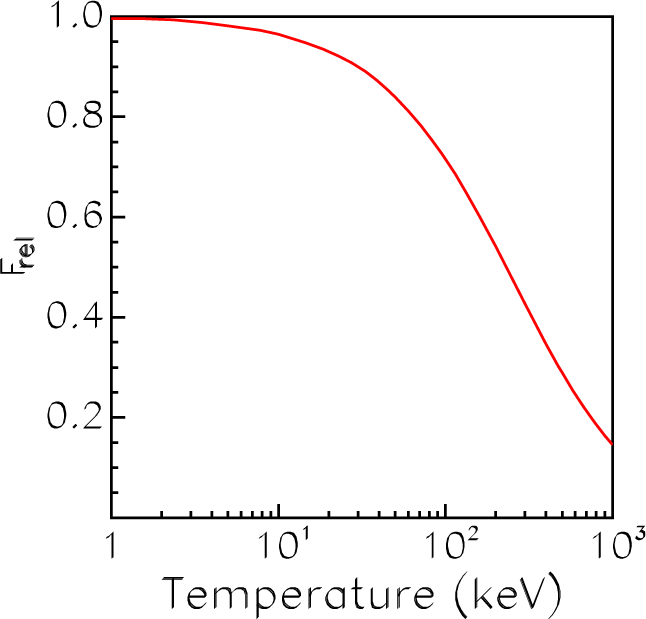
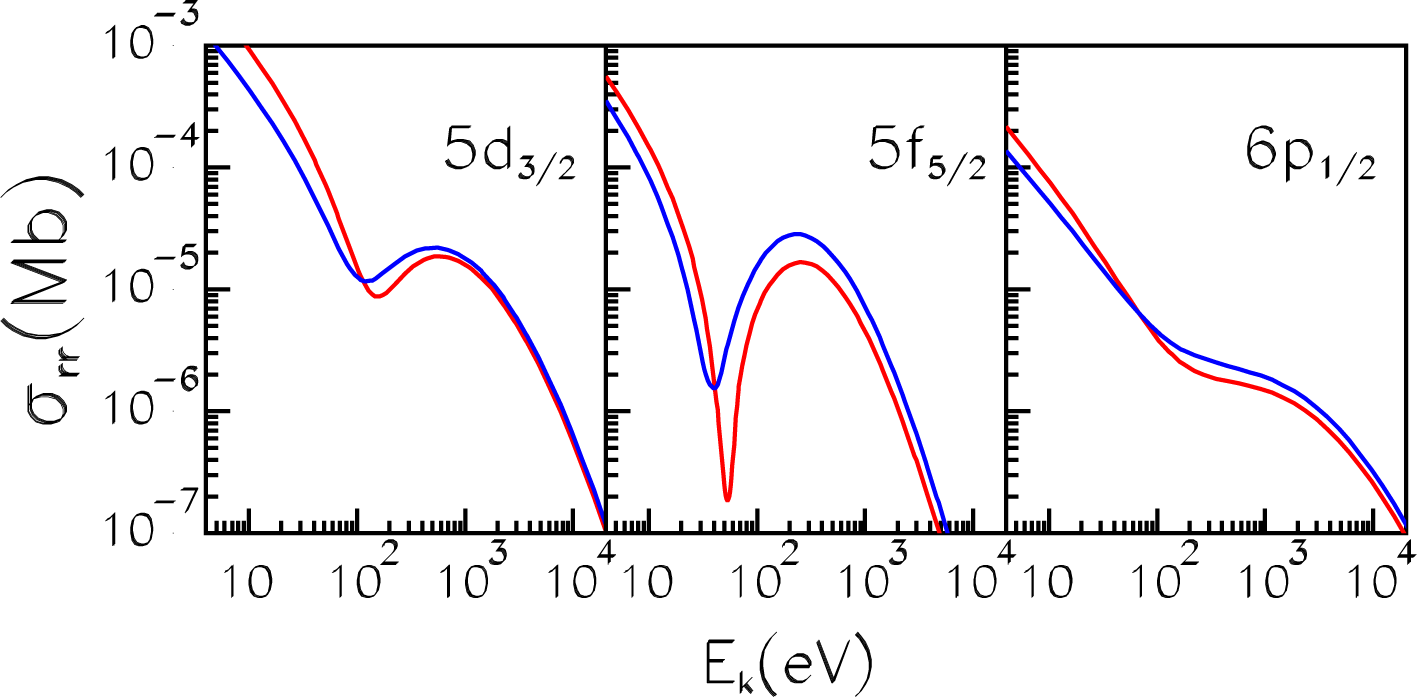
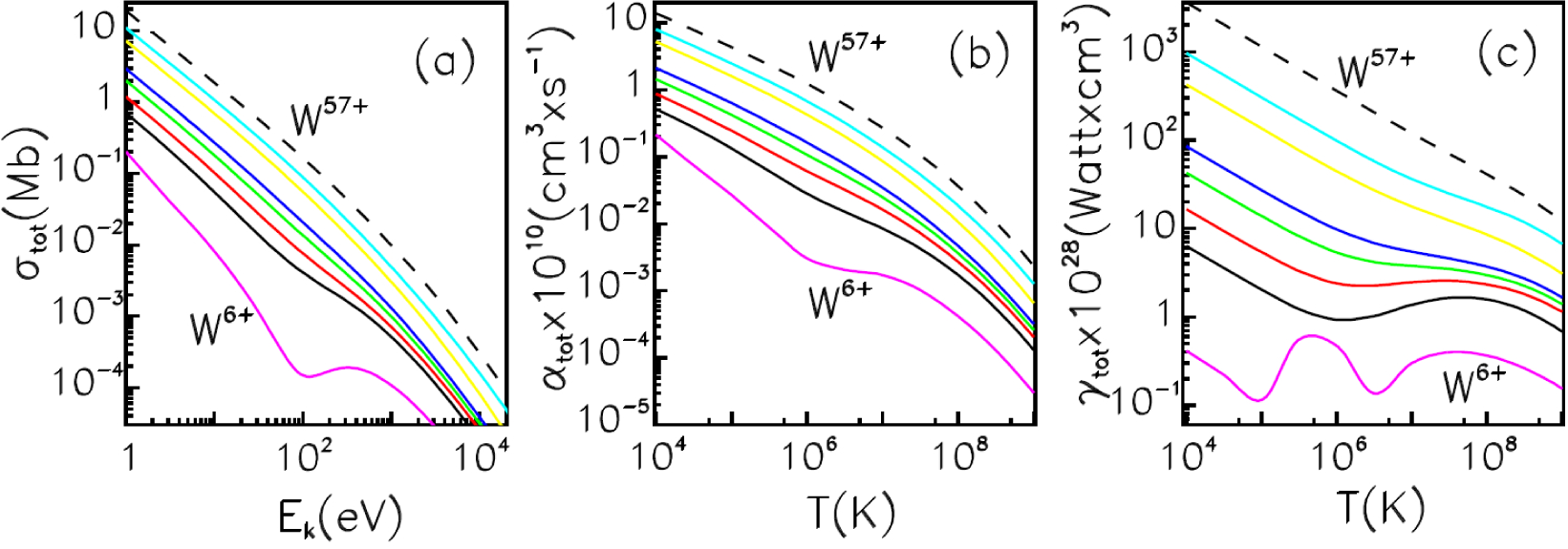
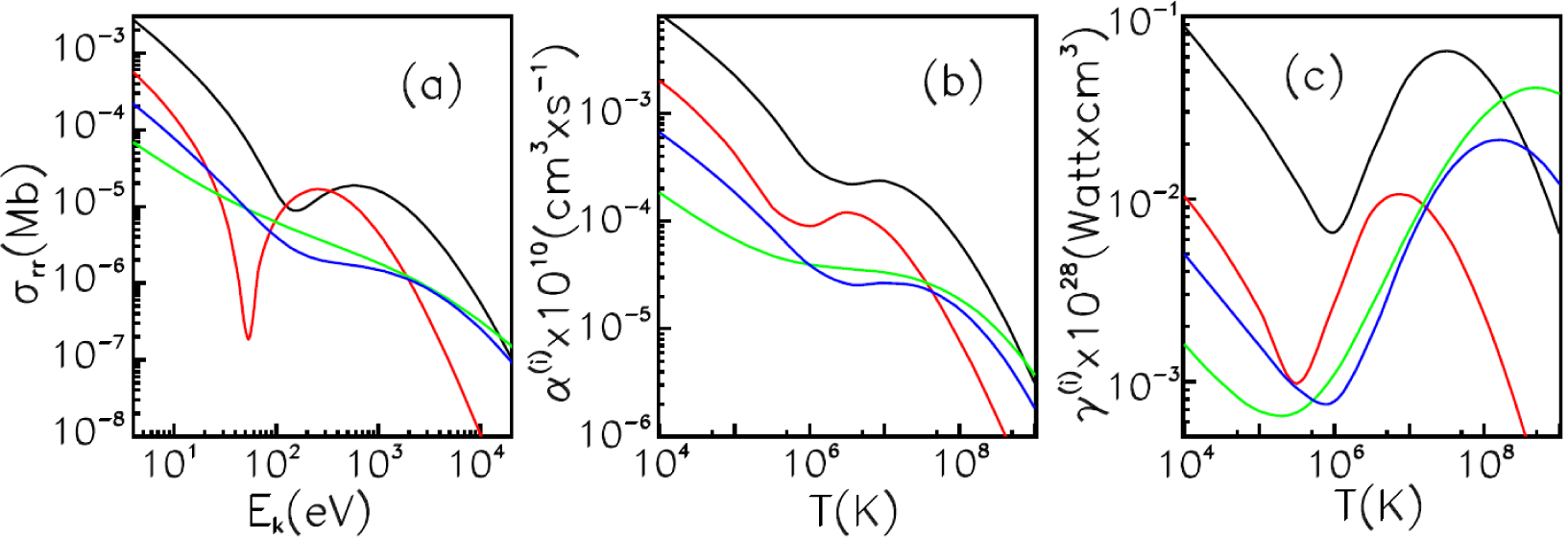
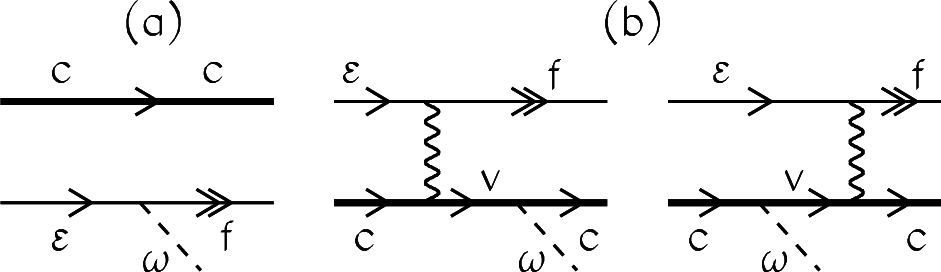
4.1. Average-Atom Model
To study the electron structure of ions in LTE plasmas, the code PLASMASATOM has been designed on the basis of our computer program complex RAINE [
16–
18]. The code PLASMASATOM is based on the average-atom model. The model has been applied in the Los Alamos code INFERNO [
19,
20], code PURGATORIO [
21] and the more advanced code PARADISIO [
22].
In the average-atom model, the plasma is taken to consist of the neutral Wigner–Seitz (WS) cells [
34,
35]. Each of them contains a nucleus with a charge
Z and
Z bound and continuum electrons. The bound wave function and its derivative coincide with those of a neighboring atom. The continuum density is finite at the WS cell boundary and merges into the uniform free-electron density outside the cell. Therefore, we treat an isolated neutral cell in a local thermodynamic average sense, neglecting the interaction of the cell with other ones. The radius of the WS cell
