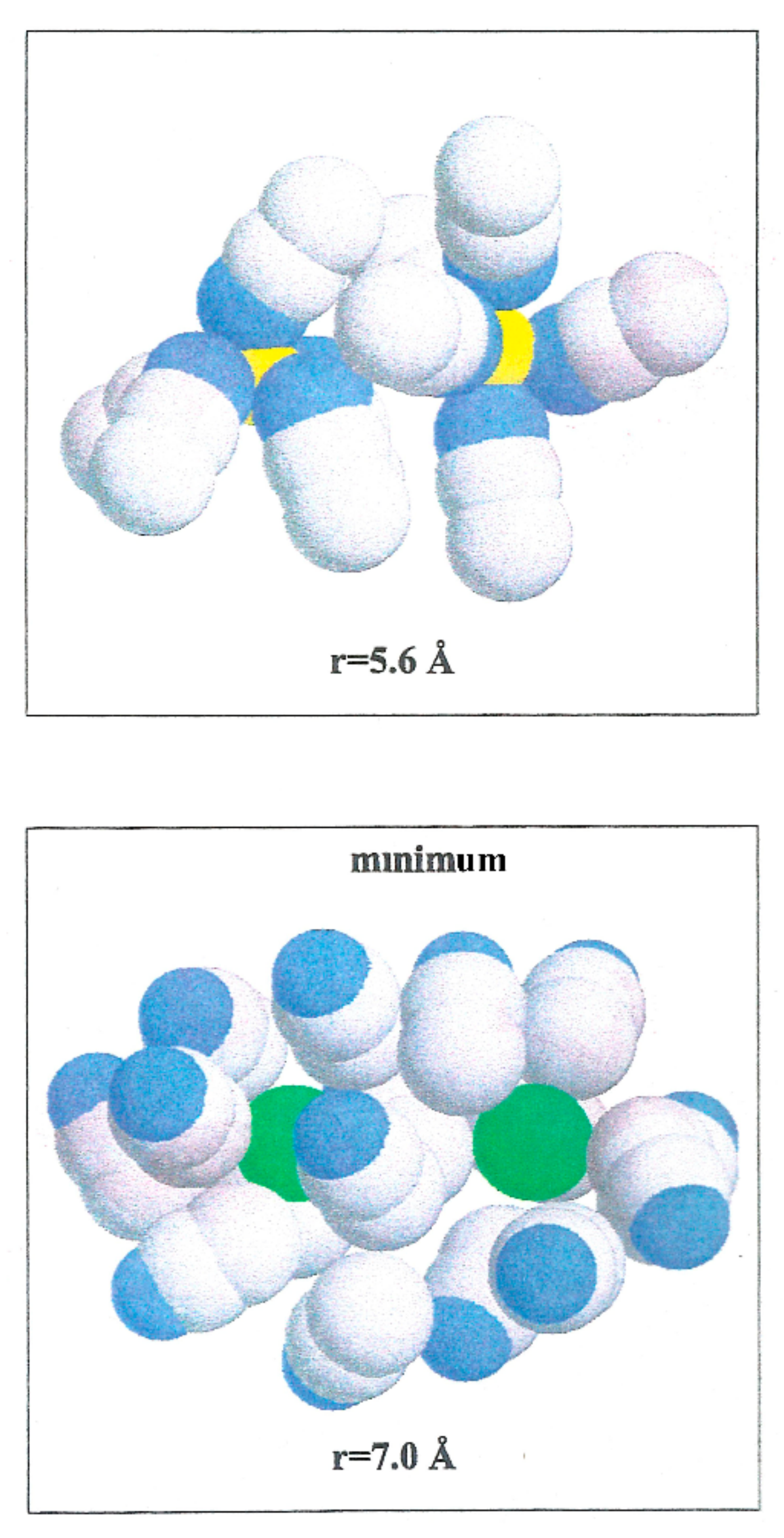
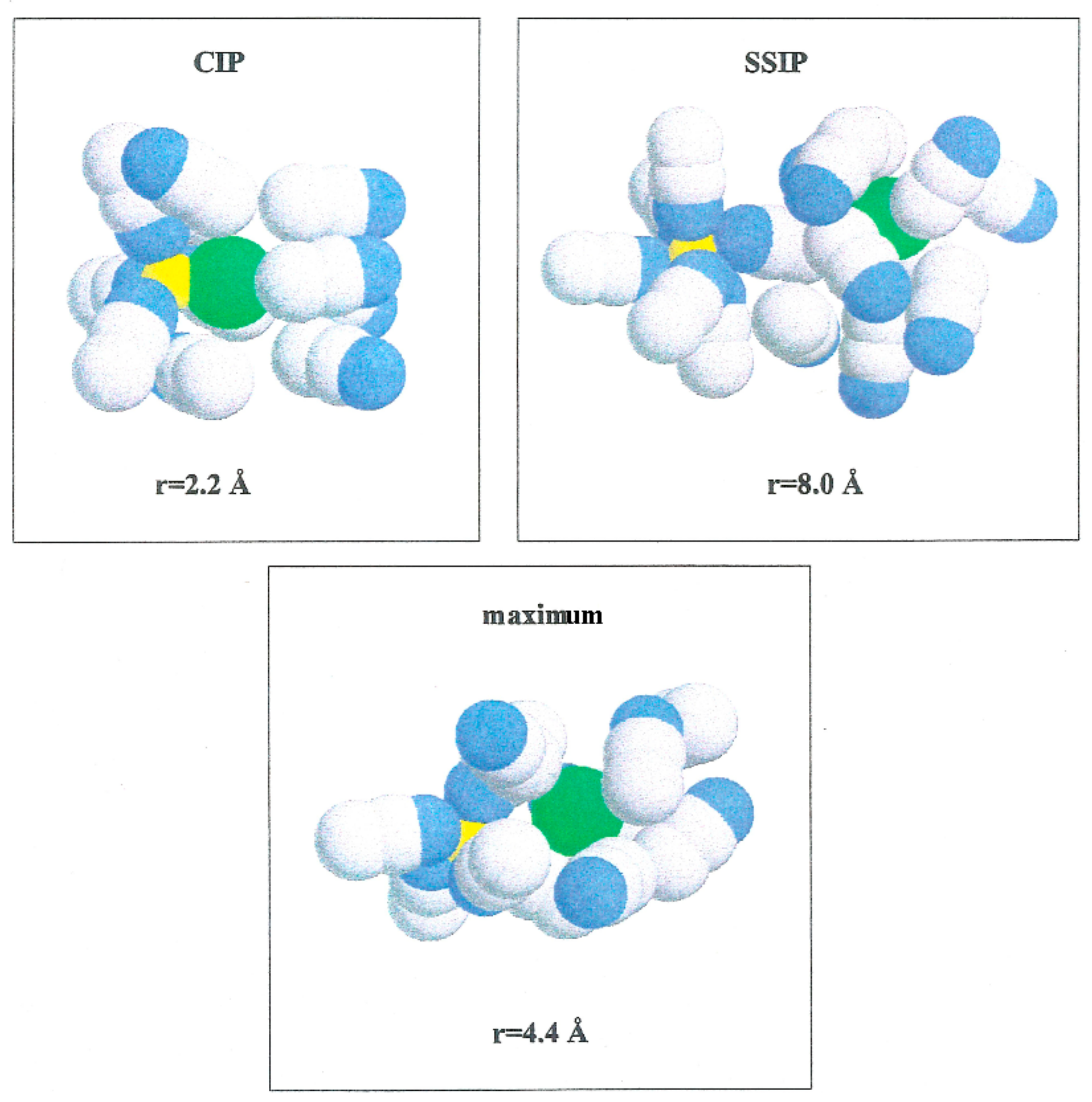
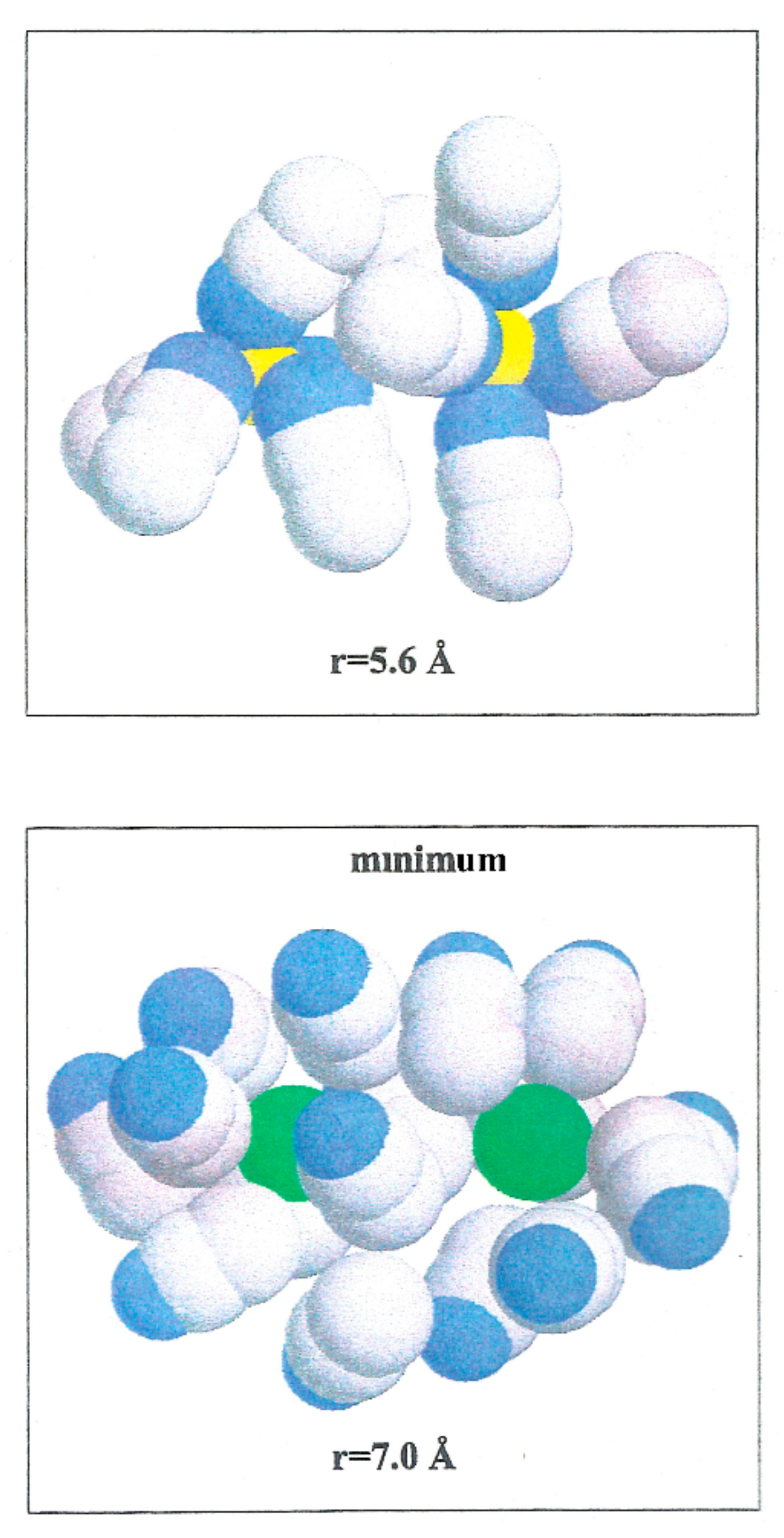
W(r) shows a noticeable minimum at 2.2 Å and lower one at 8.0 Å. The contact ion pair (CIP) region corresponding to the first minimum, and for the solvent separated ion pair (SSIP) region the second one.
W(r) shows a maximum at
r = 4.4 Å. In the CIP region, the ion pairs are enclosed by acetonitrile particles, with the corresponding nitrogen atoms geared to the Li
+ and their methyl groups in the direction of Br
−, as illustrated in
Figure 6. The SSIP region values are attributed to attractive forces produced by solvent molecules between ion pairs.