RWS is determined from the material density and atomic weight.
In the following studies, we used the relativistic DS method where an electron is assumed to satisfy the system of the Dirac central-field equations:
Here,
E is the total electron energy, and
V (
r) is the electron potential energy. The potential with the exchange term in the local density approximation may be written as:
where
ρ(
r) is the total electron density:
The bound density contribution
ρb(
r) is written as:
where the summation is over all bound states, and the Fermi–Dirac factor
fi(
εi, μ) is given by the Fermi distribution:
Here,
εi = 1
− E < 0 is the electron binding energy,
μ is the chemical potential and
kβT is the temperature. The occupation number of the i-th level is determined by:
Bound electron wave functions are normalized so that:
The continuum density contribution
ρc(
r) has the form:
Here,
ε =
E − 1 > 0, and the associated Fermi–Dirac factor
f(
ε, μ) is given by:
Continuum wave functions are normalized per unit energy interval, so that:
The chemical potential
μ appearing in
Equations (28) and (
32) is determined provided that the cell with the radius
RWS is electrically neutral:
The sum over
κ in
Equation (31) converges slowly. Because of this, to eliminate the need of a direct calculations of the sum, we transform
Equation (31) by the method, which makes it possible to perform the summation over
κ not to infinity, but only for a few values of |
κ| ≤ |κmax| [
36]. In the case of the DS method and for our notations,
Equation (31) may be rearranged by the following way. Where the influence of the potential
V (
r) is negligible, the continuum wave function normalized according to
Equation (33) may be written as:
where δ(
r) is the phase shift,
,
is the normalization factor and
jℓ and
yℓ are the spherical Bessel functions of the first and second kind, respectively.