The stability of the CIP and SSIP regions is indicated by the association constants [
25]. For the CIP and SSIP state we have the following expressions
where
is the Avogadro’s number and
is a cut-off parameter that we have chosen as the position of the first potential of mean force
W(r) maximum, and
is some value of
r for which it is considered that the diffusive motion is a good approximation. Moreover, the equilibrium constant is giving by
we calculate the association constants
and
and the equilibrium constant, as shown in
Table 6. We found that
. The value obtained from the equilibrium constant (
) indicates much less stable configurations in the SSIP region compared to the CIP region.
W(r) for Li
+-Li
+ is repulsive and decreases monotonically to zero. Even though the acetonitrile molecules located near the ion pairs have their nitrogen atoms toward Li
+, as shown in
Figure 7, the electrostatic interaction N-Li
+ is less important than
Fd.
The Br−-Br− pair conformation is steadied at a separation corresponding to the minimum (r = 7.0 Å) of W(r) by the acetonitrile molecules enveloping it. Their methyl atoms are oriented toward the Br− ions due to an attractive interaction. The structure is stabilized by the methyl atoms of the liquid molecule in the central area.
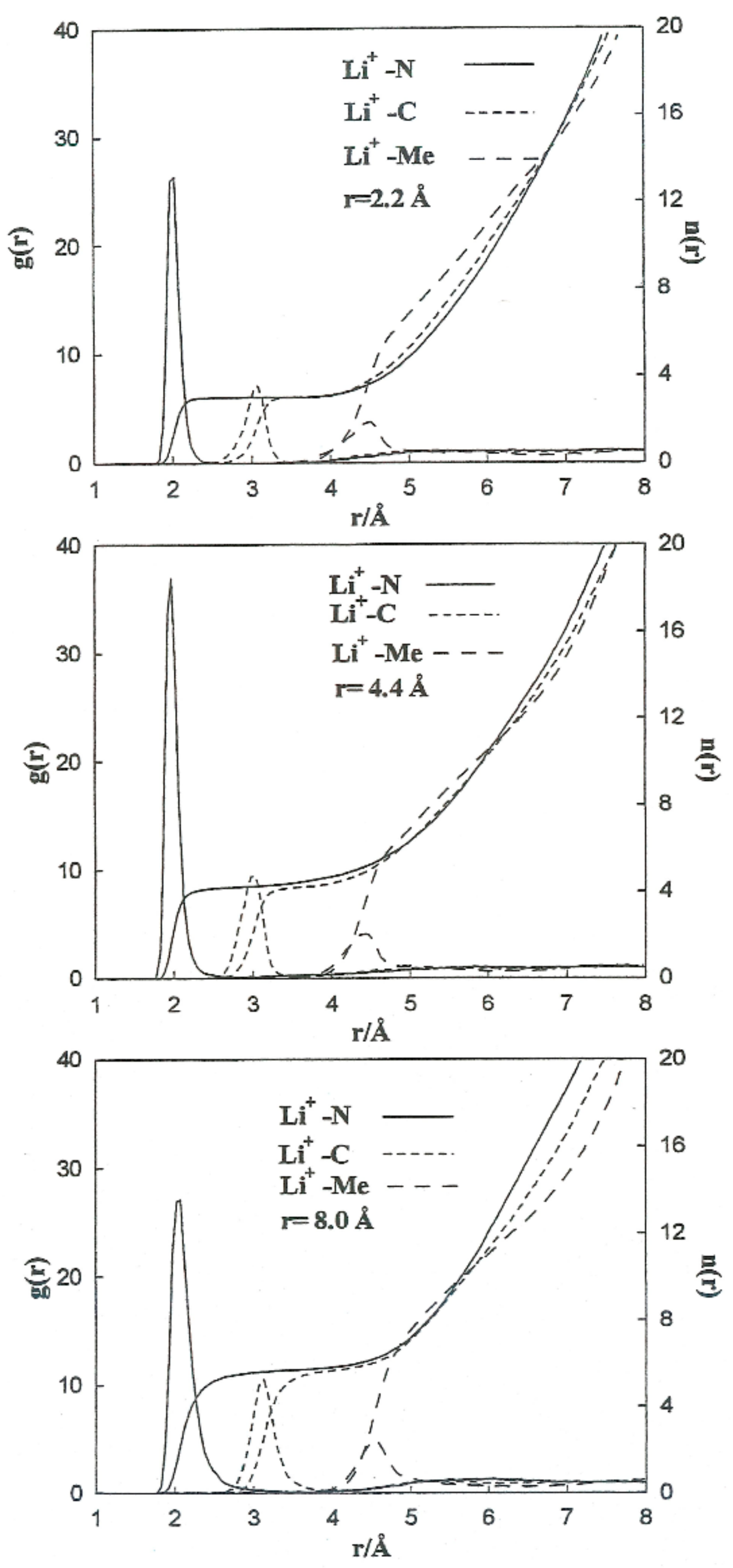
To carry out a more quantitative analysis of the distribution of the liquid molecules around the ionic particles, we determined the radial ion–solvent distribution functions (left axis) and their corresponding coordination numbers (right axis) for different interionic separations.