Setting in
Equations (35) δ = 0, we arrive to functions
G(
r) and
F (
r) for a free wave:
Further, we replace the summation over
κ in
Equation (31) by two sums:
In addition, to accelerate the sum convergence, we subtract from the real density
the relevant density for a free wave
in each
κ-term of the first sum in the right part of
Equation (37). To compensate these terms and to include the second sum in
Equation (37), we add the total sum of such terms for
±1
≤ κ ≤ ±∞. Then, the expression for
ρc(
r) takes the form:
Terms in the second line of
Equation (38) compensate the sum over
±1
≤ κ ≤ ±|κmax| for a free wave, which we have subtracted, as well as include approximately the sum over
±(|
κmax| + 1)
≤ κ ≤ ±∞. It is easy to check that:
Taking into consideration
Equations (33), (
39) and (
40), we arrive at the following expression for the continuum density:
It is instructive to evaluate how many terms have to be taken into account in sum over
κ in
Equation (41). We present in
Table 5 results obtained with various values of |
κmax| for the iron ion at temperature 100 eV and
RWS = 2.672 a.u. Data of
Table 5 demonstrate differences in a third or fourth significant digit of the
εi,
Ni and
μ magnitudes obtained in calculations having regard to |
κmax| = 10 and |
κmax| = 15. This means that the difference between the two calculations is less than 0.5%. We checked that further increasing |
κmax| has no influence on the results. Therefore, the value |
κmax| = 10 is slightly lacking to give a high accuracy, while |
κmax| = 15 is quite enough. Consequently, adoption of
Equation (41) for
ρc(
r) permits one to restrict |
κmax| = 15, while the direct summation in
Equation (31) requires several tens of
κ-terms to reach the same accuracy.
The integral over
ε in
Equation (41) is evaluated up to
εmax, where
εmax is chosen so that the Fermi–Dirac factor is small:
where δ
ε = 10
−8. The integrand is calculated for 10
3 equidistant points
ε in the interval [0
− εmax]. This energy grid is used for calculation of the integral in
Equation (41) by the Simpson method. Calculations where the continuum density is based on
Equations (31)–(
42) will be refereed to as the DS-DS model.
To simplify and significantly accelerate the computational procedure, we also elaborated another version of code PLASMASATOM, where
ρc is evaluated within the framework of the semi-classical Thomas–Fermi (TF) approximation according to [
34]. In this case, a continuum density is written in atomic units as follows:
Here,
I1/2(
b, x) is the incomplete Fermi integral:
where:
and
Vex(
r) is the exchange term of the potential
V (
r). Calculations where the continuum density is based on
Equations (43)–(
46) will be refereed to as the DS-TF model.
In the both models, the SCF values of
V (
r),
ρ(
r) and
μ are found by the iterative method. The process starts from calculations for a neutral atom by the DS method without regard for a temperature. The initial potential
V 0(
r) constructed from the DS bound wave functions allows us to determine the initial density. Two initial values of the chemical potential
μ0 and
μ′
0 have to be specified so that:
Further, on the
n-th iteration, we calculate a root of
Equation (34) μn. Knowledge of a new value of
μn permits finding Fermi–Dirac factors
fi(
εi, μ) and
f(
ε, μ), then new densities
ρb(
r),
ρc(
r) and
ρ(
r), which permit, in turn, to determine a new potential
V n+1(
r). The iterative process is accomplished when the following condition is fulfilled:
The accuracy of SCF calculations is chosen as δV = 10−5.
In a general case, the iterative process is unstable. Because of this, the initial potential for the (
n + 1)-th iteration
V (n+1)i(
r) is determined using the initial and final potentials for previous
n-th and (
n − 1)-th iterations in the following manner. If the iteration number (
n + 1) is odd, the initial potential is determined as:
where the mixing coefficient
A is prescribed within the limits 0.2
≤ A ≤ 0.9.