Figure 8 and
Figure 9 show the radial distribution functions,
Ion-Me,
Ion-C and
Ion-N, obtained in the situation of the Li
+-Br
− ion pair for the interionic separations corresponding to
rCIP,
rSIP and the maximum value of
W(r).
Table 7 summarizes the positions of the maximum and minimum radial distribution functions for the various cases analyzed. The corresponding coordination numbers are given in
Table 8. A coordination number (
) of 23 for a free bromide ion in acetonitrile is large. This value may be attributed to the deficiency in the interaction potentials used. Nevertheless, other studies indicate that the solvation shell of the Br-anion can have a maximum number of molecules of acetonitrile bigger than 10 [
9]. It should also be pointed out that in aqueous solutions coordination numbers increase when the concentration decrease [
26].
3.3. Li+-Br− Interconversion and Friction Kernels
The friction kernels
depend on the ionic species and interionic distances [
27].
We determined the transmission coefficient (
) of the barrier at 4.4 Å according to the Grote–Hynes theory of chemical reactions in a solution. In this model, the reaction coordinate develops by a global Langevin equation. The transmission coefficient (subscript
GH or
Kr stand for Grote–Hynes or Kramers) can be obtained as:
where
is the barrier frequency, obtained by extrapolating an inverted parabola with an apex equivalent to the maximum PMF value.
(reactive frequency) is the result of the next expression:
It is follows that