If the iteration number (
n + 1) is even, the initial potential is calculated using the following scheme (see [
16] and the references therein):
If the iterative process diverges just the same, the mixing coefficient A should be increased.
4.2. Comparison with Previous Calculations
Our results were verified by comparing with calculations [
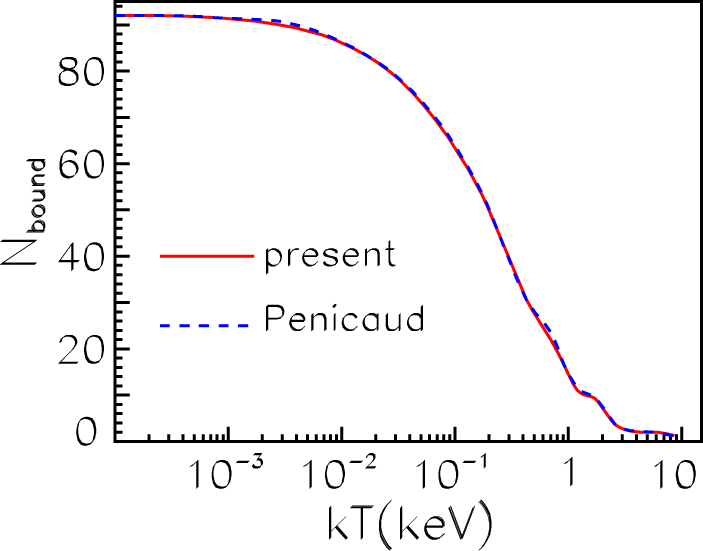
34] for iron. The bound, continuum and total densities for the iron ion in plasmas with the normal density at
kβT = 100 eV are shown in
Figure 10.
As is seen from
Figure 10, our results (red curves) are very close to the results from [
34] (blue curves). The slight differences occur in
ρb in ranges of maxima and minima where electron wave functions are usually very sensitive to all details of calculations and in
ρc near the very WS boundary.
Next, we compare our final results for the iron ion with calculations [
34]. Presented in
Table 6 are the spectrum of binding energies
εi, level populations
Ni (
Equation (29)), the chemical potential
μ, the ion charge
q and a number of bound electrons
Nbound in the resulting ion:
Our data were obtained using the DS-TF and DS-DS models. One can see that the results calculated by the DS-TF model correlate with data from [
34], where the same model has been adopted. Our binding energies are in excellent agreement with those from [
34]. The largest difference for the binding energy of the valence 4
s level is likely to be due to different boundary conditions. However, the difference in level occupation numbers reaches
∼12%. In addition, data of
Table 6 allow one to compare results obtained within the DS-DS and DS-TF models. As is seen, there is a minor difference between the two calculations. The largest difference (
∼5%) occurs for the binding energy of the valence 4
s shell. Values of
μ obtained in the two models differ by ≲0.3%, and values of the charge
q only by
∼0.2%. It should be noted that all levels and the chemical potential become lower, as well as the charge increases when passing from the DS-TF model to the DS-DS one.
As is well known, the DS and TF continuum densities diverge drastically, the DS density ρc(r) being an oscillating function, while the TF ρc(r) is a quite smooth function. Nevertheless, the results obtained using the DS-DS and DS-TF models are very close to each other.