is the characteristic frequency for the unstable motion at the transition state. For the coefficients involved in the previous theory regarding the Li
+-Br
−, Li
+-Li
+, and Br
−-Br
− systems, the initial values of the friction kernels (
are summarized in
Table 9. The initial time values of the
kernels are not only dependent on the ionic species but also show noticeable changes with the interionic separation. This interionic separation dependence of
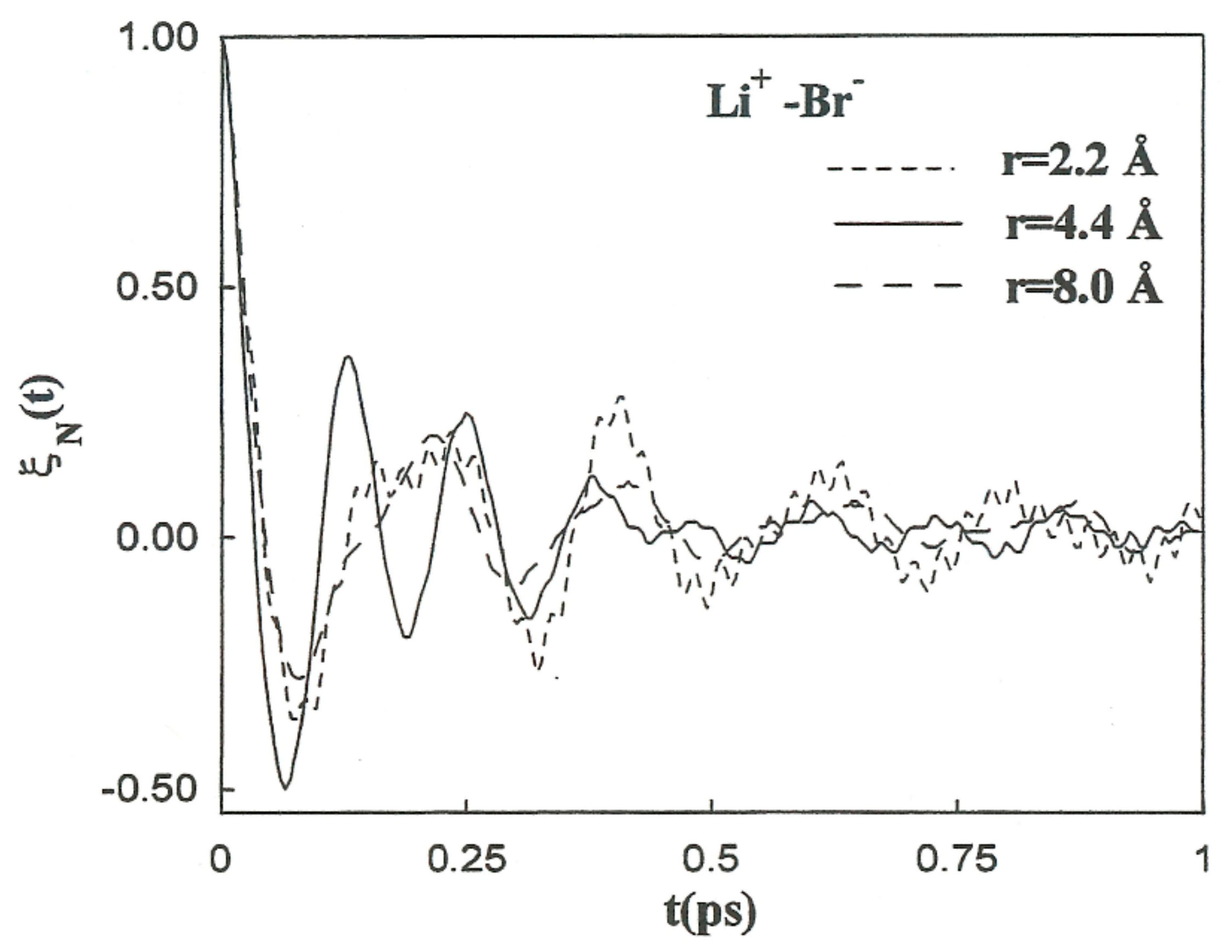
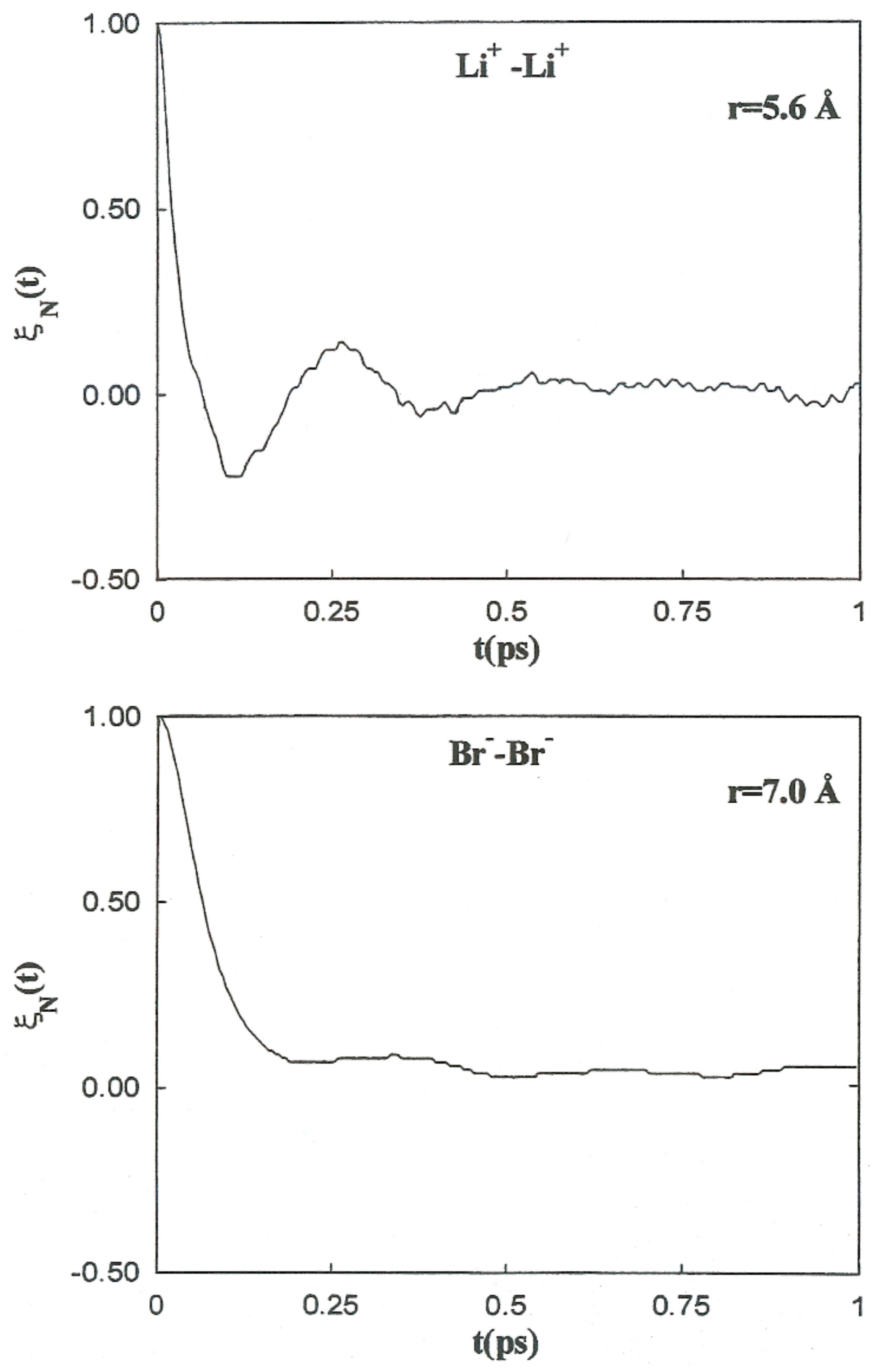
is strongly related to the magnitude of the changes in the solvent structure around the ion pairs as a function of these separations. The normalized friction kernels (
) related through all cases are displayed in
Figure 10 and
Figure 11. There are features common to the functions of both systems, all of which show a rapid and very similar initial decay before 0.2 ps. A lengthy decay follows, characterized for each individual system.
The coefficients involved in the Li
+-Br
− system are summarized in the
Table 10.
, gives us a temporal measure of the transition state in the presence of the solvent. We determined that, for acetonitrile, .
The correlation time of the friction of the solvent acting on the ion pair (
is defined by the value of the integral of the normalized kernel function:
In the case of acetonitrile, the correlation time is shorter than the reaction time.
A polarization caging regime is verified because a negative amount for the nonadiabatic barrier frequency (
was found [