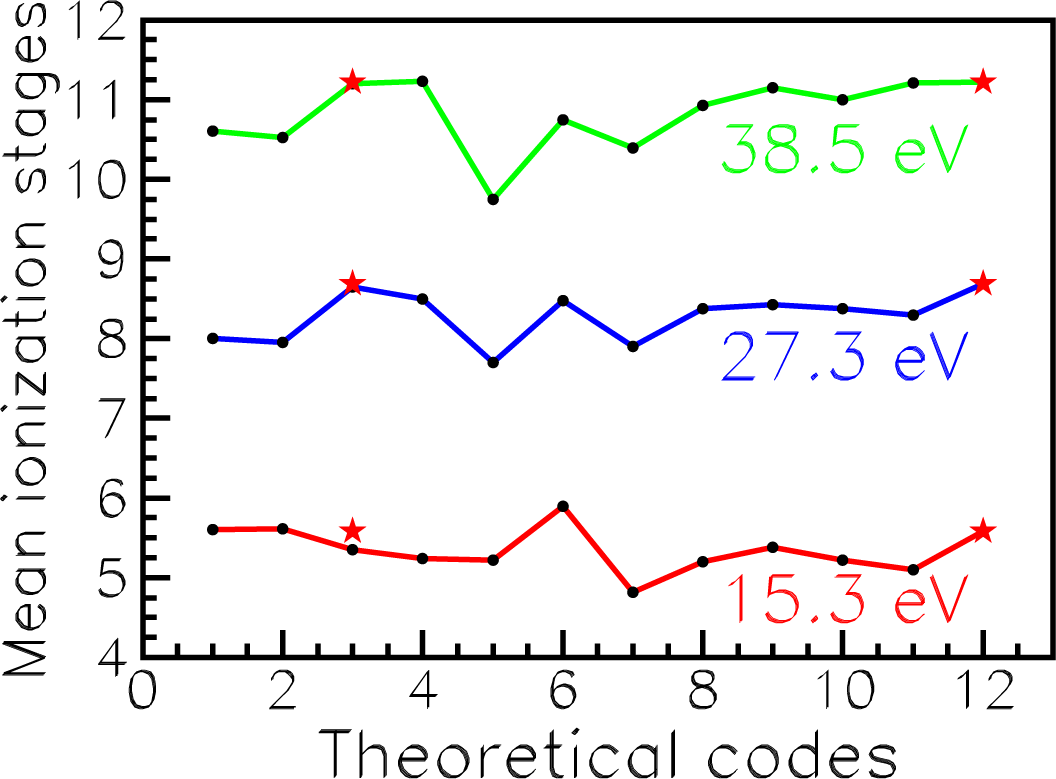
In addition, we compare our ion charge
q for iron in three cases listed in
Table 7 with mean ionization stages
< q > obtained by eight groups from Los Alamos, Livermore and with the data from Opacity Project (OPAC collaboration). The results have been calculated with different eleven codes to prepare LULI (Laboratoire pour L’Utilization des Lasers Intenses) 2010 experiments [
37]. The difference in the mean ionization stage obviously implies the discrepancy between the frequency-dependent opacity. Therefore, the data are of importance for astrophysics.
As is shown in
Figure 11, our values of
q (red asterisks
versus codes with numbers 12 and 3) correlate well with previous calculations. Besides, the data of
Table 7 show that our results are in excellent agreement with the best results [
37] of
< q > from OP. The largest difference is 4%.
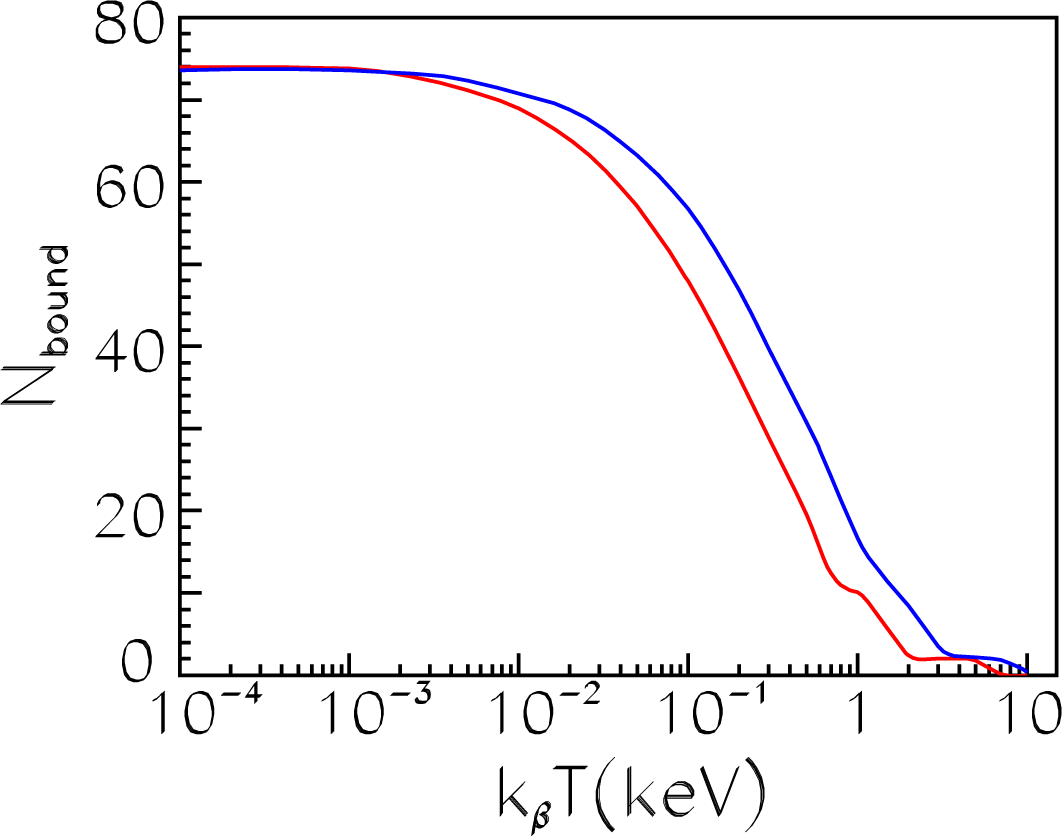
We also compared results for the heavy uranium ion with those obtained in [
22] using code PARADISIO. In
Figure 12, the number of bound electrons
Nbound (
Equation (53))
versus a temperature is presented for the uranium ion in plasmas with the density
ρ = 0.01 g/cm
3 (
Nion=2.5·10
19 cm
−3). As is seen, our calculation (red curve) is in excellent agreement with the previous results (blue dashed curve) in the wide temperature range 0.1 eV
≤ kβT ≤ 10 keV.
4.3. Results for Tungsten Ions
New calculations were performed within the DS-DS model for the tungsten ion in laser plasmas of two densities
ρ1 = 1.93 g/cm
3 (the ion density
Nion = 6.3
× 10
21cm
−3) and
ρ2 = 0.01g/cm
3 (
Nion = 3.3
× 10
19cm
−3). The spectrum of energies
εi and level occupation numbers
Ni are given in
Table 8 for the tungsten ion in plasmas with densities
ρ1 and the associated value of
RWS = 6.339 a.u. at temperatures 100 eV and 1,000 eV, as well as for
ρ2 and
RWS = 36.635 a.u. at temperature 100 eV. Comparing data for
ρ1 at different temperatures, it may be noted that the ion compresses when the plasmas temperature increases,
i.e., the levels become deeper and the outer shell occupation numbers decrease. As is seen from comparison data for various densities at
kβT = 100 eV, the ion compresses when the plasmas density decreases. As is seen from
Table 8, high electron states contribute significantly at the higher density and lower temperature. For example, the 5
f, 5
g, 6
s, 6
p, 6
d, 6
f and 7
d levels have occupation numbers from
∼0.1–
∼0.5 in the case of
ρ1 and
kβT = 100 eV. Occupation numbers for all levels decrease with the temperature increasing and the density decreasing.
The temperature dependence of
Nbound is presented in
Figure 13 for the two densities. The blue curve refers to
ρ1 and the red curve to
ρ2. A comparison between the two curves gives an idea of the plasmas density dependence. As is seen, the red and blue curves are not too different, even though the associated densities differ by
∼200-times. Increasing of a plasmas density shifts the curve
Nbound(
kβT) to higher temperatures.
To study tungsten impurities in fusion plasmas, we use the non-linear SCF screening model [
39,
40] for the calculation of the screening impurity potential. In the model, impurities in plasmas are considered as neutral pseudo-atoms.
RWS is assumed to be large. The chemical potential
μ is found before the SCF calculations on the basis of the prescribed values of the plasmas electron density
Ne and temperature
kβT using the following expression [
40]:
We consider the typical fusion plasmas density Ne = 1014cm−3 and the low temperature range 1 eV ≤ kβT ≤ 5 eV. The SCF potential V (r) and the total electron density ρ(r) are found by the iterative process described above. The DS-DS model is used.