28]. In this regime, the solvent immediately catches the solute in a well or “polarization cage” of solvent molecules. Hence, the motion of solvent molecules is extremely crucial. A limiting case of the polarization caging regime is when
and designated as adiabatic. In such circumstances, the predictions of the Grote–Hynes theory are reduced to Kramers doublets. Our results confirm that for the Li
+-Br
− ion pair,
.
Lastly, we computed the full rate constants for the dissociation (
) and association (
) processes, given by [
27,
29]:
whereby
is the reduced mass of the ion pair,
is the transmission coefficient,
is the interionic separation at the transition state (i.e., the top barrier),
and
is an arbitrary interionic distance beyond the outer boundary of the SSIP region, where there is no further interactivity. The results are summarized in
Table 10. We have obtained a
rate constant that is roughly smaller than
. This is due to the greater stability of the CIP complex for the present model system since a value of
was obtained for
. Our overall constant rate
corresponds to a relaxation time of approximately 835 ps. Values for
were found. In such circumstances, the predictions of the Grote–Hynes theory are reduced to Kramers doublets. From the transmission coefficient values (
) results confirm the case called regime adiabatic and for the Li
+-Br
− ion pair,
. A polarization caging regime is verified because a negative amount for the nonadiabatic barrier frequency (
was found.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}