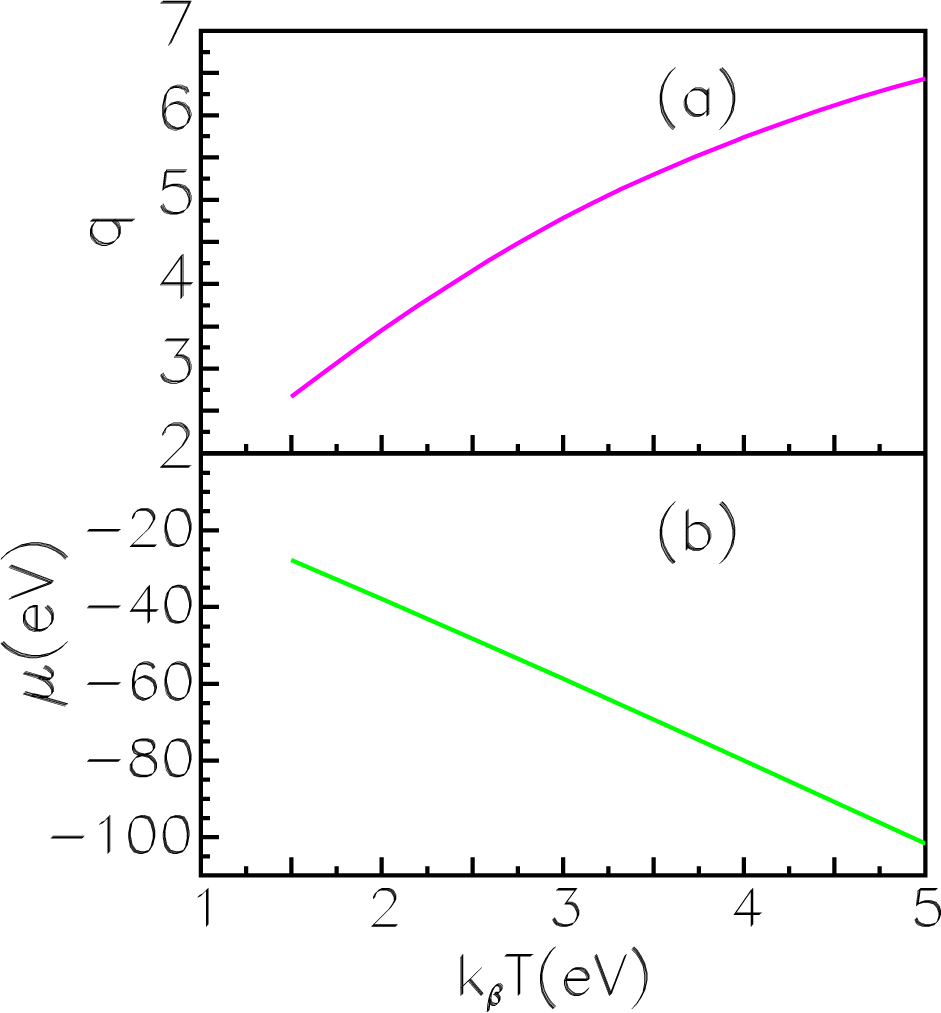
In
Figure 14, the ion charge
q (14a) and the chemical potential
μ (14b) are displayed. As is evident, values of the charge and chemical potential change noticeably when the temperature increases. In
Table 9, we present spectra obtained for the tungsten ion at various values of temperature. The case
kβT = 0 refers to the usual DS calculations for a free neutral tungsten atom. One can see that the increasing of temperature causes all binding energies of inner levels to become lower by approximately the same value. Outer levels also become lower, relatively to a greater extent. Consequently, the energy spectrum depends considerably on a plasma temperature, the changes being different for inner and outer levels. Calculations showed that only valence 5
d3/2 and 5
d5/2 states have large occupation numbers, while other excited states involved in calculations with regard to a temperature, for example 5
f, 6
d, 6
f, 7
d, 7
s and 7
p, have zero occupation numbers. The 6
s and especially 6
p states have very small occupation numbers (≲ 0:1), which decrease when temperature increases.
The data of
Table 9 demonstrate different results for the free neutral atom (
kβT = 0) and for calculations with regard to a temperature. Nevertheless, this was just calculations for a free neutral atom, which were adopted as the initial data, as for instance in [
41], where the non-LTE calculations in the collisional radiative model were performed. The average ionization stage
< q > = 2.07 was obtained in [
41] for the tungsten ion at the electron density
Ne = 10
14 cm
−3 and
kβT = 2 eV. It was also shown that the largest contributions were made by transitions 5
d36
s1 → 5
d36
p1 and 5
d4 → 5
d36
p1. This means that the 5
d, 6
s and 6
p states are of primary importance in [
41] as in our calculations. We obtained the ionization stage
q = 3.45. Therefore, we believe that our results could be used as initial data in more sophisticated calculations rather than data for a free neutral atom. This may considerably change the results of these calculations.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}